Strong lattice anharmonicity and glass-like lattice thermal conductivity in nitrohalide double antiperovskites: A case study based on machine-learning potentials Download PDF
Yuan Li
1

,
Weidong Zheng
2
,
Jin Huan Pu
1,3,*
,
Qi Wang
4,5,*
,
Jian-Hua Jiang
4,5,6,*
*Correspondence to:
Jin Huan Pu, Institute of Thermal Science and Technology, Shandong University, Jinan 250061, Shandong, China.
E-mail: j.pu@sdu.edu.cn
Qi Wang, School of Biomedical Engineering, Division of Life Sciences and Medicine, University of Science and Technology of China, Hefei 230026, Anhui, China. E-mail: qiw@ustc.edu.cn
Jian-Hua Jiang, School of Biomedical Engineering, Division of Life Sciences and Medicine, University of Science and Technology of China, Hefei 230026, Anhui, China. E-mail: jhjiang3@ustc.edu.cn
Qi Wang, School of Biomedical Engineering, Division of Life Sciences and Medicine, University of Science and Technology of China, Hefei 230026, Anhui, China. E-mail: qiw@ustc.edu.cn
Jian-Hua Jiang, School of Biomedical Engineering, Division of Life Sciences and Medicine, University of Science and Technology of China, Hefei 230026, Anhui, China. E-mail: jhjiang3@ustc.edu.cn
Thermo-X. 2025;1:202501. 10.70401/tx.2025.0001
Received: April 24, 2025Accepted: July 04, 2025Published: July 10, 2025
Abstract
Antiperovskites have attracted significant interest in the field of energy conversion in recent years. While extensive research has focused on the magnetism, ionic conductivity and superconductivity of antiperovskites, their thermal properties including lattice anharmonicity and thermal transport remain less explored compared to their well-studied perovskite counterparts. Recently, nitrohalide double antiperovskites have been successfully synthesized. In this work, we investigate the thermal transport properties of nitrohalide double antiperovskites
Keywords
Lattice thermal conductivity, machine-learning potentials, molecular dynamics, double antiperovskite
1. Introduction
Perovskite materials, as prominent thermoelectric materials, excel in solar cells, microchips and thermoelectric applications due to their excellent photoelectric and tunable thermal properties[1-7]. Antiperovskites, the electrically inverted counterparts of perovskites, remain relatively underexplored in materials science, especially regarding their thermal properties. Recently, a new family of antiperovskite materials, double antiperovskites, has shown significant potential in energy-related applications. For example, X6SOA2 (X = Na, K; A = Cl, Br and I) exhibits stable zT (dimensionless figure of merit) over a wide temperature range, indicating outstanding thermoelectric performance suitable for energy storage and harvesting applications[8,9]. Li6OS(BH4)2 demonstrates stable cyclic performance and long lifespan, making it a promising novel solid-state electrolyte[10]. Simulations reveal that BiPCa3[11] and Mg3BiN[12] possess high zT values at elevated temperatures with good thermal stability, rendering them promising candidates for thermoelectric devices. Experimental measurements show that Ca3AO (A = Pb, Sn) also exhibits great potential for thermoelectric applications due to its large thermoelectric power and low electrical resistivity[13]. IIt has been demonstrated that both perovskite materials including oxide perovskite SrTiO3[14], double perovskites Cs2NaYbCl6[15] and Cs2AgBiBr6[16], whose strong lattice anharmonicity mainly originates from octahedral rotations and tilting, loosely bonded cations, or strong four-phonon scattering, and antiperovskite materials such as Li3OCl[17,18], Rb3X(Se & Te)I[19], and X(Ba & Sr)3BiN[20], where strong anharmonicity mainly arises from weak bonding, exhibit pronounced anharmonicity due to their unique crystal structures. Given their similar lattice structures, double antiperovskite materials are expected to exhibit strong anharmonicity as well. While extensive research has been devoted to thermoelectric materials for optimizing device performance[21,22], fundamental studies on the thermal properties of double antiperovskites remain scarce.
Many theoretical and experimental studies have been conducted to understand the thermal transport properties of strongly anharmonic crystals. Zhao et al.[23] investigated thermal transport in A3BO (A = K, Rb; B = Br, Au) using first-principles calculations combined with the Boltzmann transport equation (BTE), revealing strong quartic phonon scattering and a resultant weak temperature dependence of lattice thermal conductivity. Wang et al.[24] employed first-principles-based machine-learning potentials (MLP), the Wigner transport equation (WTE), and experimental measurements to study the thermal properties of halide double perovskites Cs2AgB(II)X5 (B(II): Pd or Pt; X: Cl or Br), demonstrating that unique atomic vibrations and atom rattling contribute to their strong anharmonicity. Liu et al.[25] examined thermal transport in Cu6Te3S through density functional theory (DFT) calculations and identified glass-like thermal conductivity caused by anharmonic vibrations of Cu atoms and overall weak bonding. In theoretical computational studies of strongly anharmonic materials, lattice dynamics calculations combined with the BTE or WTE based on first-principles are commonly used to obtain mode-resolved phonon transport properties. Although replacing first-principles calculations with MLP partially enhances the capacity to handle complex crystals, challenges remain due to the lower symmetry of strongly anharmonic materials, the breakdown of perturbation theory, and the cumbersome process of obtaining temperature-dependent high-order interatomic force constants (IFCs) and solving phonon scattering rates beyond three-phonon processes. Molecular dynamics (MD) simulations using MLP derived from first-principles calculations can capture full lattice anharmonicity while balancing computational accuracy and efficiency. These approaches have been applied extensively to thermal property calculations of materials such as metal-organic frameworks[26-28], water[29-33], and graphene[34-39].
Since the introduction of neural network potentials in 2007[40], MLPs have achieved great success in materials science. Leveraging data from first-principles calculations, methods including Gaussian approximation potential[41], moment tensor potential[42,43], neuroevolution potential (NEP)[44,45], recursive embedded-atom neural network[46,47] and various others employ diverse atomic environment descriptors and machine learning models to achieve both high accuracy and fast computational speed. Recently, the integration of high-performance graphics processing units (GPUs) has further accelerated MD simulations. For example, the NEP within the GPUMD package achieves a computational speed of approximately 1 × 107 atom steps per second on a single V100 GPU, outperforming other MLPs[26,48] and providing a powerful tool for investigating thermal transport properties in complex crystals[49].
In this work, we develop accurate machine-learning NEPs for the recently synthesized stable double antiperovskites Li6NII2 and Li6NBrBr2[50] using an active learning (AL) strategy[49] combined with DFT calculations. By incorporating thermal expansion and phonon anharmonic renormalization effects, we find that both harmonic and temperature-dependent phonon dispersion relations exhibit a significant portion of optical phonons with imaginary frequencies, indicating the failure of perturbation theory in describing phonon transport properties of these double antiperovskites. The calculated acoustic phonon branches of both materials at 300 K across the entire Brillouin zone remain below 2.5 THz, demonstrating low phonon energies and ultralow acoustic phonon group velocities. Large-scale MD simulations with long runtimes confirm the thermodynamic stability of Li6NII2 and Li6NBrBr2 and reveal confined Li-ion movements among lattice points within a single conventional unit cell. Thermal transport in these materials is further studied using both homogeneous nonequilibrium molecular dynamics (HNEMD) and EMD methods. Ultralow lattice thermal conductivities with glass-like temperature-dependent behavior are observed for both materials. Detailed phonon spectral analysis confirms that these thermal transport characteristics arise from the confined Li-ion motions, suppressed contributions, and weak temperature dependence of acoustic phonons.
2. Computational Models and Details
2.1 Construction of neuroevolution potentials with active learning
Previous studies have demonstrated the excellent performance of the NEP approach in investigating thermal transport in complex perovskite materials. In this work, we construct accurate NEPs for both Li6NII2 and Li6NBrBr2 based on first-principles calculations using an AL strategy[49]. DFT calculations were performed with the projector augmented wave (PAW) method[51] and the Perdew-Burke-Ernzerhof (PBE) generalized gradient approximation functional[52], implemented in the Vienna Ab initio Simulation Package (VASP)[53]. Initially, ab initio molecular dynamics trajectories of a 2 × 2 × 2 primitive cells for both materials were obtained at 300 K using VASP[54] under an isothermal-isobaric (NPT) ensemble with a timestep of 1 fs and 10,000 steps. Only the Г point was used for k-point integration, and the energy cutoff was set to 500 eV (convergence tests for the cutoff energy are presented in Figure S1). Configurations for the initial training dataset were sampled every 25 steps, resulting in a total of 400 configurations. Additional configurations were obtained through independent MD simulations at various temperatures using the AL strategy[55,56] with a timestep of 1 fs for both Li6NII2 and Li6NBrBr2. Furthermore, configurations subjected to strains ranging from -3% to 3% were also included following the same AL procedure implemented in the PYNEP framework[49]. The selection distance threshold for new configurations was set at 0.025. This select-and-train process was iterated until no new structures were selected after 2 ns of MD simulation. To further enhance the stability of the NEPs, manually generated uniaxial strain, biaxial strain, and rattled configurations were added to the training set. Ultimately, 356 and 320 configurations were selected to train the NEPs for Li6NII2 and Li6NBrBr2, respectively (the distribution of sampled structures obtained via PYNEP is shown in Figure S2). Accurate virials, energies, and forces for these configurations were computed via precise DFT calculations using a 7 × 7 × 7 k-point mesh and a total energy convergence criterion of 10-8 eV. The radial and angular cutoffs and other hyperparameters used for NEP training are summarized in Table S1.
Our NEPs for both Li6NII2 and Li6NBrBr2 exhibit excellent agreement with DFT results in predicting energies, atomic forces, and virials for both training and testing datasets, as illustrated in Figure 1, Figure S3 and Figure S4, respectively. For the Li6NII2 training dataset, the root mean square errors (RMSE) for energies, atomic forces, and virials are 0.39 meV, 14.41 meV/Å and 2.64 meV per atom, respectively. The convergence trends of each term in the loss function as well as phonon dispersion validation are presented in Figure S5 and Figure S6.
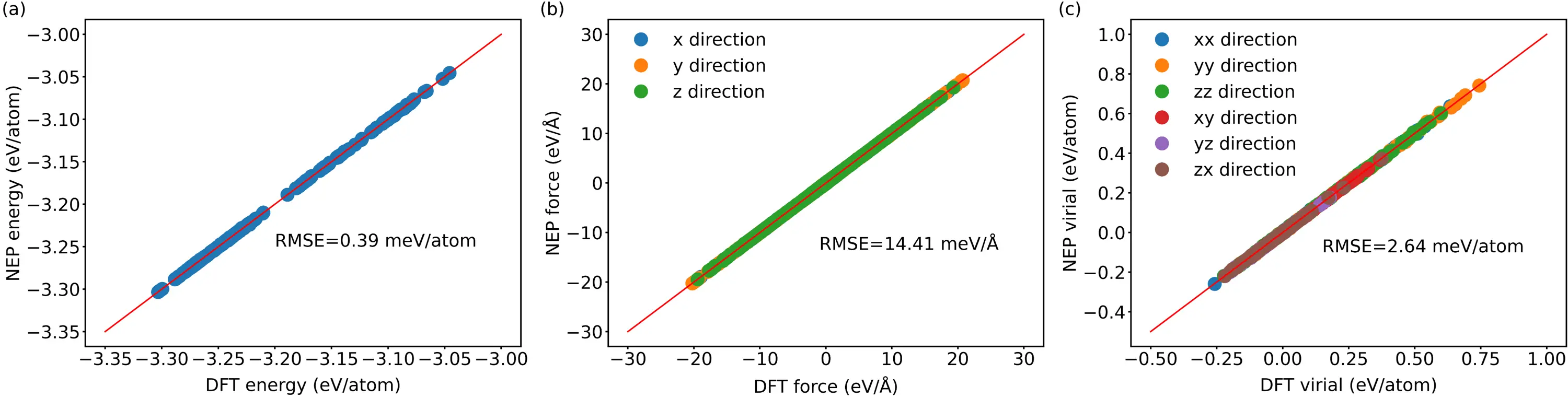
Figure 1. For Li6NII2, (a) Energy; (b) Force; (c) Virial calculated by NEP as compared to the relevant results calculated from DFT in the training dataset. The solid lines in (a), (b) and (c) are the identity function applied to guide the eyes. NEP: neuroevolution potential; RMSE: root mean square errors.
2.2 Crystal structures and phonon dispersion relations
Determining temperature-dependent lattice constants is fundamental for accurately describing thermal transport in strongly anharmonic materials. In this study, we extracted the temperature-dependent lattice constants of both Li6NII2 and Li6NBrBr2 using our NEPs combined with NPT (MD simulations. The calculated lattice constants forLi6NII2 and Li6NBrBr2 are 9.714 Å and 9.131 Å, respectively, which are close to the experimental values of 9.536 Å and 8.925 Å at room temperature[50]. The primitive and conventional unit cells of both materials are presented in Figure 2a and Figure S7, respectively, while the temperature-dependent lattice constants are shown in Figure 2b and Figure S8.
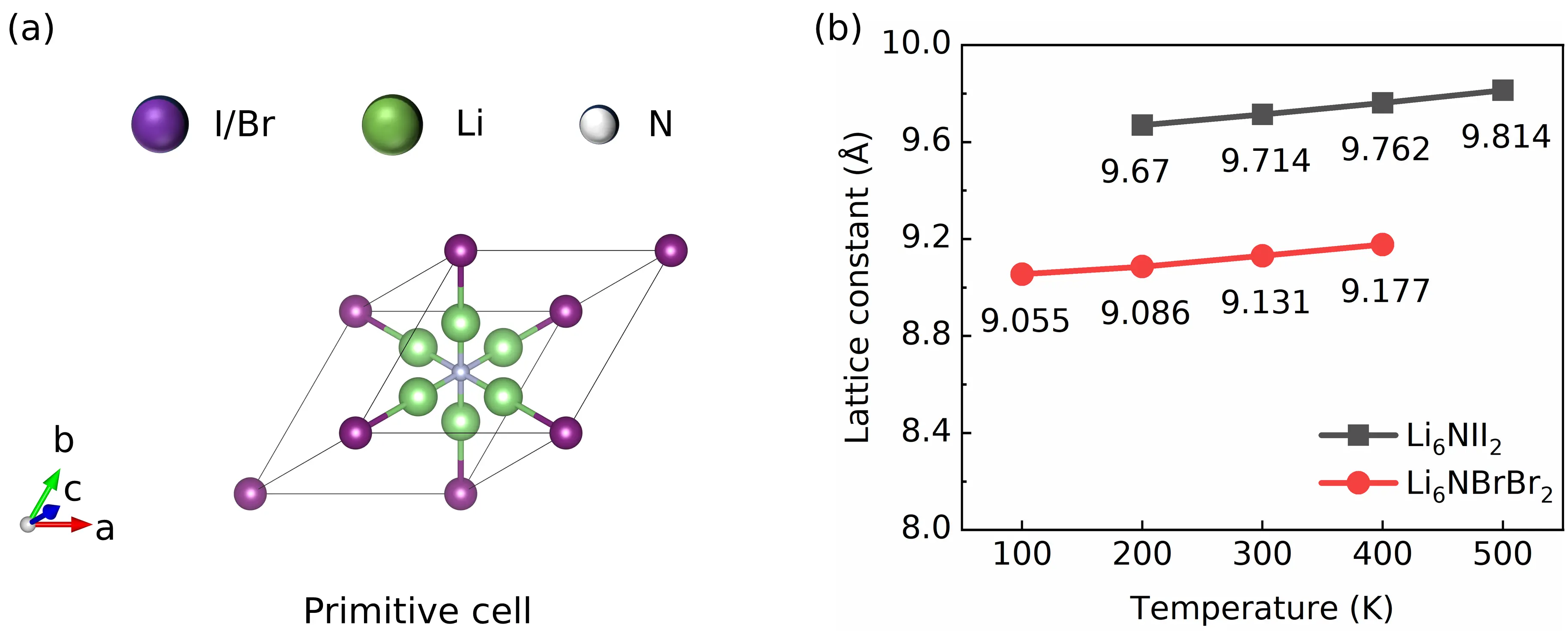
Figure 2. (a) Primitive cell of Li6NII2 or Li6NBrBr2; (b) The temperature-dependent lattice constants of Li6NII2 and Li6NBrBr2.
Figure 3a and Figure 3b depict the harmonic phonon dispersion relations of Li6NII2 and Li6NBrBr2, calculated using the finite displacement method and DFT. A 5 × 5 × 5 k-point mesh, an energy convergence criterion of 10-8 eV, and 2 × 2 × 2 supercells were employed. Large portions of both acoustic and optical phonons with imaginary frequencies appear in the harmonic phonon dispersions. Anharmonic renormalized IFCs at finite temperatures were also obtained using the temperature-dependent effective potential method[57] and trajectories from isothermal (NVT) ensemble MD simulations with NEPs for both materials (details are provided in Figure S9). The resulting anharmonic phonon dispersion relations are shown in Figure 3c and Figure 3d, revealing clear temperature dependencies. Although acoustic phonon branches become stable at finite temperatures, optical phonon branches exhibit large imaginary frequencies (approximately -6 THz) across the entire Brillouin zone, indicating the failure of perturbation theory to accurately describe phonon transport in both Li6NII2 and Li6NBrBr2.
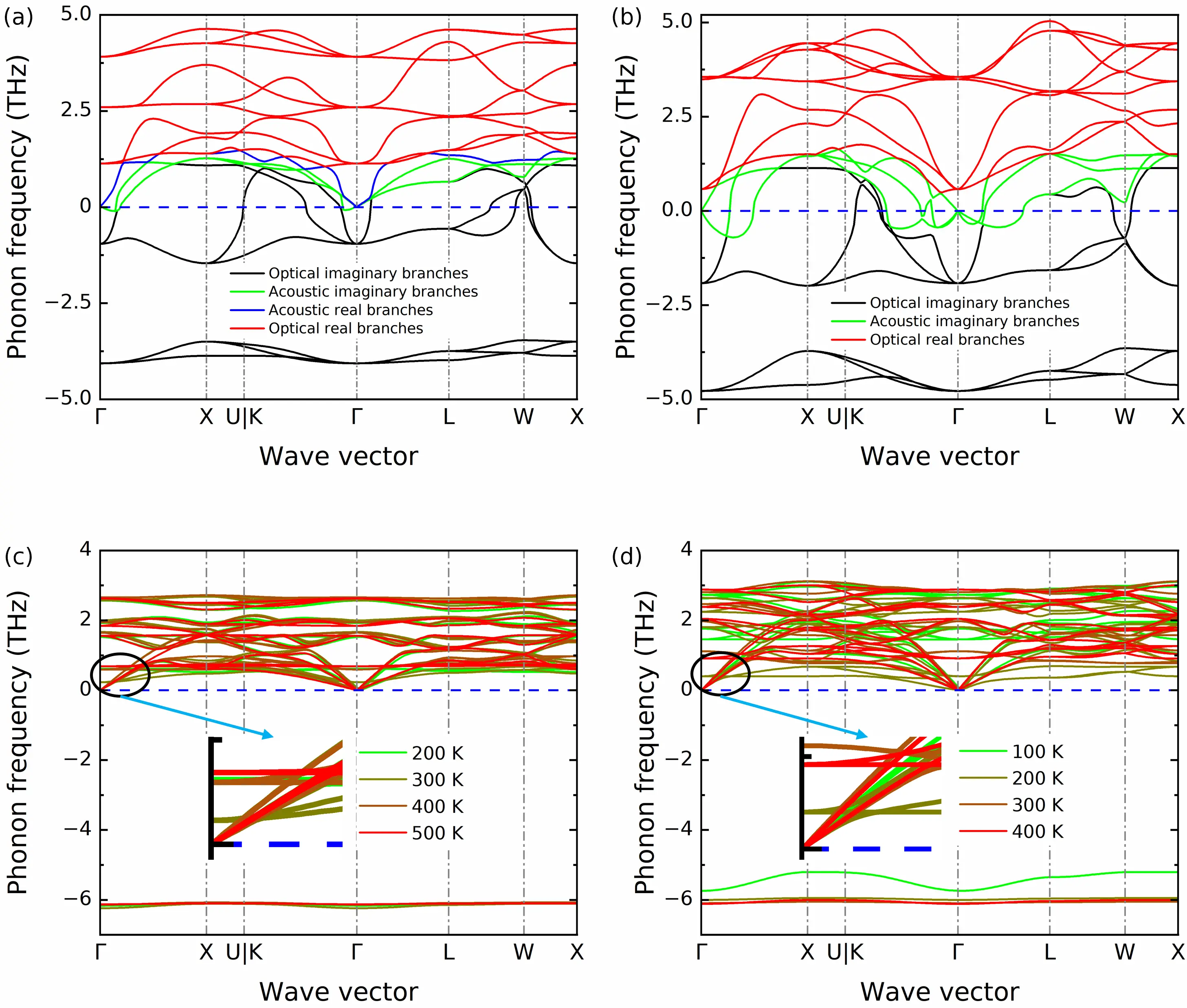
Figure 3. Harmonic phonon dispersion of (a) Li6NII2 and (b) Li6NBrBr2; The temperature-dependent phonon dispersion of (c) Li6NII2 from 200 to 500 K and (d) Li6NBrBr2 from 100 to 400 K.
To clarify the origin of optical phonons with imaginary frequencies, we further analyzed the vibrational patterns of atoms in both materials. The atomic vibrational modes corresponding to the optical phonons with imaginary frequencies are shown in Figure 4a, Figure 4b, and Figure S10. Notably, only Li atoms participate in these imaginary phonon modes for both compounds. Additionally, atomic trajectories from NVT MD simulations are presented in Figure 4c and Figure S11. Unlike the vibrations of N, Br, and I atoms near their equilibrium positions, Li atoms exhibit stochastic movements among adjacent Li lattice sites confined within a single octahedron. These stochastic Li-ion motions invalidate perturbation theory, suggesting that MD simulations are more appropriate for calculating the thermal conductivities of such materials. Accordingly, we evaluate thermal conductivity using EMD and HNEMD methods, and analyze frequency-dependent phonon contributions via the spectral heat current (SHC) method[58]. Additional verifications of thermodynamic stability and related discussions for both materials are provided in Figure S12, Figure S13, Figure S14 and Figure S15.
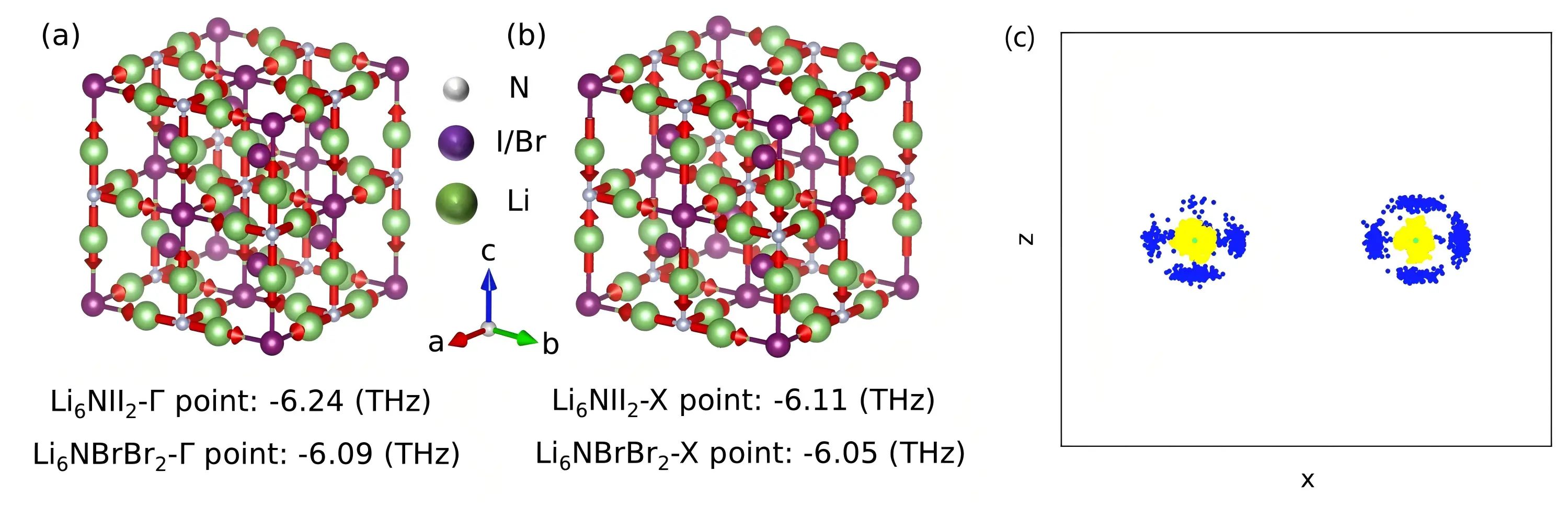
Figure 4. Phonon vibrational modes of Li6NII2 and Li6NBrBr2 at the (a) Γ and (b) X points (red arrows indicate the atomic vibrational directions); (c) The projection on the xz plane of two adjacent Li-ion stochastic movements in the conventional cell at 300 K. Blue: Li-ion hopping trajectories; Yellow: Li-ion atomic vibration trajectories; green: Li-ion equilibrium position).
3. Results and Discussion
3.1 Room-temperature thermal properties using molecular dynamics
Figure 5a and Figure 5b present the EMD results at 300 K, obtained using orthogonal supercells sized 5.8 nm × 5.8 nm × 5.8 nm and 5.4 nm × 5.4 nm × 5.4 nm for Li6NII2 and Li6NBrBr2, respectively (convergence tests with respect to supercell size are shown in Figure S16 and Figure S17). Figure 5c and Figure 5d show that for both double antiperovskites, a supercell with a characteristic lattice length of approximately 5 nm is required to converge the thermal conductivity based on EMD at 300 K. Forty-two independent simulations were conducted for each material. Each simulation began with a 300 ps equilibration stage, followed by a 10 ns production stage performed separately under the NVT and microcanonical (NVE) ensembles. The mean thermal conductivity values along the x, y, and z directions converge over the correlation time. The final reported thermal conductivities are averaged over these three directions throughout the entire correlation period. The predicted thermal conductivities of Li6NII2 and Li6NBrBr2 at 300 K are 0.367 W/mK and 0.536 W/mK, respectively. Results and convergence tests for EMD simulations at other temperatures are presented in Figures S18, Figure S19, Figure S20 and Figure S21.
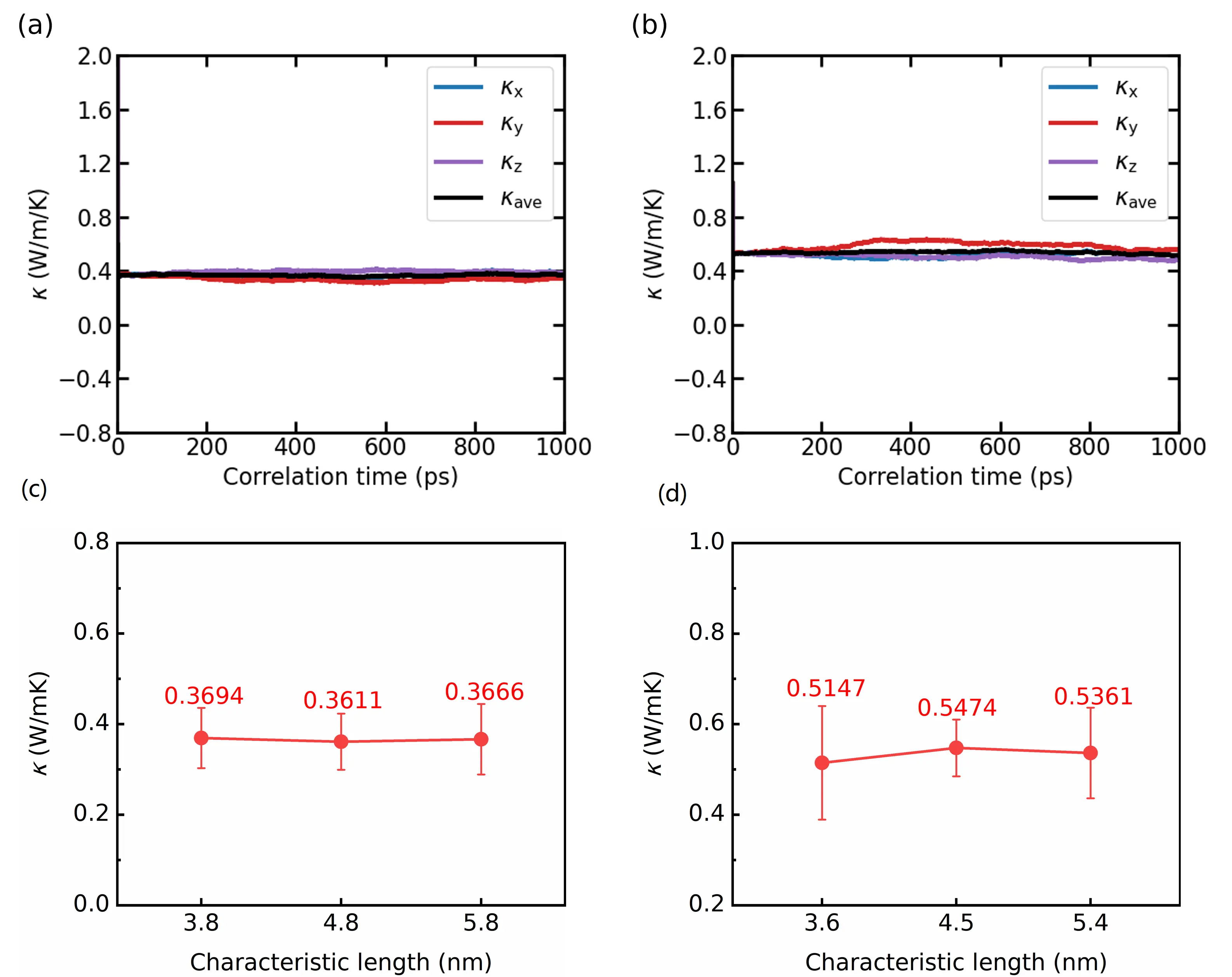
Figure 5. Running lattice thermal conductivity as a function of correlation time using the EMD method for (a) Li6NII2 and (b) Li6NBrBr2 at 300 K along the x, y and z directions, as well as their averages; Lattice thermal conductivity κ as a function of the characteristic length for (c) Li6NII2 and (d) Li6NBrBr2 at 300 K from the EMD simulations. EMD: equilibrium molecular dynamics.
Figure 6a and Figure 6b show the HNEMD results at 300 K, obtained using orthogonal supercells of the same sizes as above for 5.8 nm × 5.8 nm × 5.8 nm and 5.4 nm × 5.4 nm × 5.4 nm for Li6NII2 and Li6NBrBr2, respectively (convergence tests with respect to supercell size are included in the Supplementary Materials).Figure 6c and Figure 6d indicate that a supercell with a characteristic length of about 5 nm is also required to converge the thermal conductivity from HNEMD at 300 K. Given the isotropic nature of both materials, lattice thermal conductivity was computed along the y-direction using HNEMD. The simulations involved a 300 ps equilibration stage followed by a 6 ns production stage under the NVT ensemble at 300 K. The driving force magnitude was optimized around 2.0×10-4 Å-1 for both materials to maintain the system within the linear response regime and ensure a sufficiently large signal-to-noise ratio. Thermal conductivities were averaged over the last 2 ns of the production stage, based on eight independent simulations, resulting in values of 0.35 W/mK and 0.531 W/mK for Li6NII2 and Li6NBrBr2, respectively. These ultralow thermal conductivities are comparable to those of metal oxides (Bi3Ir3O11: 1 W/mK)[59], nitride perovskite (LaWN3: 2.98 W/mK)[60], double perovskites (Cs2AgBiBr6: 0.33 W/mK[16], Cs2PTi6: 0.15 W/mK[61], and Cs2SnI6: 0.29 W/mK[62]) and antiperovskites (K3BrO: 0.73 W/mK[23], K3ITe: 0.18 W/mK[63], and K3ISe: 0.6 W/mK[63]) at room temperature. This comparison highlights the strong lattice anharmonicity of both nitrohalide double antiperovskites. Further details of HNEMD simulations are provided in Figure S22, Figure S23, Figure S24 and Figure S25.
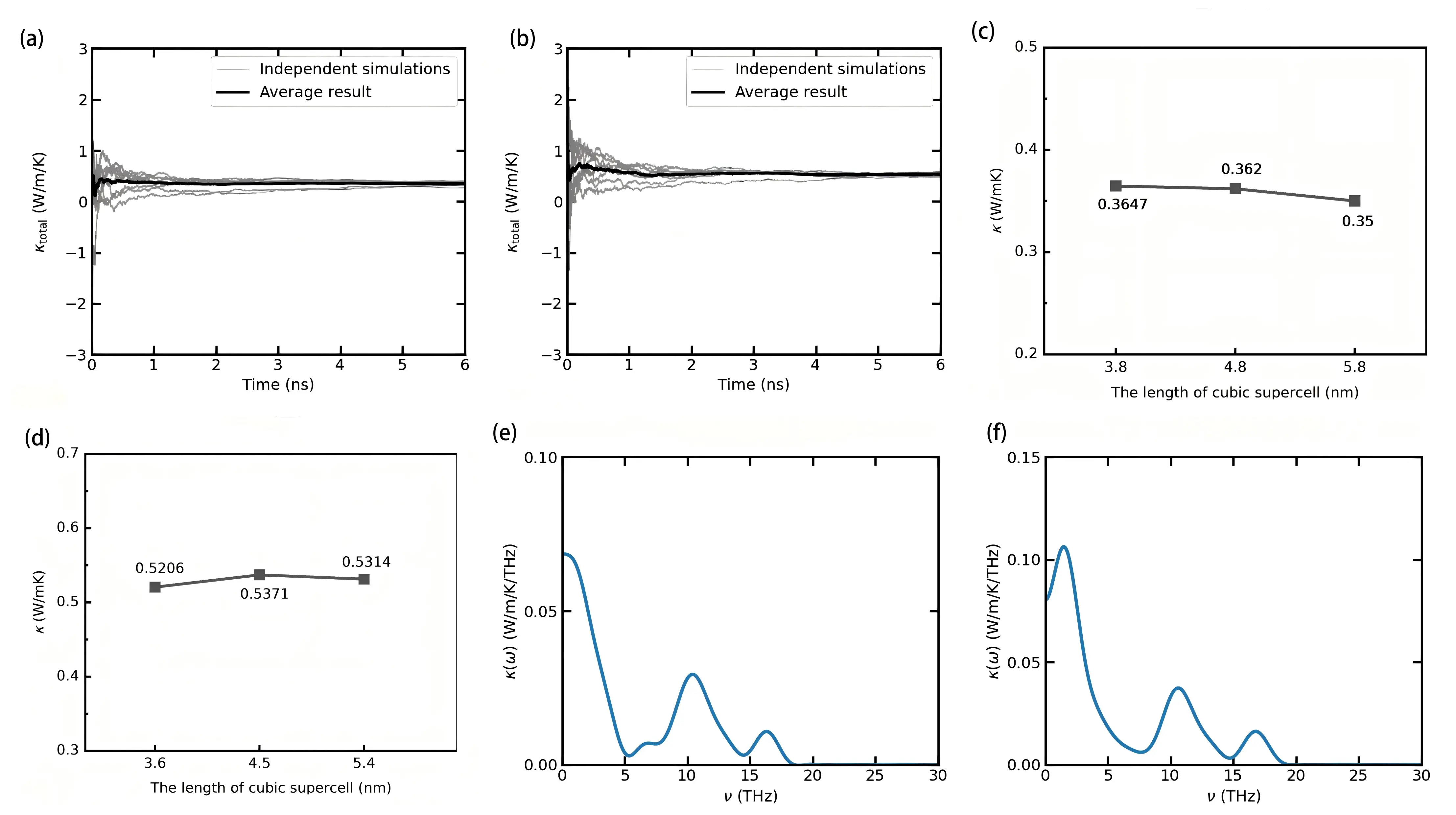
Figure 6. Running lattice thermal conductivity as a function of simulation time using the HNEMD method for (a) Li6NII2 and (b) Li6NBrBr2 at 300 K along the y direction, as well as their average; Lattice thermal conductivity κ as a function of the characteristic length for (c) Li6NII2 and (d) Li6NBrBr2 at 300 K from the HNEMD simulations; Spectral lattice thermal conductivity as a function of phonon frequency for (e) Li6NII2 and (f) Li6NBrBr2 at 300 K. HNEMD: homogeneous nonequilibrium molecular dynamics.
Additionally, we calculated the spectral lattice thermal conductivity of both Li6NII2 and Li6NBrBr2 using the HNEMD-based SHC method, as shown in Figure 6e and Figure 6f. The dominant phonon contributions to thermal conductivity arise from phonons with frequencies below 5 THz and around 11 THz.
3.2 Temperature-dependent thermal transport properties
Figure 7a and Figure 7b show the temperature-dependent lattice thermal conductivities of Li6NII2 and Li6NBrBr2, obtained using both EMD and HNEMD methods. The results from the two methods exhibit good agreement. Compared with other antiperovskites such as Li3OCl and Li3OBr[18], as well as Li3HS, Li3HSe and Li3HTe[64], both Li6NII2 and Li6NBrBr2 exhibit significantly lower lattice thermal conductivities and reduced sensitivity to temperature over the studied range. For Li6NII2, the thermal conductivity remains below 0.4 W/mK from 200 K to 500 K, with a total variation of only 16.44% across this temperature range. Li6NBrBr2 displays slightly higher values, decreasing from 0.64 W/mK (0.59 W/mK) to 0.51 W/mK (0.54 W/mK) as temperature increases from 100 K to 400 K, based on EMD (HNEMD) results. The temperature dependence of thermal conductivity for Li6NII2 and Li6NBrBr2 follows a T-0.13 and T-0.11 trend. These temperature-insensitive and ultralow thermal conductivities indicate the glass-like thermal transport behavior of both materials. Figure 7c and Figure 7d display the spectral thermal conductivities of both compounds at different temperatures. Phonons with frequencies below 5 THz are the main contributors to thermal transport, which aligns with the observed flattening of phonon branches above 3 THz and the near-zero phonon group velocities, as shown in the temperature-dependent phonon dispersions and group velocity plots in Figure S26 and Figure S27. An additional substantial contribution comes from phonons near 11 THz. As revealed in Figure 3, Figure S10 and Figure S28, the vibrations of N, I, Br, and some Li atoms correspond to well-defined phonon modes, while the stochastic motion of a subset of Li atoms gives rise to imaginary phonons. Because no distinct phonon branches exist near 11 THz, the thermal conductivity in this region can be attributed to the stochastic motion of Li atoms and their interactions with surrounding atoms. The defining lattice feature of both Li6NII2 and Li6NBrBr2 is the stochastic hopping of Li atoms among neighboring lattice sites, confined within a single octahedron. This behavior violates the assumptions of perturbation theory, which requires atoms to vibrate around their equilibrium positions, allowing potential energy to be expanded in terms of small displacements. The hopping distances of Li atoms can reach 4.689 Å in Li6NII2 and 4.408 Å in Li6NBrBr2, which renders perturbation theory inapplicable. In other materials such as (BA)2PbI4[65], Cs2SnI6[66,67], Cs2PbI2Cl2[68], LaWN3[51] and antiperovskites like Li3OCl[17] and Na3FS[69], strong lattice anharmonicity generally originates from octahedral rotations, tilting, weakly bonded cations (rattler atoms), soft phonon branches, and significant four-phonon scattering. In contrast, the anharmonicity in Li6NII2 and Li6NBrBr2 is dominated by suppressed acoustic phonons and the presence of imaginary phonons. Mode-projected phonon eigenvector analysis indicates that these imaginary branches span the entire Brillouin zone and result from the stochastic motion of Li atoms. This invalidates the use of conventional transport formalisms such as the phonon Boltzmann transport equation and the Wigner transport equation[70] in describing phonon transport in these materials. Instead, MD-based simulations and spectral analysis show that phonons below 5 THz and around 11 THz make the largest contributions to thermal conductivity. The significant contribution of phonons above 3 THz, especially those near 11 THz, is mainly associated with the stochastic motion of Li atoms.
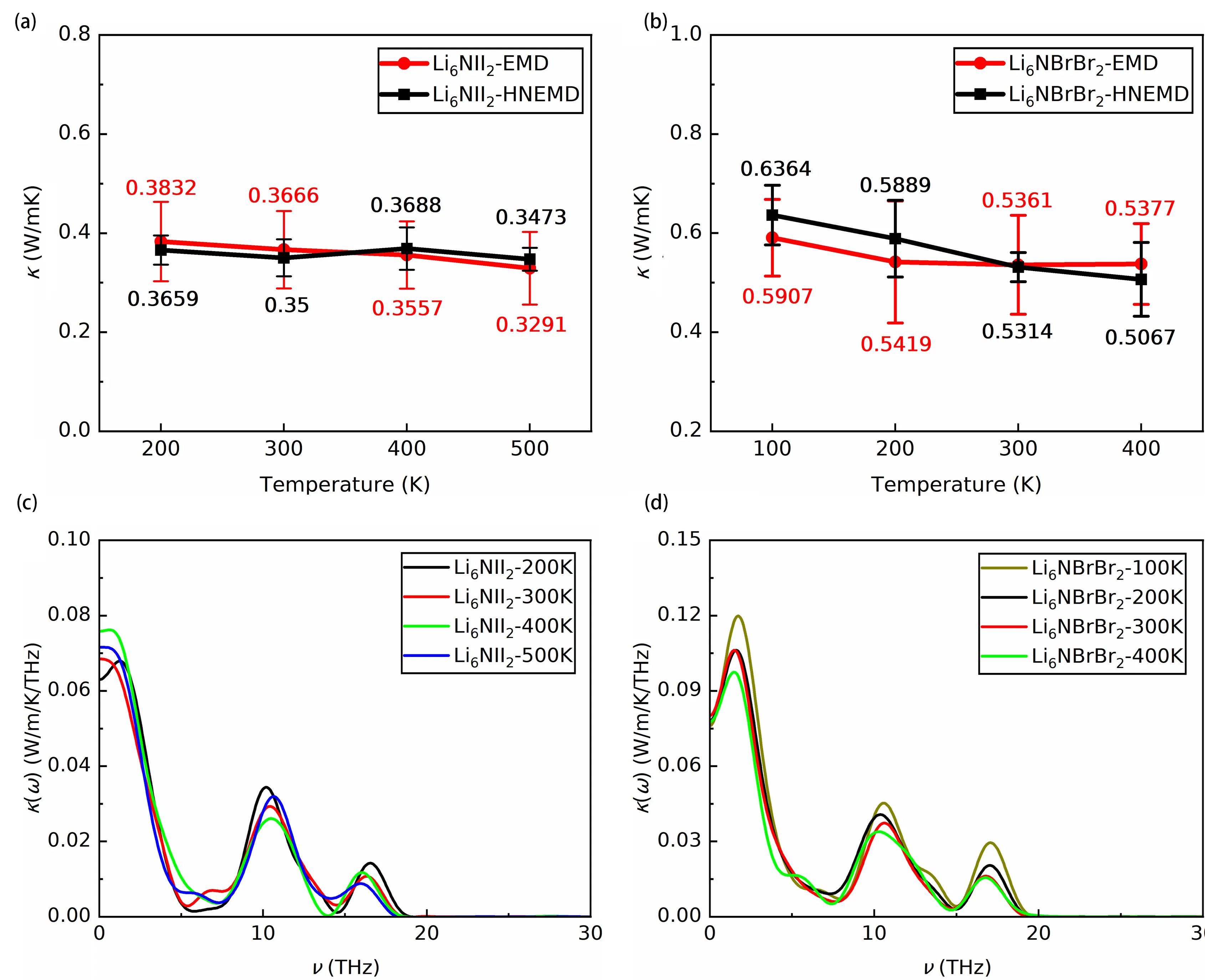
Figure 7. The lattice thermal conductivity of (a) Li6NII2 from 200 to 500 K and (b) Li6NBrBr2 from 100 to 400 K; Corresponding spectral thermal conductivity of (c) Li6NII2 and (d) Li6NBrBr2. EMD: equilibrium molecular dynamics; HNEMD: homogeneous nonequilibrium molecular dynamics.
Given their ultralow lattice thermal conductivities across a wide temperature range and their composition from earth-abundant elements, Li6NII2 and Li6NBrBr2 are promising candidates for low-cost thermoelectric applications, particularly at room temperature. The stochastic motion of Li ions confined within a single conventional unit cell may also be tuned to enhance carrier mobility in thermoelectric devices, thereby improving overall energy conversion efficiency.
4. Conclusions
In summary, we investigated phonon transport in nitrohalide double antiperovskites Li6NII2 and Li6NBrBr2 using first-principles machine-learning potentials. The confined stochastic motion of Li ions within a single conventional unit cell leads to the emergence of imaginary phonon modes throughout the entire Brillouin zone and causes the breakdown of perturbation theory. Both equilibrium and nonequilibrium molecular dynamics simulations confirm that Li6NII2 and Li6NBrBr2 exhibit ultralow lattice thermal conductivities with glass-like temperature dependence. Spectral thermal conductivity analysis reveals that phonons with frequencies below 5 THz and around 11 THz dominate the overall thermal transport. Notably, the significant contribution near 11 THz arises from the stochastic, confined movement of Li ions. This study uncovers unconventional atomic dynamics and a distinct phonon transport mechanism in double antiperovskites, offering valuable insights for the design of low-thermal-conductivity materials for thermoelectric applications.
Supplementary materials
The supplementary material for this article is available at: Supplementary materials.
Acknowledgements
Qi Wang acknowledges the support from the start-up funding of the Suzhou Institute for Advanced Research at the University of Science and Technology of China.
Authors contribution
Li Y: Data curation, formal analysis, investigation, methodology and writing-original draft.
Zheng W: Data analysis and interpretation.
Pu JH: Resources and supervision.
Wang Q: Conceptualization, formal analysis, methodology, resources, software, supervision, writing-review and editing.
Jiang JH: Supervision, writing-review and editing.
Conflict of interest
Jian-Hua Jiang is an Editorial Board member of Thermo-X. The other authors declared that there are no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
The data that support the findings of this study are available within the article and its supplementary materials. Other questions regarding this study can be directed to the corresponding authors.
Funding
This study was supported by the Natural Science Foundation of Shandong Province (Grant No. ZR2023QE223)
Copyright
© The Author(s) 2025.
References
-
1. Lai KT, Antonyshyn I, Prots Y, Valldor M. Anti-perovskite Li-battery cathode materials. J Am Chem Soc. 2017;139(28):9645-9649.
[DOI] -
2. Deng Z, Ni D, Chen D, Bian Y, Li S, Wang Z, et al. Anti-perovskite materials for energy storage batteries. InfoMat. 2022;4(2):e12252.
[DOI] -
3. Zheng J, Fang H, Fan L, Ren Y, Jena P, Wu Y. Antiperovskite K3OI for K-ion solid state electrolyte. J Phys Chem Lett. 2021;12(30):7120-7126.
[DOI] -
4. Xia W, Zhao Y, Zhao F, Adair K, Zhao R, Li S, et al. Antiperovskite electrolytes for solid-state batteries. Chem Rev. 2022;122(3):3763-3819.
[DOI] -
5. Rani U, Kamlesh PK, Shukla A, Verma AS. Emerging potential antiperovskite materials ANX3 (A = P, As, Sb, Bi; X = Sr, Ca, Mg) for thermoelectric renewable energy generators. J Solid State Chem. 2021;300:122246.
[DOI] -
6. Basit A, Khan SA, Murtaza G, Mehmood A, Khenata R, Omran SB, et al. Electronic, optical and thermoelectric properties of XNMg3 (X = P, As, Sb, Bi) compounds. Mater Sci Semicond Process. 2016;43:69-74.
[DOI] -
7. Quan LN, Rand BP, Friend RH, Mhaisalkar SG, Lee TW, Sargent EH. Perovskites for next-generation optical sources. Chem Rev. 2019;19(12):7444-7477.
[DOI] -
8. Rani U, Soni Y, Kamlesh PK, Pachori S, Verma AS. Fundamental theoretical design of Na-ion and K-ion based double antiperovskite
X6SOA2 (X = Na, K; A = Cl, Br and I) halides: Potential candidate for energy storage and harvester. Int J Energy Res. 2021;45(9):13442-13460.
[DOI] -
9. Rani U, Kamlesh PK, Agarwal R, Kumari J, Verma AS. Electronic and thermo-physical properties of double antiperovskites X6SOA2 (X = Na, K and A = Cl, Br, I): A non-toxic and efficient energy storage materials. Int J Quantum Chem. 2021;121(19):e26759.
[DOI] -
10. Islam MM, Maruf AA, Pokharel J, Zhou Y. Superhalogen-based Li-rich double antiperovskite Li6OS(BH4)2 as solid electrolyte. MRS Commun. 2022;12(6):1140-1146.
[DOI] -
11. Rani U, Kamlesh PK, Joshi TK, Singh R, Al-Qaisi S, Gupta R, et al. Electronic structure, theoretical power conversion efficiency, and thermoelectric properties of bismuth-based alkaline earth antiperovskites. J Mol Model. 2023;29(10):329.
[DOI] -
12. Hu T, Hu Y, Shang W, Li L, Feng C, Zhou P, et al. Thermoelectric properties of lead-free anti-perovskites X3BN (B = Bi, Sb, X = Mg, Ca, Sr): A theoretical study based on first-principles calculations and machine learning interatomic potential. AIP Adv. 2024;14(4):045119.
[DOI] -
13. Okamoto Y, Sakamaki A, Takenaka K. Thermoelectric properties of antiperovskite calcium oxides Ca3PbO and Ca3SnO. J Appl Phys. 2016;119(20):205106.
[DOI] -
14. Wang Q, Zeng Z, Chen Y. Revisiting phonon transport in perovskite SrTiO3: Anharmonic phonon renormalization and four-phonon scattering. Phys Rev B. 2021;104(23):235205.
[DOI] -
15. Cappai A, Melis C, Marongiu D, Quochi F, Saba M, Congiu F, et al. Strong anharmonicity at the origin of anomalous thermal conductivity in double perovskite Cs2NaYbCl6. Adv Sci. 2024;11(9):2305861.
[DOI] -
16. Klarbring J, Hellman O, Abrikosov IA, Simak SI. Anharmonicity and ultralow thermal conductivity in lead-free halide double perovskites. Phys Rev Lett. 2020;125(4):045701.
[DOI] -
17. Chen MH, Emly A, Van Der Ven A. Anharmonicity and phase stability of antiperovskite Li3OCl. Phys Rev B. 2015;91(21):214306.
[DOI] -
18. Sattar MA, Javed M, Benkraouda M, Amrane N. The structural stability, lattice dynamics, electronic, thermophysical, and mechanical properties of the inverse perovskites A3OX: A comparative first-principles study. Int J Energy Res. 2021;45(3):4793-4810.
[DOI] -
19. Zeng S, Shen Q, Guo L, Zhao Y, Huang H, Li G, et al. Remarkable thermoelectric efficiency of cubic antiperovskites Rb3X(Se & Te)I with strong anharmonicity. J Mater Chem A. 2023;11(44):24047-24056.
[DOI] -
20. Zeng S, Yan X, Shen Q, Tu Y, Huang H, Li G. Low lattice thermal conductivities and good thermoelectric performance of hexagonal antiperovskites X(Ba & Sr)3BiN with quartic anharmonicity. Phys Chem Chem Phys. 2023;25(39):26507-26514.
[DOI] -
21. Zhou WX, Wu CW, Cao HR, Zeng YJ, Xie G, Zhang G. Abnormal thermal conductivity increase in β-Ga2O3 by an unconventional bonding mechanism using machine-learning potential. Mater Today Phys. 2025;52:101677.
[DOI] -
22. Chen XK, Zhang Y, Luo QQ, Jia PZ, Zhou WX. Strain-driven anisotropic enhancement in the thermal conductivity of KCaBi: the role of optical phonons. Int J Heat Mass Transfer. 2025;236:126364.
[DOI] -
23. Zhao Y, Lian C, Zeng S, Dai Z, Meng S, Ni J. Quartic anharmonicity and anomalous thermal conductivity in cubic antiperovskites A3BO (A = K, Rb; B = Br, Au). Phys Rev B. 2020;101(18):184303.
[DOI] -
24. Wang Q, Zeng Z, Zhao P, Chen C, Ouyang N, Mao J, et al. B-site columnar-ordered halide double perovskites: Breaking octahedra motions induces strong lattice anharmonicity and thermal anisotropy. Chem Mater. 2023;35(4):1633-1639.
[DOI] -
25. Liu Z, Zhang W, Gao W, Mori T. A material catalogue with glass-like thermal conductivity mediated by crystallographic occupancy for thermoelectric application. Energy Environ Sci. 2021;14(6):3579-3587.
[DOI] -
26. Ying P, Liang T, Xu K, Zhang J, Xu J, Zhong Z, et al. Sub-micrometer phonon mean free paths in metal-organic frameworks revealed by machine learning molecular dynamics simulations. ACS Appl Mater Interfaces. 2023;15(30):36412-36422.
[DOI] -
27. Fan D, Ozcan A, Lyu P, Maurin G. Unravelling abnormal in-plane stretchability of two-dimensional metal-organic frameworks by machine learning potential molecular dynamics. Nanoscale. 2024;16(7):3438-3447.
[DOI] -
28. Wieser S, Zojer E. Machine learned force-fields for an Ab-initio quality description of metal-organic frameworks. npj Comput Mater 2024;
[DOI] -
29. Xu K, Hao Y, Liang T, Ying P, Xu J, Wu J, et al. Accurate prediction of heat conductivity of water by a neuroevolution potential. J Chem Phys. 2023;158(20):204114.
[DOI] -
30. Cheng B, Engel EA, Behler J, Dellago C, Ceriotti M. Ab initio thermodynamics of liquid and solid water. Proc Natl Acad Sci USA. 2019;116(4):1110-1115.
[DOI] -
31. Wohlfahrt O, Dellago C, Sega M. Ab initio structure and thermodynamics of the RPBE-D3 water/vapor interface by neural-network molecular dynamics. J Chem Phys. 2020;153(14):144710.
[DOI] -
32. Monserrat B, Brandenburg JG, Engel EA, Cheng B. Liquid water contains the building blocks of diverse ice phases. Nat Commun. 2020;11(1):5757.
[DOI] -
33. Zhang L, Wang H, Car R, Weinan E. Phase diagram of a deep potential water model. Phys Rev Lett. 2021;126(23):236001.
[DOI] -
34. Mortazavi B, Podryabinkin EV, Roche S, Rabczuk T, Zhuang X, Shapeev AV. Machine-learning interatomic potentials enable first-principles multiscale modeling of lattice thermal conductivity in graphene/borophene heterostructures. Mater Horiz. 2020;7(9):2359-2367.
[DOI] -
35. Mortazavi B, Podryabinkin EV, Novikov IS, Roche S, Rabczuk T, Zhuang X, et al. Efficient machine-learning based interatomic potentialsfor exploring thermal conductivity in two-dimensional materials. J Phys Mater. 2020;3(2):02LT02.
[DOI] -
36. Chan H, Narayanan B, Cherukara MJ, Sen FG, Sasikumar K, Gray SK, et al. Machine learning classical interatomic potentials for molecular dynamics from first-principles training data. J Phys Chem C. 2019;123(12):6941-6957.
[DOI] -
37. Mortazavi B, Podryabinkin EV, Novikov IS, Rabczuk T, Zhuang X, Shapeev AV. Accelerating first-principles estimation of thermal conductivity by machine-learning interatomic potentials: A MTP/ShengBTE solution. Comput Phys Commun. 2021;258:107583.
[DOI] -
38. Rowe P, Csányi G, Alfè D, Michaelides A. Development of a machine learning potential for graphene. Phys Rev B. 2018;97(5):054303.
[DOI] -
39. Singh A, Li Y. Machine learning potentials for graphene. In: Proceedings of the ASME 2022 International Mechanical Engineering Congress and Exposition; 2022 October 30-November 3; Columbus, USA. New York: American Society of Mechanical Engineers; 2022. p. V003T03A036.
[DOI] -
40. Behler J, Parrinello M. Generalized neural-network representation of high-dimensional potential-energy surfaces. Phys Rev Lett. 2007;98(14):146401.
[DOI] -
41. Bartók AP, Payne MC, Kondor R, Csányi G. Gaussian approximation potentials: The accuracy of quantum mechanics, without the electrons. Phys Rev Lett. 2010;104(13):136403.
[DOI] -
42. Novikov IS, Gubaev K, Podryabinkin EV, Shapeev AV. The MLIP package: moment tensor potentials with MPI and active learning. Mach Learn Sci Technol. 2021;2(2):025002.
[DOI] -
43. Shapeev AV. Moment tensor potentials: A class of systematically improvable interatomic potentials. Multiscale Model Simul. 2016;14(3):1153-1173.
[DOI] -
44. Fan Z, Zeng Z, Zhang C, Wang Y, Song K, Dong H, et al. Neuroevolution machine learning potentials: Combining high accuracy and low cost in atomistic simulations and application to heat transport. Phys Rev B. 2021;104(10):104309.
[DOI] -
45. Fan Z. Improving the accuracy of the neuroevolution machine learning potential for multi-component systems. J Phys Condens Matter. 2022;34(12):125902.
[DOI] -
46. Zhang Y, Xia J, Jiang B. REANN: A PyTorch-based end-to-end multi-functional deep neural network package for molecular, reactive, and periodic systems. J Chem Phys. 2022;156(11):114801.
[DOI] -
47. Zhang Y, Xia J, Jiang B. Physically motivated recursively embedded atom neural networks: incorporating local completeness and nonlocality. Phys Rev Lett. 2021;127(15):156002.
[DOI] -
48. Li Z, Wang J, Dong H, Zhou Y, Liu L, Yang JY. Mechanistic insights into water filling effects on thermal transport of carbon nanotubes from machine learning molecular dynamics. Int J Heat Mass Transfer. 2024;235:126152.
[DOI] -
49. Fan Z, Wang Y, Ying P, Song K, Wang J, Wang Y, et al. GPUMD: A package for constructing accurate machine-learned potentials and performing highly efficient atomistic simulations. J Chem Phys. 2022;157(11):114801.
[DOI] -
50. Mi R, Hu S, Xiao Z. Rock-salt-ordered nitrohalide double antiperovskites: Theoretical design and experimental verification. Chem Mater. 2022;34(20):9098-9103.
[DOI] -
51. Kresse G, Joubert D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys Rev B. 1999;59(3):1758.
[DOI] -
52. Perdew JP, Burke K, Ernzerhof M. Generalized gradient approximation made simple. Phys Rev Lett. 1996;77(18):3865.
[DOI] -
53. Kresse G, Furthmüller J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys Rev B. 1996;54(16):11169.
[DOI] -
54. Kresse G, Furthmüller J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput Mater Sci. 1996;6(1):15-50.
[DOI] -
55. Wang B, Li K, Zhang W, Sun Y, Zhou J, Sun Z. Thermal transport of GeTe/Sb2Te3 superlattice by large-scale molecular dynamics with machine-learned potential. J Phys Chem C. 2025;129(13):6386-6396.
[DOI] -
56. Wan J, Li G, Guo Z, Qin H. Thermal transport in C6N7 monolayer: a machine learning based molecular dynamics study. J Phys Condens Matter. 2024;37(2):025301.
[DOI] -
57. Hellman O, Steneteg P, Abrikosov IA, Simak SI. Temperature dependent effective potential method for accurate free energy calculations of solids. Phys Rev B. 2013;87(10):104111.
[DOI] -
58. Fan Z, Dong H, Harju A, Ala-Nissila T. Homogeneous nonequilibrium molecular dynamics method for heat transport and spectral decomposition with many-body potentials. Phys Rev B. 2019;99(6):064308.
[DOI] -
59. Ji J, Liu Z, Dai J, Qiu D, Yang J, Xi J, et al. Delocalized Bi-tetrahedral cluster induced ultralow lattice thermal conductivity in Bi3Ir3O11. Mater Today Phys. 2023;32:101005.
[DOI] -
60. Tong Z, Zhang Y, Pecchia A, Yam C Y, Zhou L, Dumitrică T, et al. Predicting the lattice thermal conductivity in nitride perovskite LaWN3 from ab initio lattice dynamics. Adv Sci. 2023;10(9):2205934.
[DOI] -
61. Sajjad M, Mahmood Q, Singh N, Larsson JA. Ultralow lattice thermal conductivity in double perovskite Cs2PtI6: A promising thermoelectric material. ACS Appl Energy Mater. 2020;3(11):11293-11299.
[DOI] -
62. Bhui A, Ghosh T, Pal K, Singh Rana K, Kundu K, Soni A, et al. Intrinsically low thermal conductivity in the n-Type vacancy-ordered double perovskite Cs2SnI6: Octahedral rotation and anharmonic rattling. Chem Mater. 2022;34(7):3301-3310.
[DOI] -
63. Zeng S, Guo L, Huang Z, Shen Q, Zhao Y, Huang H, et al. Anomalous thermal conductivity and high thermoelectric performance of cubic antiperovskites K3IX(Se & Te). Chem Mater. 2024;36(1):211-218.
[DOI] -
64. Gao S, Broux T, Fujii S, Tassel C, Yamamoto K, Xiao Y, et al. Hydride-based antiperovskites with soft anionic sublattices as fast alkali ionic conductors. Nat Commun. 2021;12(1):201.
[DOI] -
65. Menahem M, Dai Z, Aharon S, Sharma R, Asher M, Diskin-Posner Y, et al. Strongly anharmonic octahedral tilting in two-dimensional hybrid halide perovskites. ACS Nano. 2021;15(6):10153-10162.
[DOI] -
66. Maughan AE, Ganose AM, Candia AM, Granger JT, Scanlon DO, Neilson JR. Anharmonicity and octahedral tilting in hybrid vacancy-ordered double perovskites. Chem Mater. 2018;30(2):472-483.
[DOI] -
67. Pandey T, Du MH, Parker DS, Lindsay L. Origin of ultralow phonon transport and strong anharmonicity in lead-free halide perovskites. Mater Today Phys. 2022;28:100881.
[DOI] -
68. Zeng Z, Chen C, Zhang C, Zhang Q, Chen Y. Critical phonon frequency renormalization and dual phonon coexistence in layered Ruddlesden-Popper inorganic perovskites. Phys Rev B. 2022;105(18):184303.
[DOI] -
69. Gupta M K, Kumar S, Mittal R, Chaplot SL. Soft-phonon anharmonicity, floppy modes, and Na diffusion in Na3FY (Y = S, Se, Te): Ab initio and machine-learned molecular dynamics simulations. Phys Rev B. 2022;106(1):014311.
[DOI] -
70. Simoncelli M, Marzari N, Mauri F. Wigner formulation of thermal transport in solids. Phys Rev X. 2022;12(4):041011.
[DOI]
Copyright
© The Author(s) 2025. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Publisher’s Note
Science Exploration remains a neutral stance on jurisdictional claims in published maps
and institutional affiliations. The views expressed in this article are solely those of
the author(s) and do not reflect the opinions of the Editors or the publisher.
Share And Cite



Science Exploration Style
Li Y, Zheng W, Pu JH, Wang Q, Jiang JH. Strong lattice anharmonicity and glass-like lattice thermal conductivity in nitrohalide double antiperovskites: A case study based on machine-learning potentials. Thermo-X. 2025;1:202501. https://doi.org/10.70401/tx.2025.0001
Tips
Copy completed.
Submit a Manuscript
Author Instructions
Cite this Article
Article Metrics
0
View
0
Download
Article Updates
