The role of glia autophagy in CNS homeostasis, ageing and disease
Onur Çakıcı
1,2
,
Nektarios Tavernarakis
1,2,*

*Correspondence to:
Nektarios Tavernarakis, Institute of Molecular Biology and Biotechnology, Foundation for Research and Technology-Hellas, Heraklion 70013, Crete, Greece.
E-mail: tavernarakis@imbb.forth.gr
Geromedicine. 2026;2:202527. 10.70401/Geromedicine.2026.0019
Received: December 25, 2025Accepted: March 17, 2026Published: March 23, 2026
Abstract
Autophagy is a fundamental catabolic process that is critical for maintaining cellular homeostasis and protein quality control in the central nervous system (CNS). While neuronal autophagy has been extensively characterized, growing evidence highlights the indispensable roles of glial autophagy, specifically in astrocytes, oligodendrocytes and microglia, in CNS physiology and pathology. These glial populations employ the autophagic machinery to regulate distinct but interconnected functions: astrocytes manage metabolic support and glutamate homeostasis; oligodendrocytes rely on autophagic flux for differentiation and myelin maintenance; and microglia employ specific pathways, such as LC3-associated phagocytosis, to orchestrate immune surveillance and inflammasome regulation. Impairment of glial autophagy has been implicated in non-cell-autonomous neurodegeneration, leading to excitotoxicity, myelin damage, the emergence of senescence-associated secretory phenotypes, and persistent neuroinflammation. Dysregulation of autophagic pathways during ageing has also been implicated in the pathogenesis of multiple neurodegenerative diseases, including Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, and amyotrophic lateral sclerosis. In this review we summarize the cell-type-specific molecular mechanisms of autophagy in glia, and delineate their role in the clearance of pathogenic aggregates such as β-amyloid, α-synuclein, and mutant huntingtin. A deeper understanding of the spatiotemporal dynamics of glial autophagy and associated intercellular crosstalk is essential to fully elucidate the complex etiology of age-associated neurodegenerative conditions.
Keywords
Alzheimer’s disease, astrocytes, autophagy, excitotoxicity, neurodegeneration, oligodendrocytes, Parkinson’s disease, senescence
1. Introduction
Autophagy, derived from the Greek words αὐτό (self) and φαγεῖν (to eat), is a conserved process mediated by lysosomes that enables eukaryotic cells to decompose and recycle their internal components and organelles[1,2]. This essential mechanism for cellular quality control plays a crucial role in maintaining homeostasis by eliminating damaged organelles, misfolded proteins, and invasive pathogens, and by repurposing the resulting macromolecules to maintain energy balance and cellular renewal. Functioning continuously and in response to stress, autophagy is vital for embryonic development, survival during the neonatal stages, cellular differentiation, and adaptation to metabolic, oxidative, and inflammatory challenges[1,3].
In mammalian cells, there are three distinct but interconnected autophagy: macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA). All these processes ultimately lead to lysosomes, where cellular components are broken down[2]. In the case of microautophagy, the process is the direct engulfment of the cytosol by the invagination of the lysosomal membranes[4]. On the other hand, CMA relies on cytosolic chaperones, especially heat shock cognate protein HSC70, to recognize substrate proteins with a KFERQ-like motif. These proteins are then directed to the lysosomal receptor LAMP2A for translocation and subsequent degradation[5]. Macroautophagy, often simply referred to as autophagy, involves the sequestration of cytoplasmic material within double-membrane vesicles known as autophagosomes. These autophagosomes merge with lysosomes to form autolysosomes, where cellular material is broken down and recycled[6-8].
The autophagic process unfolds through a sequence of meticulously orchestrated phases governed by autophagy-related (ATG) proteins that have been conserved throughout evolution[9-13]. Autophagy is triggered by either the suppression of the mTOR or the activation of AMP-activated protein kinase (AMPK) when cells experience nutrient deprivation or various stress conditions. This activation stimulates the ULK1–ATG13–FIP200–ATG101 complex, which initiates the formation of a membrane precursor, the phagophore[6-8]. The class III phosphatidylinositol 3-kinase complex I, consisting of VPS34, Beclin 1, ATG14, and AMBRA1, produces phosphatidylinositol-3-phosphate (PI3P). This molecule attracts downstream effectors WIPI2 and DFCP1 to nascent phagophores[9-13]. These effectors facilitate the assembly of the ATG12–ATG5–ATG16L1 complex and the attachment of LC3 (microtubule-associated protein light chain 3) to phosphatidylethanolamine, leading to the creation of membrane-bound LC3-II, a vital marker and structural component of the autophagosomal membrane[14,15].
Phagophore expansion is driven by lipid transfer, a process facilitated by ATG2 and ATG9, the latter of which acts as a scramblase to modify the phospholipid distribution across the membrane leaflets[16-20]. The autophagosome membrane is sealed by the endosomal sorting complexes required for transport machinery, which performs the membrane fission necessary to close the vesicle[21,22]. Once sealed, the autophagosome undergoes a maturation process involving the dissociation of ATG components and removal of PI3P, preparing it for fusion with lysosomes through the actions of Rab GTPases, SNARE proteins, and tethering complexes. After fusion, lysosomal hydrolases degrade the internal contents, releasing metabolites such as amino acids, fatty acids, nucleotides, and sugars into the cytoplasm for reuse in biosynthesis and energy production[23,24].
Autophagy plays a vital role in the central nervous system (CNS) by ensuring cellular homeostasis and safeguarding neurons[3]. Astrocytes, the most abundant glial cells in the brain, exhibit a wide array of autophagic activities that extend beyond simple self-preservation processes. In astrocytes, autophagy facilitates the removal of neurotransmitters, supports metabolic interactions with neurons, and helps manage neuroinflammation through interactions with endothelial cells, microglia, and oligodendrocytes[4]. Impaired astrocytic autophagy can lead to brain instability, heightened oxidative stress, and the initiation of neuroinflammatory responses[7].
A growing body of evidence has linked impaired autophagy to a range of neurological conditions, including Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), amyotrophic lateral sclerosis (ALS), and stroke, all of which are characterized by continuous neuronal damage and degeneration[5]. Traditionally, research on neurodegeneration has focused on neurons; however, glial cells, including astrocytes, microglia, oligodendrocytes, and ependymal cells, constitute the majority of the brain cell population[25]. Particularly astrocytes, are now recognized as important players in the disease process. A lack of adequate autophagy in astrocytes may exacerbate the accumulation of toxic protein aggregates, interfere with the metabolic support provided to neurons, and increase the inflammatory response[4,7].
Glial cells are involved in the pathophysiology of numerous neurodegenerative diseases and, act as a double-edged sword based on the severity of the injury and the specific nature of the disease[26]. On one hand, they can aid brain repair by performing phagocytosis, offering metabolic support, and releasing growth factors. However, they can also inflict harm by triggering excessive neuroinflammatory responses[26,27]. Considering the role of autophagy in both adaptive and innate immunity[28,29], autophagy is vital for glial functions related to controlling inflammatory signalling, eradicating pathogens, sustaining immune modulators, and modifying antigen presentation in various neurodegenerative diseases. Nonetheless, current studies have only begun to uncover how autophagy influences the regulation of glial cells and their effects on neurodegenerative disorders.
2. Autophagy in Glia
Although glial cells constitute at least half of the brain’s cellular composition, the role of autophagy in glial regulation remains largely unexplored. Glial cells are essential for maintaining neuronal homeostasis and overall brain function, influencing brain development, synaptic activity, metabolism, and recovery following injury. Recent research has indicated that autophagy is critical for glial cell formation[30,31]. However, the majority of studies on neurodegeneration, nerve damage, and aging have predominantly focused on neurons when investigating autophagy. Disruptions in the production or differentiation of glial cells can lead to structural and functional abnormalities in the brain. There is limited evidence concerning the role of autophagy in regulating glial function under both normal and pathological conditions[32,33]. The investigation of autophagy modulation as a therapeutic strategy in glia is constrained by an incomplete understanding of the ATG proteins and pathways involved in development and neurodegenerative diseases. Furthermore, there is a lack of understanding regarding the specificity of pharmacological agents that inhibit or enhance autophagy in glial cells.
As the understanding of the essential role of astrocytic autophagy in maintaining CNS health and addressing diseases expands, pharmaceutical strategies aimed at restoring or enhancing this process have demonstrated significant therapeutic potential. By targeting autophagic pathways, it may be possible to eliminate harmful aggregates, re-establish metabolic equilibrium, and reduce neuroinflammation[7]. In the following sections, we examine the mechanisms, functions, and implications of glial autophagy, emphasizing its increasing significance in maintaining brain homeostasis and mitigating neurodegenerative diseases.
2.1 Autophagy in astrocytes
Glial cells constitute approximately 50% of the total cellular population within the human brain, exhibiting significant heterogeneity across different brain regions[34,35]. Among this population, astrocytes account for approximately 20% and have traditionally been characterized as essential “housekeeping” cells responsible for neuronal support[36]. These cells function to maintain the extracellular milieu by regulating ionic and osmotic balance, facilitating neurotransmitter uptake, modulating neurovascular coupling, preserving blood-brain barrier (BBB) integrity, and ensuring synaptic maturation and sustained function[37,38]. This functional versatility is underpinned by significant physiological diversity[39,40]. Under pathological conditions, astrocytes undergo reactive astrogliosis, a process involving profound morphological and transcriptomic alterations determined by the specific nature and severity of the injury[41]. Whether these reactive phenotypes exert protective or deleterious effects remains a subject of debate, a complex issue that is further complicated by the difficulty of distinguishing astrocytic contributions from those of microglia, given the extensive molecular signalling and crosstalk between these cell types during neurodegeneration[42,43].
As the predominant glial cell type in the CNS, astrocytes are integral not only to synaptic architecture and metabolic support but also to the modulation of immune responses via the secretion of inflammatory mediators[41,44,45]. While the specific mechanisms of astrocytic autophagy are less defined than those in neurons, emerging evidence indicates that this pathway is vital for coordinating astroglial physiology. In vitro studies have demonstrated that autophagy is essential for viability and the clearance of protein aggregates, particularly when proteasomal degradation is compromised[46]. A paradigmatic example of this critical function is observed in Alexander disease, a rare astrogliopathy caused by dominant mutations in the gene encoding glial fibrillary acidic protein (GFAP). In this condition, mutant GFAP, an intermediate filament exclusively expressed in astrocytes, accumulates in cytoplasmic inclusions known as Rosenthal fibers, triggering proteasomal inhibition and stress pathway activation[47-49]. Consequently, autophagy is upregulated in an attempt to degrade these aggregates, suggesting a compensatory protective mechanism against proteotoxicity[50,51].
In addition to protein quality control, autophagy is a critical regulator of astrocyte differentiation and development. In the embryonic cortex, deletion of Atg5 impairs the differentiation of neural stem cells into astrocytes by inhibiting the SOCS2-JAK-STAT signalling axis, whereas Atg5 overexpression promotes astrogenesis[52]. A similar dependency is observed in the adult hippocampus, where autophagic inhibition restricts astrocyte differentiation[53]. Furthermore, autophagy maintains mitochondrial integrity during neuroinflammation; autophagic sequestration of fragmented mitochondria prevents excessive reactive oxygen species (ROS) production and cell death[54]. Disruption of core autophagy genes, such as ATG5 or ATG7, precipitates neurodegeneration in animal models, highlighting the necessity of this pathway for CNS survival[55,56].
In the specific context of ischemic stroke, astrocytic autophagy exhibits a complex, dual role that is contingent upon the intensity of activation and the cellular environment. Excessive autophagy, driven by factors such as circHECTD1 upregulation or zinc accumulation, can exacerbate astrocyte injury. Conversely, moderately regulated autophagy is indispensable for survival, as its inhibition promotes apoptosis[57]. This delicate balance suggests that while unchecked autophagy may be detrimental, basal autophagic activity is required to withstand ischemic stress, oxidative damage, and inflammation. Thus, modulating astrocytic autophagy represents a potential therapeutic strategy, provided that the intervention can restore equilibrium rather than simply suppressing or hyperactivating the pathway[57-60].
2.2 Autophagy in oligodendrocytes
Oligodendrocytes, the primary myelinating glia of the CNS, undergo profound structural and functional remodelling throughout their lifespan, a process driven by extensive protein synthesis, lipid production, and membrane compaction. Accounting for approximately 75% of the glial population in the human neocortex[61], these cells act as fundamental architects of white matter, which is composed of glial cells along with myelinated and unmyelinated axons[62,63]. However, white matter is highly susceptible to aging, exhibiting volume reductions of up to 28% and accumulating abnormalities beginning in middle age[62,64]. Given that both developmental myelination and regenerative remyelination rely strictly on the differentiation of oligodendrocyte precursor cells (OPCs)[65,66], autophagy has emerged as a critical regulatory mechanism governing oligodendrocyte function and resilience throughout the lifespan[67-69].
The lysosome-dependent catabolic pathway of autophagy is indispensable for oligodendrocyte differentiation, survival, and the physical process of myelination[70]. The transition from OPCs to mature oligodendrocytes is marked by a robust upregulation of autophagic flux, characterized by increased levels of conjugated ATG5 and LC3B-II, decreased p62, and a significant expansion of autophagosomes and autolysosomes within distal processes. Autophagic markers, including ATG5 and LC3B, localize to developing myelin sheaths, suggesting that autophagosomes generated in expanding myelin are trafficked to the soma for degradation. This activity is essential for development; Atg5 is required to increase autophagosome puncta and elevate LC3-II levels in processes, and appropriate cytoplasmic levels of this protein are necessary to ensure proper myelin compaction[70]. Disruption of this autophagic machinery has severe consequences for myelin architecture. Conditional deletion of Atg5 in OPCs precipitates apoptosis, inhibits differentiation, and results in significant hypomyelination, with surviving cells exhibiting cytoplasmic retention, swollen inner tongues, and myelin outfoldings. Similarly, pharmacological inhibition impairs myelination in OPC–dorsal root ganglion co-cultures, whereas autophagy induction via Tat-beclin1 or Atg5 overexpression enhances differentiation and myelin segment formation[70].
Mechanistically, autophagy is vital for the turnover of myelin basic protein (MBP). In the absence of autophagy, MBP fails to integrate into the myelin sheath and instead accumulates as high-molecular-weight aggregates within the cytoplasm. This is compounded by the downregulation of endocytic recycling proteins, preventing the salvage of damaged MBP[71]. Furthermore, autophagy deficiency disrupts mitochondrial quality control (mitophagy), leading to compromised oxidative phosphorylation and energy deficits in white matter. The convergence of proteotoxicity, mitochondrial dysfunction, and cellular loss results in “precocious aging-related demyelination”, characterized by thinner myelin (increased G-ratios) and axonal loss[71]. Consequently, animals lacking macroautophagy display age-dependent neurodegeneration, behavioral impairments, seizures, and premature mortality[72,73].
In addition to development, autophagy functions as a critical housekeeping mechanism in mature oligodendrocytes to preserve CNS integrity[74]. Inactivation of autophagy in adult myelinating cells leads to morphological defects such as myelin decompaction and splitting, disproportionately affecting large-caliber axons. This disruption alters protein homeostasis, increasing proteolipid protein (PLP) while reducing MBP and CNP, and compromises neuronal support, as evidenced by increased hippocampal caspase-3 activation. In tamoxifen-inducible plpCreERT2; atg5f/f models, selective deletion of Atg5 in adults causes severe pathology in aged mice, including axonal degeneration, impaired memory, and microglial activation indicative of myelin debris clearance[74]. Aging exacerbates these vulnerabilities, as autophagic efficiency naturally declines across the oligodendroglial lineage, contributing to cognitive, motor, and sensory impairments[75-80]. Aging myelin exhibits structural degeneration, such as balloon-like expansions and paranodal pilling, whereas OPCs lose differentiation potential and mature oligodendrocytes reduce metabolic support via downregulated MCT1 transporters and lipid synthesis[81].
Crucially, the age-related decline in autophagy drives OPC dysfunction beyond simple myelination failure; it induces premature cellular senescence. Autophagy-deficient OPCs accumulate senescence markers P21 and P16INK4A, adopting a senescence-associated secretory phenotype (SASP). These cells secrete elevated levels of chemokines CCL3 and CCL5, which bind to CCR5 receptors on glutamatergic neurons. This aberrant signalling suppresses synaptic transmission, neuronal excitability, and long-term potentiation. Thus, while autophagy loss directly damages myelin, the resulting SASP-driven chemokine signalling is the primary driver of neuronal plasticity deficits and cognitive decline[71]. Finally, autophagy is integral to recovery from neurodegenerative injury. Conditional Atg5 ablation impedes recovery following spinal cord injury[82], and disruptions in this pathway impair myelin plasticity in multiple sclerosis, precipitating demyelination[30]. Conversely, Atg7 deletion leads to progressive motor decline and neurodegeneration[72]. These findings highlight the critical necessity of elucidating oligodendroglial autophagy mechanisms to develop targeted therapies for demyelinating and neurodegenerative disorders.
2.3 Autophagy in microglia
Microglia, the resident innate immune cells of the CNS, are myeloid-lineage cells derived from embryonic erythromyeloid progenitors in the yolk sac that migrate into the developing brain[83,84]. As essential guardians of CNS homeostasis, these cells continuously monitor the brain parenchyma to facilitate neurotransmission, regulate blood flow, and provide metabolic support. Their housekeeping functions include the phagocytic clearance of apoptotic cells, myelin debris, and the sculpting of neuronal circuits via synaptic pruning[85-88]. Microglial populations exhibit significant spatiotemporal heterogeneity in density and transcriptomic profiles, with individual cells capable of persisting for decades[86,89-91]. Under conditions of injury or stress, microglia undergo rapid proliferation and morphological transformation into an ameboid state. Recent advancements in transcriptomic analyses have revealed that microglia exhibit a complex spectrum of activation states. Particularly in the context of neurodegeneration, these cells undergo a transformation into a specialized disease-associated microglia (DAM) phenotype, which is a state heavily reliant on intact autophagic flux for the clearance of pathological aggregates. However, this state can become neurotoxic if the autophagic capacity is chronically overwhelmed[90,92,93].
Autophagy functions as a critical regulator of cellular homeostasis within myeloid lineages, governing innate and adaptive immune responses, phagocytosis, and antigen presentation[94-100]. Although research specifically targeting microglial autophagy is nascent, existing evidence indicates it is indispensable for lysosomal degradation and physiological responsiveness. Genetic ablation of autophagic pathways disrupts CNS stability; specifically, the deletion of Atg7 impairs synaptosome degradation, resulting in defective connectivity and an accumulation of dendritic spines, highlighting the role of this pathway in synaptic maintenance[101]. Furthermore, microglia-specific Atg7 deletion leads to the intracellular accumulation of phagocytosed myelin and exacerbates multiple sclerosis–like pathology, distinguishing its role from Ulk1-dependent pathways[102]. Consistently, autophagy-deficient microglia drive dysregulated oligodendrocyte homeostasis, characterized by increased cell density and myelination markers, thereby increasing susceptibility to lethal seizures[73].
Autophagic flux is intrinsically linked to microglial activation states. The inhibition of autophagy via Atg7 deletion, Atg5 knockdown, or pharmacological blockade precipitates a shift toward a pro-inflammatory phenotype, characterized by ameboid morphology and the overproduction of cytokines such as IL-1β, IL-6, and TNF[103,104]. These autophagy-deficient cells display transcriptomic profiles resembling aged microglia enriched in neurodegenerative pathways[102,104]. In vitro, Atg5 deletion exacerbates TNF-α-induced neurotoxicity, a potentially reversible state using rapamycin or AKT inhibitors, suggesting that autophagy modulates polarization during inflammation[103]. Moreover, disrupted autophagy triggers NLRP3-dependent inflammation, contributing to pathology reminiscent of PD[105].
In the context of AD, autophagy is markedly upregulated in DAM, evidenced by elevated LC3-II and phosphorylated ULK1[106]. This upregulation facilitates the compaction of amyloid plaques and the clearance of amyloid-beta (Aβ) fibrils and α-synuclein (α-syn)[107-110]. Conversely, the loss of autophagy compromises these protective mechanisms; Atg7-deficient microglia fail to sustain the DAM signature (downregulating Clec7a, Trem2, and Tyrobp) and instead adopt a senescence-associated microglial (SAM) phenotype. This transition results in diffuse, toxic plaques and dystrophic neurites[108]. Therapeutic intervention with senolytics, such as dasatinib and quercetin, can eliminate these senescent cells, restoring plaque compaction and reducing neurotoxicity, thereby underscoring the necessity of autophagy for maintaining the protective DAM identity[108].
Finally, aging in the brain is characterized by “inflamm-aging”, a low-grade chronic inflammation driven by the accumulation of undigested cellular waste and organelles[111]. This decline in lysosomal clearance is mirrored by reduced mitophagy, which correlates with human ageing and age-associated disorders[112,113]. Atg7 deficiency is linked to elevated levels of inflammatory cytokines and inflammasome activation in macrophages, processes that are normally regulated by autophagy[114,115]. While the specific contributions of glial autophagy to the aging process require further elucidation, bolstering these pathways via lifestyle or pharmacological interventions offers a potential strategy to mitigate age-related neurodegeneration.
2.4 Inter-glial crosstalk: A coordinated autophagic network
A critical broader principle emerging in neurobiology is that glial cells do not operate in isolation; rather, their autophagic capacities are intricately linked through constant intercellular communication. The functional state of one glial population directly affects the metabolic and autophagic burden placed upon another. Recent comprehensive literature has elaborated the significance of these complex, overarching glial networks and their collective impact on brain health in the context of astrocytes[116].
Inter-glial communication is crucial for coordinating autophagic processes that sustain homeostasis and promote repair in the CNS. Particularly, the dynamic signaling between astrocytes and microglia is pivotal in regulating neuroinflammation and debris clearance. Microglia, as primary injury sensors, secrete cytokines, such as IL-1α, TNF, and C1q, which induce a neurotoxic and reactive state in astrocytes. This reactive transformation severely impairs astrocytic autophagic flux and reduces their capacity to metabolically support neurons. Consequently, overwhelmed astrocytes release fragmented mitochondria and misfolded proteins that microglia uptake for degradation through LC3-associated phagocytosis (LAP), demonstrating a reciprocal regulation of autophagy between these glial populations[117,118].
Similarly, microglia-oligodendrocyte interactions rely on coordinated autophagic functions for effective myelin repair. Upon myelin damage, microglia need to clear myelin debris via autophagy to enable OPCs to differentiate and remyelinate axons. Failure of microglial autophagy results in the persistence of myelin debris, which obstructs OPC maturation and stalls remyelination. Such failure acts as a dominant catalyst for network-wide dysfunction, emphasizing how autophagic deficits in one glial population adversely impact others. Notably, microglia-derived transglutaminase-2 signals through the G protein-coupled receptor ADGRG1 on OPCs, promoting their proliferation and enhancing remyelination efficacy in demyelinating conditions, further illustrating the importance of microglia-to-oligodendrocyte autophagic and signaling communication[119,120].
In addition to these direct cellular interactions, glial crosstalk influences neuroinflammatory pathways that exacerbate or ameliorate disease progression. Activated glial cells, including microglia and astrocytes, release proinflammatory cytokines that not only promote neurotoxicity but also impair BBB integrity, amplifying neuroinflammation. The intercellular signaling among glia shapes the inflammatory milieu and metabolic conditions within the CNS microenvironment. This complex glial interplay is instrumental in determining outcomes in neurodegenerative diseases and aging-related neuroinflammaging, where dysregulated autophagy and senescence contribute to chronic inflammation and neuronal dysfunction[117,121-123].
In summary, the autophagic capacities of individual glial populations are tightly and dynamically coupled through intercellular signaling pathways. The functional impairment in one glial type increases the metabolic and autophagic burden on others, precipitating a cascading effect that compromises CNS homeostasis and repair mechanisms. Understanding these mechanisms opens avenues for targeted therapies aimed at modulating glial autophagy and their communication networks to mitigate neuroinflammation and promote myelin repair.
3. Glial Autophagy in Neurodegenerative Diseases
Under physiological conditions, autophagy functions as a cornerstone of cellular homeostasis; however, its dysregulation is increasingly implicated in the onset and progression of neurodegenerative pathologies. The impairment of autophagic pathways precipitates the accumulation of misfolded proteins and damaged organelles, thereby inducing cellular stress that ultimately culminates in neuronal toxicity. This dependence is substantiated by experimental evidence demonstrating that the ablation of core autophagy genes, such as ATG5 or ATG7, is sufficient to drive progressive neurodegeneration in animal models, thereby underscoring the indispensability of this pathway for survival[55,56,124]. Historically, investigations have predominantly focused on neuronal autophagy. However, the contribution of glial cells to the neurodegenerative landscape has recently emerged as a critical area of inquiry. Given that glial cells are fundamental to brain development and function, understanding the mechanisms by which they maintain proteostasis and the consequences of their failure, remains a vital, albeit relatively unexplored, frontier. Emerging evidence suggests that glial autophagy significantly modulates disease trajectories[58-60]. Consequently, this review examines the distinct autophagic roles of astrocytes, oligodendrocytes, and microglia, and delineates how these processes influence the pathology of major neurodegenerative disorders, including AD, PD, HD, and ALS. To conceptualize the shared and distinct mechanisms of glial autophagic failure across these diverse pathologies, Table 1 provides a comparative synthesis of the primary protein aggregates, specific autophagic defects, and the resulting glial and neuronal consequences in AD, PD, HD, and ALS.
Table 1. Comparative synthesis of glial autophagic dysregulation in major neurodegenerative diseases.
| Disease | Glial Cell Type | Primary Pathogenic Aggregate | Specific Autophagic Defect/Mechanism | Resulting Glial Phenotype | Impact on Neurons/CNS |
| AD | Astrocytes | Extracellular Aβ | APOE4-mediated reduction in autophagic flux; TFEB/FOXO3A dysregulation. | Pro-inflammatory “A1” phenotype; loss of neurotrophic support. | Aβ-mediated neuronal death; increased tau phosphorylation. |
| Microglia | Extracellular Aβ, Tau | Atg7 deficiency; impaired LAP/LANDO; accumulation of p62. | SAM; NLRP3 inflammasome hyperactivation. | Dystrophic neurites; diffuse toxic plaques; chronic neuroinflammation. | |
| Oligodendrocytes | Aβ42 | Atg5/Atg7 silencing; p62 accumulation. | Defective remyelination; accumulation of myelin debris. | Axonal degeneration; loss of neuronal synchrony. | |
| PD | Astrocytes | Extracellular α-synuclein | Disrupted BAG3-dependent clearance; ATP13A2/LRRK2GBA1 mutations impairing lysosomes. | Astrogliosis; mitochondrial dysfunction; oxidative stress. | Reduced dopaminergic neuron survival; excitotoxicity. |
| Microglia | Extracellular α-synuclein | Impaired synucleinphagy; Atg5 deletion driving PDE10A-cAMP-NLRP3 axis. | Hyper-inflammatory state; exosomal α-syn release. | Accelerated dopaminergic neuron loss; enhanced protein spreading. | |
| Oligodendrocytes | Extracellular α-synuclein | Impaired autophagic flux (decreased LC3-II, increased p62). | Formation of GCIs; myelin swelling. | Axonal disorganization; early white matter pathology. | |
| HD | Astrocytes | mHTT | mHTT co-localizes with TPC2, blocking NAADP-induced Ca2+ release and autophagic flux. | Loss of GLT-1 transporter; impaired glutamate uptake. | Severe excitotoxicity; non-cell-autonomous neuronal death. |
| Microglia | mHTT | Unknown specific autophagic defect; activation by mHTT. | Secretion of chemokines (CCL3, CCL4, CCL5). | Activation of neuronal mTORC1 via CCR5, suppressing neuronal autophagy. | |
| ALS | Astrocytes | SOD1, TDP-43 | Impairment of mTOR/ULK1/Beclin-1/p62/LC3B pathway; reduced LC3B-II. | Secretion of pro-inflammatory cytokines (IL-1β, TNF-α, IL-6); loss of GLT-1. | Secretion of TGF-β1 suppresses neuronal autophagy; motor neuron death. |
AD: Alzheimer’s disease; PD: Parkinson’s disease; HD: Huntington’s disease; ALS: amyotrophic lateral sclerosis; mHTT: mutant huntingtin; SOD1: Cu/Zn superoxide dismutase; TDP-43: TAR DNA-binding protein 43; APOE: apolipoprotein E; TFEB: transcription factor EB; BAG3: BCL2-associated athanogene 3; NLRP3: NOD-like receptor family pyrin domain containing 3; PDE: phosphodiesterase 10A; mTOR: mechanistic target of rapamycin; ULK: unc-51 like autophagy activating kinase 1; SAM: senescence-associated microglia; GCIs: glial cytoplasmic inclusions; GLT-1: glutamate transporter 1; CCL: C–C motif chemokine ligand; TNF: tumor necrosis factor; IL: interleukin; CNS: central nervous system; CCR: C–C motif chemokine receptor; TGF: transforming growth factor.
3.1 AD
AD represents the predominant form of dementia, manifesting as cognitive impairments, including memory loss and learning deficits[125]. The condition is classically distinguished by neuropathological hallmarks comprising extracellular Aβ plaques and intracellular neurofibrillary tangles composed of hyperphosphorylated tau[126]. Furthermore, neuroinflammation has been identified as a vital pathological element that influences both the onset and progression of the disease[127,128]. Specifically, reactive astrocytes frequently aggregate near Aβ plaques; their accumulation, in conjunction with tau phosphorylation, correlates with clinical progression and cognitive deterioration seen in patients[129-132].
Regarding cellular mechanisms, disruption of autophagic flux in AD results in the buildup of autophagic vesicles filled with undigested matter within dystrophic neurites. This accumulation indicates a failure in autophagosome maturation and lysosomal fusion, subsequently hindering the clearance of toxic proteins such as Aβ and tau[59,133]. Concurrently, reduced expression of Beclin-1 is observed and associated with increased Aβ burden and neurodegeneration[59,134]. Crucially, the accumulation of the autophagy adaptor SQSTM1/p62 in AD models serves as further evidence of this dysfunction[59,135]. Highlighting the centrality of lysosomal clearance in disease mechanisms, rare mutations in genes required for endosomal-lysosomal function, such as CHMP2B, disrupt autophagy and present features overlapping with AD pathology[136]. Additionally, the age-related reduction in neuronal autophagic capacity further aggravates tau pathology[137].
3.1.1 Astrocytes
In AD, astrocytes collaborate with microglia to mitigate the accumulation and toxicity of extracellular Aβ by actively internalizing and degrading the protein. However, reactive astrocytes aggregate around Aβ plaques, and their altered function significantly influences the progression of the disease[129-132]. Astrocytes serve as the primary source of apolipoprotein E (APOE), a critical mediator of Aβ uptake and degradation, and they secrete specific Aβ-degrading proteases (Figure 1A)[33,138-140]. The APOE4 isoform, identified as the strongest genetic risk factor for late-onset AD, impairs astrocytic clearance of Aβ42 relative to the APOE3 isoform[33,141-143]. Consistent with this deficit, APOE4-expressing astrocytes exhibit reduced autophagy flux under both basal and stimulated conditions, which compromises their ability to degrade Aβ[144,145].
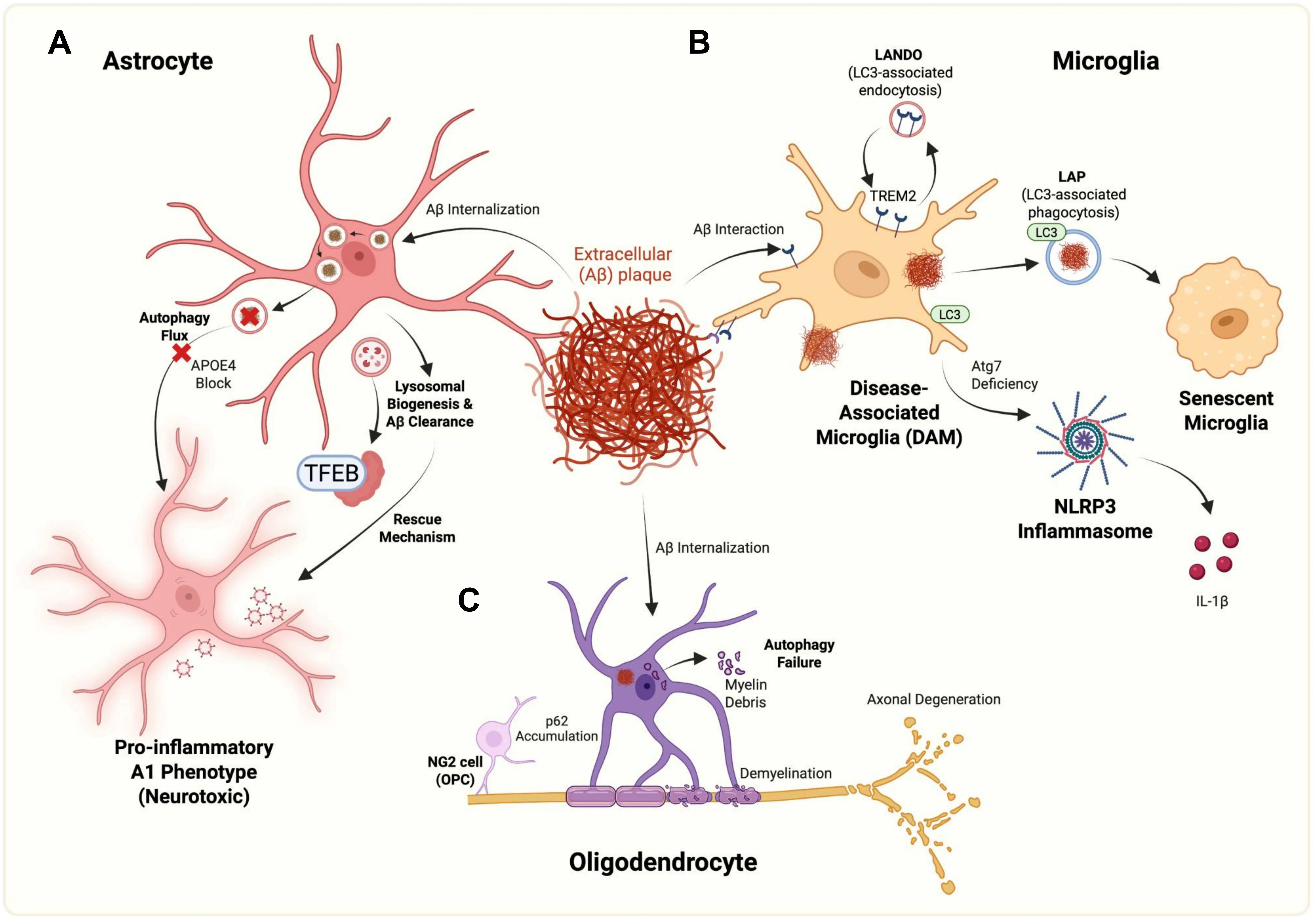
Figure 1. Dysregulated glial autophagy and cross-talk in Alzheimer’s disease. (A) Astrocytic response: Astrocytes internalize Aβ to facilitate clearance, a process heavily reliant on APOE isoforms. APOE4 carriers exhibit impaired autophagic flux and reduced Aβ degradation compared to APOE3. Overexpression of TFEB can rescue this phenotype by promoting lysosomal biogenesis and Aβ trafficking. Under chronic stress, astrocytes adopt a neurotoxic A1 phenotype, characterized by downregulated autophagy genes (SQSTM1, MAP1LC3B) and loss of neurotrophic support; (B) Microglial mechanisms: Disease-associated microglia utilize LC3-associated endocytosis to recycle TREM2 receptors and LC3-associated phagocytosis to clear Aβ plaques. Disruption of autophagy (e.g., Atg7 deficiency) leads to NLRP3 inflammasome hyperactivation and IL-1β release, driving cells toward a SAM phenotype; (C) Oligodendrocyte pathology: NG2 precursor cells are recruited to plaques to engulf Aβ. However, autophagy failure results in the accumulation of p62 and myelin debris, leading to myelin deterioration and axonal degeneration. Created in BioRender.com. APOE: apolipoprotein E; TFEB: transcription factor EB; SAM: senescence-associated microglial.
Fortunately, this impaired autophagy in APOE4 astrocytes is partially reversible via pharmacological activation of autophagy pathways. The upregulation of transcription factor EB (TFEB), a master regulator responsible for lysosomal biogenesis, autophagosome formation, and autophagosome-lysosome fusion, restores lysosomal function and enhances the uptake and degradation of Aβ[60,146-148]. APOE4 disrupts this regulation; for example, APOE4-expressing glioblastoma cells fail to induce specific autophagy genes under starvation conditions compared to APOE3 cells[144]. In vivo studies have demonstrated that TFEB overexpression in the hippocampal astrocytes of APP/PSEN1 mice increases lysosomal activity and reduces amyloid plaques, while exogenous TFEB in primary astrocytes promotes Aβ uptake, trafficking, and degradation through the endolysosomal system[148]. TFEB’s capacity to regulate genes essential for autophagosome and lysosome biogenesis further underscores its central role in this process[149].
Direct evidence supporting astrocytic autophagy in Aβ processing reveals that internalized Aβ1–42 localizes predominantly to LC3-positive autophagosomes, whereas the deletion of Atg5 disrupts this LC3–Aβ co-localization and impairs Aβ clearance[150]. Autophagy also facilitates the degradation of extracellular Aβ by promoting the release of insulin-degrading enzyme, levels of which are reduced in Atg7-deficient mice[151]. Furthermore, astrocytes display “autophagy plasticity”, evidenced by the dynamic upregulation of SQSTM1 and LC3B in response to Aβ, which helps maintain metabolic stability and proteostasis under proteotoxic stress[135]. When autophagy is inhibited during Aβ exposure, astrocytes experience increased cell death, mitochondrial dysfunction, and oxidative stress. Highlighting this protective role, in vivo astrocyte-specific disruption of autophagy exacerbates hallmark AD pathologies, including greater plaque burden, increased tau phosphorylation, reactive astrogliosis, neuronal loss, and cognitive impairments, while astrocytic LC3B overexpression ameliorates these phenotypes[135].
Transcriptomic analyses indicate that AD astrocytes adopt a proinflammatory, neurotoxic “A1” phenotype, downregulating neuronal support genes and contributing to Aβ-mediated neuronal death[152-157]. APOE4 exacerbates this shift by altering the expression of ATG, including SQSTM1, MAP1LC3B, and LAMP2, and by interfering with transcription factors, such as TFEB and FOXO3A[33,60,144,145]. Consequently, diminished autophagy and mitophagy programs weaken astrocytic neurotrophic and synaptogenic support, causing APOE4 astrocytes to fail in supporting neuronal survival[33,144,157]. Beyond Aβ clearance, autophagy-inducing agents such as trehalose or rapamycin promote tau clearance in neuronal and mouse tauopathy models[158,159]. Rapamycin also reduces tau tangles and reactive astrocytes in tauopathy models, although it remains unresolved whether these effects occur directly in astrocytes or secondarily via neuronal protection[160]. Additionally, rapamycin enhances plaque removal in APOE4 astrocytes within an in situ Aβ plaque clearance model, further supporting the therapeutic potential of autophagy activation[145,161].
3.1.2 Oligodendrocytes
AD pathology extends to oligodendrocytes and myelin integrity, where β-amyloid peptides exert complex, dual effects that are both toxic and regulatory[162,163]. Specifically, Aβ-induced signalling, mediated through the integrin β1 receptor, Src-family kinase Fyn, and Ca2+/CaMKII pathways, promotes the differentiation, maturation, and survival of oligodendrocytes. This signalling cascade enhances MBP expression and facilitates remyelination in cerebellar slices[71,162]. Despite these potential repair mechanisms, myelin deterioration remains a hallmark of aging and AD progression, significantly contributing to axonal dysfunction and cognitive decline[71,162,164]. The loss of myelin disrupts neuronal synchrony, which in turn facilitates functional disconnections, neuronal loss, and the cognitive deficits characteristic of AD[164]. Furthermore, cortical myelin damage occurs prior to clinical symptoms, and this dysfunction actually promotes Aβ deposition by causing the accumulation of the amyloid-processing machinery within axonal swellings[165,166]. Complicating matters further, Aβ-associated microglia are redirected toward sites of myelin damage, thereby linking myelin deterioration to both direct and indirect pathways of Aβ accumulation[166].
In response to pathology, OPCs, specifically NG2 cells, are recruited to amyloid plaques where they internalize and degrade Aβ42 (Figure 1C). This process results in enhanced autophagy, evidenced by the elevated expression of MAP1LC3A, MAP1LC3B, and BECN1[82,167]. However, the proliferation and differentiation of OPCs are impaired by dysregulated astrocyte-dependent glutamate–glutamine cycling, suggesting that dysfunctional glial crosstalk plays a role in AD[82]. The critical nature of autophagy is demonstrated by the conditional silencing of ATGs Atg5 or Atg7 in oligodendrocytes, which induces neurodegenerative phenotypes defined by myelin abnormalities, the accumulation of myelin debris, axonal degeneration, and neuronal death[72,168]. Such myelin dysfunction correlates with the toxic accumulation of myelin PLP[168]. Furthermore, impairment of autophagy in OPCs increases their vulnerability to ER stress and reduces cell survival, though acute inhibition of autophagy may temporarily enhance OPC survival[82,169]. Additional research indicates that the simultaneous inhibition of the pro-apoptotic gene BCL2 and autophagy increases oligodendrocyte death[170]. Finally, key genes enriched in oligodendrocytes, including LAMP2 and PIP4K2A, which act as mediators of lysosomal and autophagosome clearance, are frequently deregulated in the brains of AD patients, implicating them in the crosstalk between AD pathology and oligodendrocyte autophagy[171].
3.1.3 Microglia
Microglia function as central architects of the immune response in AD, mediating both synaptic dysfunction and neuroinflammation induced by Aβ[172]. Within these cells, autophagy serves as a critical negative regulator of cytokine production and inflammasome activation; specifically, the deletion of Atg7 skews microglia toward a proinflammatory phenotype characterized by elevated cytokine secretion[104,173]. Mechanistically, autophagic components such as LC3 associate directly with NLRP3 inflammasome aggregates to temper the release of IL-1β and IL-18[173]. Furthermore, microglia actively phagocytose axonal fragments, cellular debris, extracellular Aβ, and tau deposits. These clearance processes are tightly coordinated with autophagy through LAP and LC3-associated endocytosis (LANDO) pathways, which recruit autophagy proteins Atg5, Atg7, and Beclin1 to phagosomal membranes to facilitate lysosomal fusion and degradation (Figure 1B)[174-178]. Together, these mechanisms significantly improve the efficiency with which microglia clear pathogens and Aβ aggregates.
The LANDO pathway is particularly essential for recycling microglial Aβ receptors, such as TREM2, and its disruption is known to exacerbate neurodegeneration in AD models[175]. The direct link between microglial autophagy and AD pathogenesis is further evidenced by microglial Atg7 knockout models, which exhibit impaired Aβ degradation and increased tau pathology[104,107]. To counteract such deficits, regulatory pathways such as AMPK activation can promote microglial autophagy in response to Aβ, thereby enhancing lysosomal clearance[107]. Additionally, treatment with interferon-γ has been shown to restore autophagy in microglia by inhibiting Akt/mTOR signalling, a process that promotes Aβ clearance and leads to cognitive improvements in AD mice[179].
Microglia located adjacent to amyloid plaques often adopt a specific “disease-associated” subtype, distinguished by altered expression of lysosomal/phagocytic pathway genes and AD risk genes, including APOE, TREM2, CSTD, LPL, and TYROBP[174]. However, chronic exposure to Aβ eventually reduces microglial autophagy, indicated by the accumulation of p62 protein and decreased levels of the cargo receptor NBR1, which impairs Aβ degradation and exacerbates disease progression[180,181]. This impairment is further compounded by elevated microRNA-17, which inhibits microglial autophagy; conversely, inhibiting this microRNA restores clearance capabilities[181,182].
Defective autophagy also hinders the clearance of dysfunctional mitochondria, leading to mitochondrial fragmentation, ATP depletion, and overproduction of ROS, factors that collectively enhance neuroinflammation and AD pathology. Stimulation of mitochondrial autophagy (mitophagy) shifts microglia toward a beneficial phagocytic phenotype, reducing NLRP3/caspase-1-dependent inflammation and TNF levels through PINK1-mediated pathways, thus offering significant therapeutic promise[183,184]. Ultimately, microglial autophagy exerts protective roles by mitigating neuroinflammation, preserving synaptic function, and enhancing the clearance of Aβ and tau, underscoring its critical status as a target for therapeutic intervention in AD[104,107,172,175,179].
In AD, a recurring pattern is characterized across the three main types of glial cells. Initially, the autophagic mechanism is upregulated as a protective mechanism against Aβ and myelin debris. However, it eventually becomes overwhelmed due to ongoing proteotoxic stress and genetic susceptibilities like APOE4. This transition from adaptive clearance to autophagic failure represents a key turning point in AD pathogenesis. When astrocytic, microglial, and oligodendroglial autophagy stalls, they often shift to detrimental states, adopting neurotoxic, senescent, or pro-inflammatory characteristics, such as A1 astrocytes and SAM microglia. Consequently, the breakdown of glial autophagy in AD should not be considered merely as a secondary consequence of neurodegeneration. Instead, it functions as a primary driver of continuous inflammasome activation, impaired remyelination, and the uncontrolled progression of tau and Aβ pathology.
3.2 PD
PD ranks as the second most prevalent neurodegenerative disorder, following AD, and primarily exerts its effects on the motor system. This condition is fundamentally characterized by the substantial loss of dopaminergic neurons within the substantia nigra pars compacta, resulting in disrupted dopaminergic neurotransmission. A defining neuropathological feature is the presence of Lewy bodies, cytoplasmic inclusions that are predominantly composed of aggregated α-syn[185]. At the pathological level, PD is driven by a complex interplay of mechanisms, including mitochondrial dysfunction, oxidative stress, and impaired protein degradation processes such as autophagy, alongside neuroinflammation and glial cell activation[186].
Alongside these primary mechanisms, growing evidence highlights the significant influence of the microbiota-gut-brain axis in the development and progression of PD. According to the Braak hypothesis, α-syn pathology might originate in the enteric nervous system due to environmental factors or intestinal dysbiosis, eventually spreading to the CNS through the vagus nerve. Within this axis, autophagy acts as a crucial initial defense to eliminate misfolded proteins in the gut. However, persistent gastrointestinal inflammation and alterations in the microbiome can impair local autophagic and lysosomal processes, resulting in the unchecked buildup of α-syn aggregates in enteric neurons. When the peripheral autophagic capacity is overwhelmed, these harmful aggregates spread retrogradely to the brain. This transmission from the periphery to the center is accompanied by systemic inflammatory signals that further impair the autophagic machinery in central glial cells, thereby hastening the neuroinflammatory processes and protein spread that are characteristic of PD[187-190].
3.2.1 Astrocytes
Astrocytes serve as fundamental regulators of homeostasis within the CNS, and a growing body of evidence highlights their central and active role in the pathogenesis of PD. This involvement is mediated primarily through mechanisms governing autophagy, lysosomal degradation, mitochondrial quality control, inflammatory signalling, and proteostasis. Post-mortem examinations of PD brains have established that cytoplasmic inclusions containing α-syn accumulate not only in neurons but also in astrocytes, proving that these glial cells internalize and sequester pathological protein aggregates as the disease progresses[191,192]. Physiologically, astrocytes internalize α-syn released from neurons via a TLR4-independent endocytosis pathway (Figure 2A)[193,194]; once inside the cell, the protein is targeted to lysosomes for degradation, suggesting that astrocytes normally function to clear extracellular α-syn aggregates[195]. However, this protective capacity has limits. High levels of extracellular α-syn can activate astrocytes through TLR4-dependent inflammatory signalling, revealing that the protein itself can act as a pro-inflammatory stimulus[194].
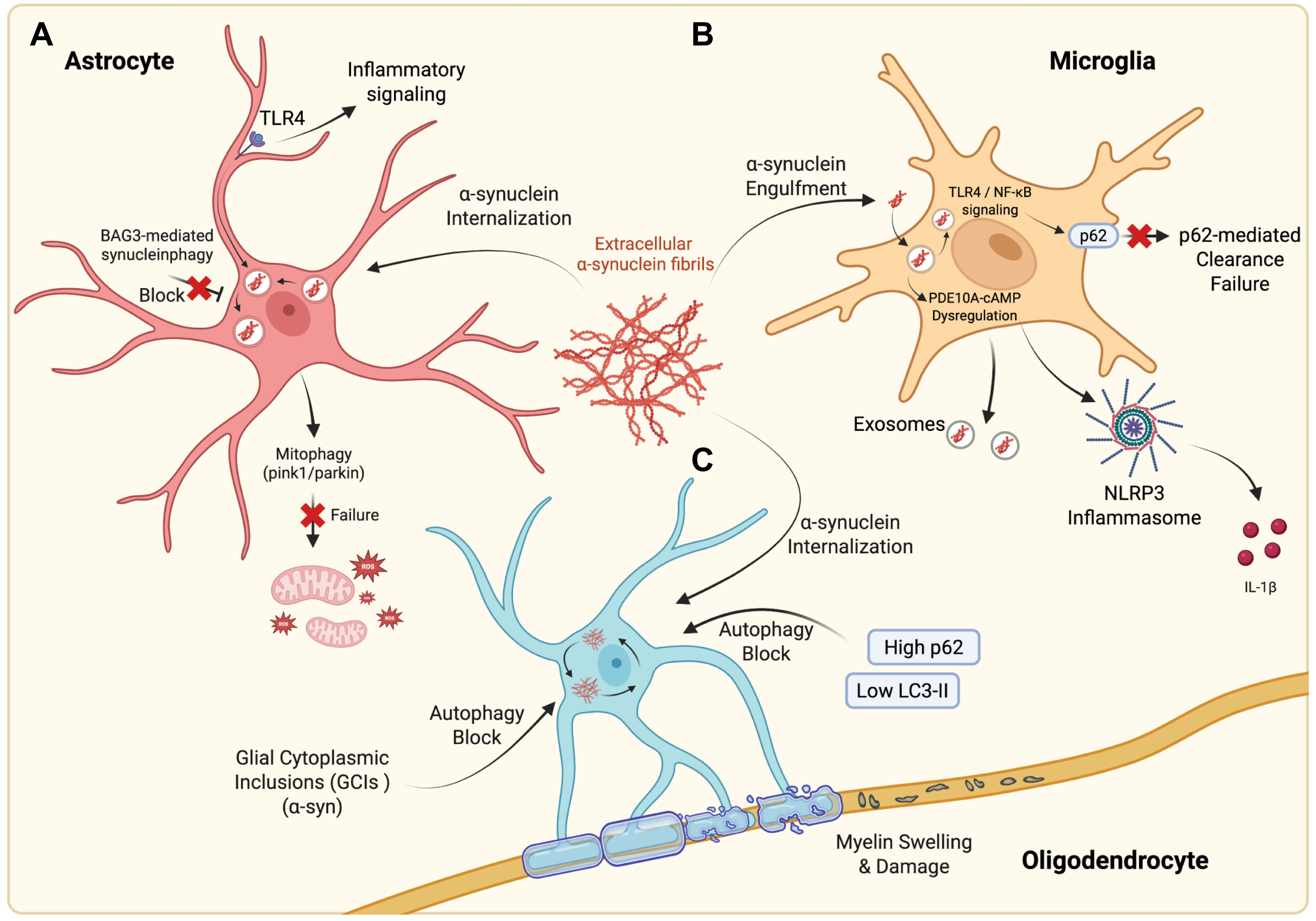
Figure 2. α-synuclein trafficking and autophagic failure in Parkinson’s disease. (A) Astrocytic synucleinphagy: Astrocytes internalize neuronal α-syn. High levels of extracellular α-syn trigger TLR4-dependent inflammatory signalling. The clearance of intracellular aggregates occurs via BAG3-mediated autophagy. Disruption of mitochondrial quality control (mitophagy), such as through Kir6.1 ablation, exacerbates ROS production and neurotoxicity; (B) Microglial clearance and spreading: Microglial uptake of α-syn activates TLR4/NF-κB pathways, which upregulate p62 to facilitate synucleinphagy. Conversely, autophagy deficiency or PDE10A-cAMP dysregulation triggers the NLRP3 inflammasome. Dysfunctional microglia may also release α-syn-loaded exosomes, contributing to the propagation of pathology; (C) Oligodendroglial inclusions: Oligodendrocytes internalize α-syn, forming GCIs. This process is driven by impaired autophagic flux, evidenced by decreased LC3-II and increased p62 accumulation, leading to myelin swelling and mitochondrial dysfunction. Created in BioRender.com. α-syn: α-synuclein; TLR: toll-like receptor; NF: nuclear factor; PDE10A: phosphodiesterase 10A; cAMP: cyclic adenosine monophosphate; GCIs: glial cytoplasmic inclusions; ROS: reactive oxygen species; BAG3: BCL2-associated athanogene 3; NLRP3: NOD-like receptor family pyrin domain containing 3.
While astrocytes express low levels of α-syn under normal physiological conditions, the landscape changes drastically during PD pathology. The accumulation of α-syn in astrocytes correlates with neuronal loss and may facilitate the spread of pathology across different brain regions[191-193,195-199]. Initially, the internalization of neuronal α-syn functions as a protective lysosomal degradation mechanism[191,193,195,196,200-206]. However, excessive accumulation eventually becomes toxic, inducing oxidative stress, mitochondrial dysfunction, secretion of pro-inflammatory cytokines, and astrogliosis, factors that ultimately compromise neuronal survival[193,195,196,203,207,208]. This toxicity is well-documented in experimental models; cultured astrocytes exposed to α-syn exhibit marked astrogliosis and glial-neuronal toxicity[193,195,196,203,207,208]. Furthermore, transgenic mice expressing mutant A53T α-syn specifically in astrocytes display robust, early astrogliosis before behavioral symptoms arise, alongside microglial activation, progressive degeneration of dopaminergic neurons in the substantia nigra, early paralysis, and a shortened lifespan[208].
Central to managing this burden is astrocytic autophagy, the modulation of which significantly impacts α-syn toxicity. In human glioblastoma cell lines, activating BAG3-dependent autophagy by reducing CRYAB facilitates α-syn clearance, whereas CRYAB overexpression in the astrocytes of A30P α-syn mice results in substantial accumulation[205]. These findings indicate that enhancing astrocytic autophagy offers therapeutic potential for both astrocytes and neurons[193,196,207,208]. Conversely, inhibiting autophagy or lysosomal pathways increases insoluble α-syn levels[195,205,209]. Indeed, autophagy inhibition in transgenic animals exacerbates aggregation, and α-synuclein overexpression disrupts autophagic flux by decreasing LC3-II levels and increasing p62 accumulation[202,205,206].
Several PD-associated genes expressed in astrocytes, including PARK2, PARK7, GBA, LRRK2, PINK1, ATP13A2, and PLA2G6, are inextricably linked to critical astrocytic functions, such as lipid metabolism, mitochondrial activity, inflammation, and lysosomal pathways[210]. Mutations in these genes disrupt homeostasis and may accelerate PD pathogenesis. ATP13A2 is particularly vital for regulating the uptake and degradation of neuronal α-syn; loss-of-function mutations in this gene impair clearance, triggering NLRP3 inflammasome activation, increased IL-1β secretion, and heightened neuroinflammation[206,211]. In co-culture models, astrocytes with ATP13A2 mutations show a reduced capacity to limit α-syn accumulation in dopaminergic neurons, thereby enhancing the propagation of the protein[212].
Similarly, LRRK2 G2019S mutations in patient-derived astrocytes lead to progressive endogenous α-syn accumulation caused by impaired CMA and macroautophagy[213]. These mutated astrocytes are less effective at clearing extracellular aggregates and can even transfer α-syn to surrounding neurons, contributing to degeneration[213]. Mechanistically, increased LRRK2 kinase activity has been shown to decrease LC3-I lipidation, interfering with autophagosome formation[214]. Furthermore, glucocerebrosidase (GCase), encoded by GBA1, is essential for astrocytic autophagy. iPSC-derived astrocytes with GBA1 mutations display astrogliosis, impaired lysosomal cathepsin activity, and α-syn aggregation[215], aligning with observations of autophagic defects (reduced LC3-I and LC3-II fragmentation) in GBA-deficient mice and primary cells[216,217]. However, some studies suggest that GCase deficiency impacts degradation differently depending on model-specific conditions[218].
Mitochondrial quality control, particularly mitophagy, acts as another critical layer of astrocyte-mediated neuroprotection. The ablation of mitophagy via Kir6.1 knockout exacerbates motor deficits and dopaminergic neuron degeneration, whereas restoring this pathway supports neuronal mitochondrial integrity[219,220]. Whether triggered by genetic or epigenetic mechanisms, impairments in autophagy and mitophagy lead to the buildup of dysfunctional mitochondria, a process closely linked to PD progression that may originate within astrocytes themselves[60,210,214,221]. These deficits compromise the astrocyte’s ability to maintain redox homeostasis and the BBB. Normally, astrocytes protect neurons by metabolizing dopamine via monoamine oxidase-B and catechol-O-methyltransferase and producing antioxidants[222,223]. Chronic impairment of these systems, particularly during aging, renders dopaminergic neurons vulnerable to stress[210,224].
Pathologically, soluble α-syn fibrils function as neurotoxic TLR4 ligands, triggering neuroinflammation in astrocytes and microglia even before neuronal pathology manifests, as seen in striatal injection models[225-232]. While TLR4 stimulation can mount a protective antioxidant response, accumulated α-syn disrupts glutamate uptake by downregulating GLAST/EAAT1 and GLT-1/EAAT2, and compromises BBB integrity by altering AQP4 localization[208]. To combat this, astrocytes utilize synucleinphagy, a selective autophagy pathway enhanced by decreased intracellular pH and glycolytic metabolism[109,233,234]. However, environmental neurotoxicants linked to PD, such as rotenone and paraquat, can hijack this system; high concentrations of paraquat initially activate but later suppress autophagic flux, causing the accumulation of toxic substances[235].
Once intracellular accumulation occurs, toxic molecules can spread to other cells, including neurons, via exosomes, passive diffusion, direct contact, or tunnelling nanotubes[207,236,237]. Collectively, these findings emphasize that the autophagy–lysosomal system, mitochondrial quality control, and metabolic adaptations in astrocytes are essential for regulating α-syn homeostasis and preventing the spread of toxic aggregates. Enhancing the astrocytic capacity for protein degradation therefore represents a promising therapeutic strategy for slowing PD progression.
3.2.2 Oligodendrocytes
Oligodendrocytes in PD undergo early and progressive alterations, particularly within the motor cortex and underlying white matter. Investigations have revealed significant axonal disorganization, enrichment of α-syn in neurofilaments, enlargement of myelinating oligodendrocytes, and increased density of their precursors. These changes occur concurrently with elevated myelin proteins and α-syn accumulation, beginning during the prodromal motor stages (Figure 2C)[238]. Such findings contrast with the classical perspective of PD as primarily a nigrostriatal degeneration, highlighting that adaptive structural remodelling occurs in oligodendrocytes and myelin prior to the onset of overt neuropathology[238,239]. Region-specific responses are evident: intracortical oligodendroglia increase myelin production around inhibitory axon collaterals, whereas oligodendrocytes in white matter motor tracts increase in both number and size to ensheath larger pyramidal axons[240]. Animal models overexpressing α-syn replicate these phenomena, demonstrating that increased myelin phospholipids precede myelin loss[241].
Mechanistically, oligodendrocytes internalize α-syn from the extracellular milieu. Subsequent mitochondrial dysfunction and oxidative stress impair autophagy, which results in the accumulation of α-syn aggregates and the formation of glial cytoplasmic inclusions (GCIs)[242-244]. This pathological aggregation further reduces autophagic efficiency, creating a cycle that perpetuates α-syn buildup. Consequently, enhancing autophagy to block α-syn accumulation in oligodendrocytes has been identified as a strategy with significant therapeutic potential[245].
3.2.3 Microglia
Microglial autophagy serves as a pivotal regulator of neuroinflammation, neuronal survival, and the pathogenesis of PD. Genetic evidence strongly links mutations in PD-associated genes, specifically SNCA, LRRK2, PRKN, and PINK1, to autophagy and mitochondrial quality control, reinforcing the critical role of autophagic pathways in disease susceptibility[246,247]. Functional studies further demonstrate that microglial Atg5 is indispensable for maintaining CNS homeostasis. Animals with a microglia-specific Atg5 deletion manifest PD-like features, including accelerated dopaminergic neuron loss and heightened inflammatory responses[105,248,249]. Furthermore, the loss of Atg5 exacerbates MPTP-induced neurodegeneration by activating the NLRP3 inflammasome[249], proving that microglial autophagy acts to restrain inflammatory cascades that contribute to neuronal damage. Conversely, impaired autophagy leads to the accumulation of phosphorylated and insoluble α-syn within microglia[109], positioning defective autophagy as a mechanistic driver of pathological α-syn burden.
To manage this burden, microglia degrade misfolded α-syn through a selective autophagy process termed “synucleinphagy”, which is mediated by p62/SQSTM1 and reduces α-syn accumulation, thereby supporting neuroprotection[109]. The activation of microglia by α-syn involves toll-like receptors (TLRs) and NF-κB signalling, both of which transcriptionally upregulate p62 to facilitate the clearance of pathogenic α-syn (Figure 2B)[109,250]. Similarly, enhancing CMA decreases α-syn levels and protects neurons from toxicity[251]. Microglial autophagy can also be triggered via the p38–TFEB pathway, which inhibits CMA-dependent NLRP3 degradation and subsequently reduces inflammatory signalling[252]. Collectively, these findings highlight the broad neuroprotective impact of microglial autophagy in PD[105,109,247-249].
Deficiency in microglial autophagy promotes profound dysfunction at behavioral, cellular, and molecular levels. Conditional Atg5 knockout results in age-dependent neurological impairments; while young mice exhibit normal behavior, by 8 months Atg5 cKO mice display gait abnormalities and cognitive deficits, indicating that microglial autophagy supports both motor coordination and cognitive learning[105]. These behavioral outcomes parallel the early and sustained degeneration of tyrosine hydroxylase-positive dopaminergic neurons and reduced striatal dopamine, with downward trends in DOPAC levels detected as early as three months[105]. This deficiency induces a robust pro-inflammatory environment marked by strong upregulation of Il1b, Nos2/Inos, and Tnf in both young and aged microglia[105]. Mechanistically, the loss of Atg5 activates NLRP3 through dysregulation of PDE10A–cAMP signalling, leading to elevated IL-1β, which stimulates macrophage migration inhibitory factor (MIF) to further amplify inflammation. Importantly, pharmacological inhibition of NLRP3 rescues microglial hyperactivation and prevents neuronal loss, demonstrating that autophagy suppresses a specific PDE10A–cAMP–NLRP3–IL1B–MIF inflammatory axis that drives neurodegeneration[105]. Under homeostatic conditions, microglia maintain CNS integrity by continuously surveying their environment via dynamic protrusions[253-255]. This surveillance supports metabolite clearance, regulates neurogenesis, and ensures overall tissue stability[256,257]. However, when exposed to endogenous or exogenous insults, microglia transition from a quiescent to an activated state, adopting macrophage-like properties and releasing inflammatory mediators[227,258-260].
Historically classified into the binary categories of M1 and M2, microglial activation is now understood to be a highly dynamic and transcriptomically varied spectrum. In the context of neurodegenerative diseases, microglia adopt specific profiles, such as the DAM phenotype. Initially, the DAM state serves as a protective mechanism, characterized by high phagocytic activity aimed at removing debris, dying cells, and neurotoxic aggregates like α-syn, a process that heavily depends on functional autophagic flux[103,261,262]. However, prolonged exposure to aggregated α-syn, along with the eventual saturation or failure of microglial autophagy, drives them into a dysfunctional, hyper-inflammatory state. In this state, microglia release excessive amounts of proteases, ROS, and pro-inflammatory cytokines, including IL-1β, TNF-α, and IL-6, which are strongly associated with the loss of dopaminergic neurons in the early stages of PD[263,264]. Furthermore, inhibiting the mTOR pathway under inflammatory conditions can restore autophagic clearance, reduce this hyper-inflammatory activation, and prevent secondary neurotoxicity[265].
Human PD studies confirm microglial involvement in the disease, with early neuropathological work identifying abundant HLA-DR-positive reactive microglia in the substantia nigra of patients[266]. Although single-cell transcriptional profiling shows substantial microglial heterogeneity across brain regions in PD, limited sample sizes currently restrict definitive conclusions[267]. Nevertheless, mechanistic studies have shown that α-syn aggregates in dopaminergic neurons drive PD pathogenesis[268], and activated microglia accelerate α-syn accumulation[269,270]. To manage this burden, microglia exposed to aggregated α-syn form F-actin–dependent intercellular networks that transfer α-syn from overloaded cells to naïve microglia for rapid degradation, thereby reducing inflammatory stress and increasing survival[271]. In parallel, however, microglia also transmit α-syn via exosomes, facilitating its spread in the CNS and contributing to PD progression[172,272]. Misfolded α-syn alters TLR expression and activates NF-κB through PKCδ-dependent mechanisms, promoting inflammatory signalling[273], yet microglia also clear α-syn through TLR4–NF-κB–p62-mediated pathways to enable synaptic phagocytosis and neuroprotection[109]. Thus, α-syn both impairs and is regulated by microglial autophagy: exosomes from neurons overexpressing α-syn inhibit microglial autophagy[274], and while α-syn accumulation increases p62/SQSTM1 expression, it suppresses autophagic flux[260]. Additionally, autophagy blockade enhances α-syn release and intercellular transfer[275]. Ultimately, while whole-brain autophagy deficiency induces neurodegeneration[55], the specific loss of microglial autophagy leads to more severe motor dysfunction, α-syn deposition, and dopamine neuron vulnerability when α-syn levels are elevated[109,260], confirming that microglial autophagy directly degrades α-syn and NLRP3 to alleviate PD pathology[276].
A central theme in the development of PD is the dependence on glial cells to initially absorb extracellular α-syn. Astrocytes, microglia, and oligodendrocytes actively take in α-syn from neurons, aiming to reduce the pathogenic load through a process called synucleinphagy, a form of selective autophagy. However, a comprehensive review of current research indicates that this common protective strategy becomes a source of non-cell-autonomous toxicity when the autophagic and lysosomal capacities of glial cells are overwhelmed. Genetic mutations, such as LRRK2 and ATP13A2, along with environmental stressors, collectively impede autophagic processes in these cells, turning them from protective agents into storage sites for aggregated α-syn. This collective failure eventually leads to mitochondrial dysfunction, significant activation of the NLRP3 inflammasome, and the exosomal spread of α-syn, which directly affects the survival of susceptible dopaminergic neurons.
3.3 HD
The expansion of a polyglutamine tract is the underlying cause of nine autosomal dominant neurodegenerative conditions, encompassing several spinocerebellar ataxias and HD[277]. HD is a fatal, genetic neurodegenerative disorder defined by symptoms that include involuntary movements, psychiatric disturbances, and cognitive deficits, for which no curative treatment currently exists[278,279]. The disease arises from an unstable expansion of CAG triplet repeats within the huntingtin (HTT) gene, which results in the formation of an abnormal polyglutamine tract at the N-terminal region of the HTT protein[280]. While the toxicity in HD stems primarily from a toxic gain of function of this mutant protein, the loss of normal HTT function also contributes to the dysfunction and eventual death of projection neurons in the caudate/putamen (known as the striatum in mice) and the cerebral cortex[281]. A defining neuropathological hallmark of the disease is the presence of mutant HTT (mHTT) aggregates, which predominantly manifest in neurons[282] but are also found in glial cells[283,284].
Disease progression is closely correlated with the selective loss of medium-sized striatal spiny neurons in the caudate nucleus, driving the progressive motor, psychiatric, and cognitive dysfunction observed in patients[281]. Within HD neurons, autophagosomes form normally; however, their capacity to recognize and sequester mHTT is compromised, thereby reducing degradation efficiency[59]. While the ubiquitin–proteasome system is capable of degrading soluble mHTT, aggregated forms of the protein require autophagy coupled to lysosomal degradation for clearance[285,286]. Unfortunately, mHTT actively impairs this process by disrupting cargo loading into autophagosomes[287] and by decreasing both autophagosome transport and degradation[288]. More broadly, polyglutamine expansions interfere with autophagy through several specific molecular mechanisms. For instance, pathologic polyglutamine tracts compete with ataxin 3 for binding to beclin 1; this interaction leads to increased ubiquitination and degradation of beclin 1, effectively abolishing autophagy activation[289]. Additional mechanisms include the inhibition of TFEB by the mutated androgen receptor, as seen in spinal and bulbar muscular atrophy[290].
HD is also characterized by astrocytosis and progressive alterations in glial cells as mHTT accumulates and modifies brain cell function[291]. Concurrently, microglia shift toward an activated phenotype during the progression of the disease[292]. When activated by mHTT, these microglia secrete chemokines, specifically CCL3, CCL4, and CCL5, that activate the CCR5 receptor (Figure 3A). This signalling cascade affects mTORC1 activation and inhibits neuronal autophagy via a non-cell-autonomous mechanism[292,293]. Although the role of microglia is increasingly recognized, there is currently no data regarding the involvement of oligodendrocyte autophagy in HD.
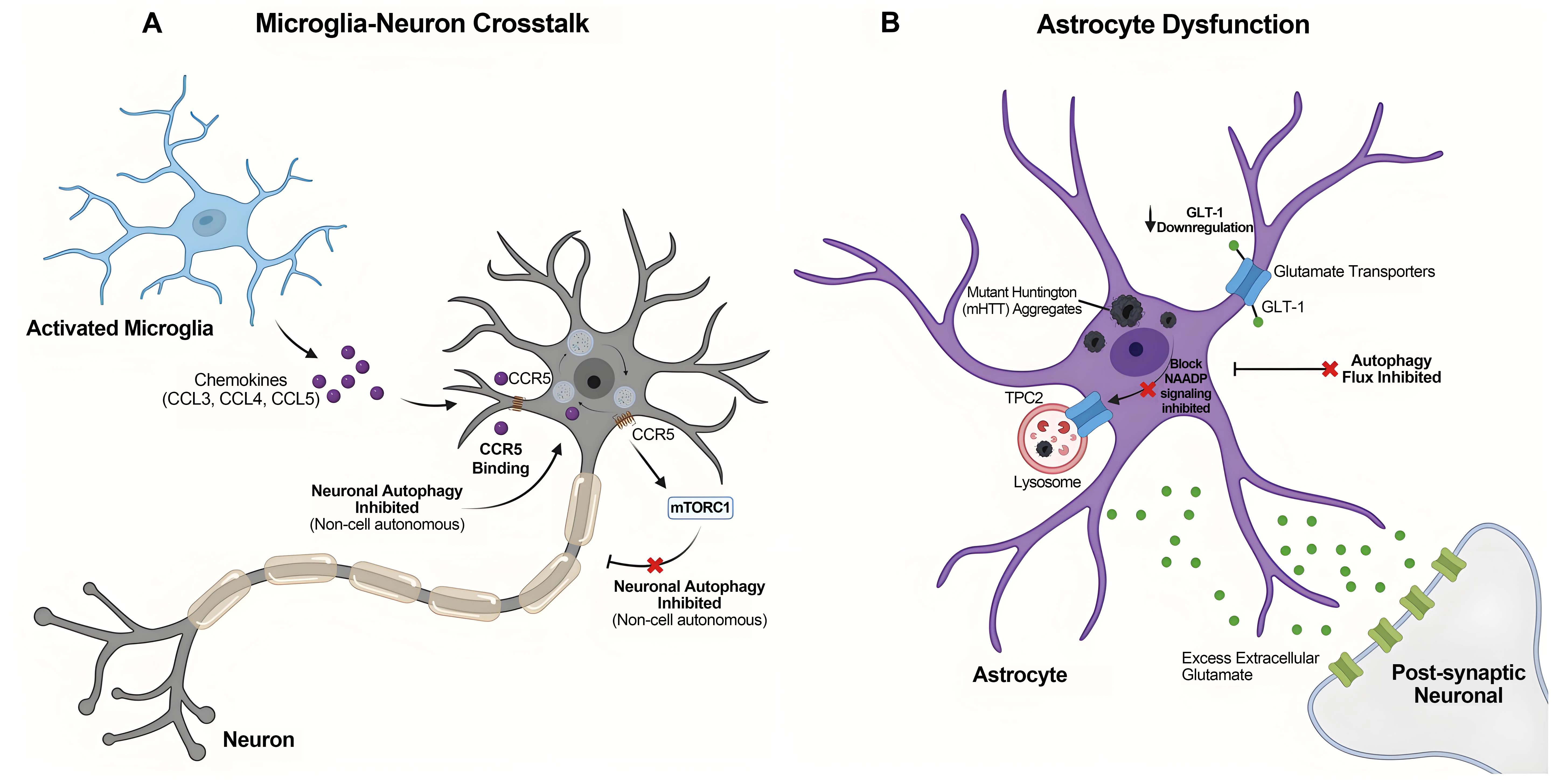
Figure 3. Non-cell-autonomous mechanisms in Huntington’s disease. (A) Microglia-neuron crosstalk: Activated microglia secrete chemokines CCL3, CCL4, and CCL5 in response to mHTT. These ligands bind to CCR5 receptors on neurons, triggering an intracellular signalling cascade that activates mTORC1. This microglial-driven activation of neuronal mTORC1 inhibits neuronal autophagy via a non-cell-autonomous mechanism, exacerbating the accumulation of aggregates within neurons; (B) Astrocytic dysfunction: The accumulation of mHTT in astrocytes leads to the loss of the GLT-1 transporter, impairing glutamate clearance and promoting excitotoxicity. mHTT specifically impairs autophagy by co-localizing with TPC2 channels on lysosomes, blocking NAADP-induced Ca2+ release and inhibiting autophagic flux. Created in BioRender.com. mHTT: mutant huntingtin; GLT-1: glutamate transporter 1; TPC: two-pore channels; NAADP: nicotinic acid adenine dinucleotide phosphate; CCL: C–C motif chemokine ligand; mTORC1: mechanistic target of rapamycin (complex 1).
3.3.1 Astrocytes
Astrocytes play a central role in the pathology of HD, particularly through their involvement in glutamate homeostasis and autophagy. In the HD brain, disease progression correlates with an increase in reactive astrocytes, where the accumulation of HTT protein is also observed[294,295]. A consistent feature of HD in both human brain tissue and animal models is the significant loss of the astrocyte-specific glutamate transporter GLT-1 (also known as EAAT2), which is essential for the clearance of extracellular excitatory glutamate[294,296-299]. This reduction of GLT-1 impairs glutamate uptake and increases excitotoxic risk, highlighting the key contribution of astrocytes to HD pathology (Figure 3B).
Experimental evidence supports a causal role for astrocytic mHTT in disease progression; mouse models selectively expressing mHTT in astrocytes develop age-dependent motor deficits and a shortened lifespan[299]. Furthermore, when mHTT is expressed in both astrocytes and neurons, neurological symptoms are exacerbated compared with expression in neurons alone[300]. These effects are predominantly mediated by reduced glutamate transporter levels and impaired glutamate uptake by astrocytes[294,296,297]. However, additional astrocyte-dependent mechanisms also contribute, including imbalances in potassium homeostasis via Kir4.1 ion channels[301] and reduced secretion of brain-derived neurotrophic factor[302].
Although astrocytes are reported to possess high proteolytic capacity, including for mHTT, which may explain why fewer aggregates are observed in astrocytes compared to neurons[303,304], astrocytic autophagy becomes progressively compromised during disease progression. For instance, the effectiveness of astrocyte LAP decreases as HD advances[305]. Additionally, the overexpression of mutant huntingtin (mHtt-Q74) in murine astrocytes impairs autophagy, a critical pathway for clearing protein aggregates, with important implications for HD pathogenesis[306]. In healthy astrocytes, nicotinic acid adenine dinucleotide phosphate (NAADP) induces autophagy by activating lysosomal two-pore channels (TPCs), particularly lysosomal calcium channels (TPC2). In mHtt-Q74–expressing astrocytes, however, this pathway is blocked because mHtt-Q74 co-localizes with TPC2, suggesting a physical interaction that disrupts channel function and lysosomal Ca2+ signalling. This interference prevents NAADP-induced autophagy and blocks the final degradative step, facilitating mHTT accumulation and cytotoxicity[306]. Additional studies further demonstrate that mHTT directly inhibits astrocytic autophagy. Transfection of astrocytes with the pEGFP-mHtt Q74 plasmid increases Ca2+ release from lysosomes, which promotes mHTT aggregation, and astrocytes overexpressing mHtt-Q74 show inhibited autophagy[306]. These findings align with broader evidence that mHTT impairs cargo recognition and endosomal sorting, functionally uncoupling autophagosome initiation from degradation despite intact autophagosome formation[59,307].
Fortunately, pharmacological activation of autophagy in astrocytes has shown protective effects in HD models. The administration of rapamycin to primary astrocytes decreases mHTT levels, restores GLT-1 expression, and ameliorates deficits in glutamate uptake[308]. Similarly, trehalose reduces the accumulation of mHTT and synuclein in primary glial cultures derived from both wild-type and mHTT mice[309]. These interventions restore astrocytic function and indirectly support neuronal survival, although some effects of rapamycin and trehalose may occur independently of autophagy[60,308,309]. Collectively, these findings demonstrate that astrocytes are not passive bystanders in HD but active contributors to disease progression through dysregulated glutamate handling, impaired autophagy, and altered lysosomal function. Disruption of astrocytic autophagy exacerbates mHTT accumulation and excitotoxic stress, whereas restoration of autophagic pathways in astrocytes represents a potential strategy to mitigate neurodegeneration in HD.
3.3.2 Microglia and oligodendrocytes
The impact of autophagy dysregulation on microglial phagocytosis and inflammation in the context of CNS senescence and disease remains unknown[177]. Similarly, the impact of autophagy dysregulation in oligodendrocytes specifically with relevance to HD remains largely unknown.
In contrast to AD and PD, where the uptake of extracellular proteins primarily triggers glial autophagic failure, HD is characterized by significant, inherent non-cell-autonomous toxicity directly caused by mHTT. The common theme is the stealthy ability of mHTT to disrupt protective processes. In astrocytes, mHTT physically obstructs TPC2, halting autophagic flux and disrupting glutamate balance. Whereas, in microglia, mHTT initiates a chemokine cascade that actively works to inhibit autophagy in nearby neurons. Consequently, glial autophagic dysfunction in HD signifies a systemic network breakdown, where glia not only lose their inherent capacity to clear aggregates but also actively release factors that undermine neuronal proteostasis. The notable absence of data on oligodendroglial autophagy in HD remains a critical conceptual gap that needs to be addressed to fully comprehend this network failure.
3.4 ALS
ALS stands as the most prevalent form of adult motor neuron disease, defined by the relentless degeneration of both upper and lower motor neurons that ultimately results in paralysis and death[310,311]. Clinically, this progressive motor neuron loss manifests as muscle weakness, paralysis, and fatal respiratory failure[311]. While approximately 90% of cases are sporadic (sALS), between 5–10% are classified as inherited or familial (fALS)[312]. These familial forms are linked to mutations in over 20 genes, most notably C9ORF72, Cu/Zn superoxide dismutase (SOD1), TAR DNA-binding protein 43 (TDP-43), and fused in sarcoma, many of which converge on pathways critical for RNA metabolism and protein homeostasis[310,313].
At the molecular level, ALS pathology is frequently characterized by the accumulation of aggregated proteins, particularly SOD1 and TDP-43, which disrupt autophagic pathways and drive neuronal toxicity[59,314]. Consequently, ATG dysfunction is recognized as a key contributor to ALS pathogenesis. Several genes genetically associated with the disease, such as SQSTM1/p62 and TBK1, encode proteins essential for autophagy receptor signalling and immune regulation[315,316]. Specifically, mutations in TBK1 impair the phosphorylation of autophagy effectors, resulting in defective mitophagy and aggrephagy. Similarly, mutations in optineurin, which functions as a cargo receptor in mitophagy, disrupt mitochondrial quality control and have been detected in both familial and sporadic ALS cases[317].
While motor neuron degeneration remains central to ALS, mounting evidence highlights that non-neuronal cells play critical roles in disease initiation and progression. Non-cell-autonomous mechanisms involving glial cells have emerged as significant contributors to pathology, particularly through their influence on neuroinflammation, proteostasis, and autophagy-dependent pathways[310,313].
3.4.1 Astrocytes
Astrocytes have become a focal point in ALS research because of their significant contribution to non-cell-autonomous neurodegeneration. Early investigations utilizing experimental rodent models expressing mutant SOD1 revealed that restricting the expression of the mutation to neurons alone[318,319] or astrocytes alone[320] was insufficient to trigger neurodegeneration. Conversely, the simultaneous expression of mutant SOD1 in both neurons and glial cells successfully recapitulated an ALS-like phenotype[321,322]. Crucially, the selective deletion of mutant SOD1 specifically from astrocytes was shown to slow disease progression and extend the lifespan of these models, confirming the critical pathogenic role of astrocytes in ALS[314,323]. This involvement was further substantiated by transplantation studies. The introduction of wild-type astrocytes into the spinal cord of mutant SOD1 rats slowed disease progression and prolonged survival; conversely, transplanting astrocytes expressing mutant SOD1 into wild-type mice induced local motor neuron degeneration and dysfunction[324]. These results demonstrate that astrocytes can exert either neuroprotective or neurotoxic effects, contingent upon their genetic and functional state.
The non-cell-autonomous toxicity mediated by astrocytes in ALS may stem from the active secretion of neurotoxic factors or the forfeiture of essential support functions. Astrocytes harboring ALS-associated mutations are known to secrete neurotoxic molecules, including pro-inflammatory cytokines (Figure 4A)[325-328]. Specifically, astrocytes reprogrammed from the peripheral blood mononuclear cells of sALS patients release abnormally high concentrations of the pro-inflammatory cytokines IL1B, TNFA, and IL6, which prove toxic to motor neurons[329]. Parallel to this, astrocytic dysfunction in ALS encompasses the loss of the glutamate transporter GLT1, leading to increased excitotoxicity[299,330], heightened oxidative stress that promotes neuronal toxicity[331,332], and defects in RNA trafficking that impede the translation of mRNAs vital for astrocyte function[333].
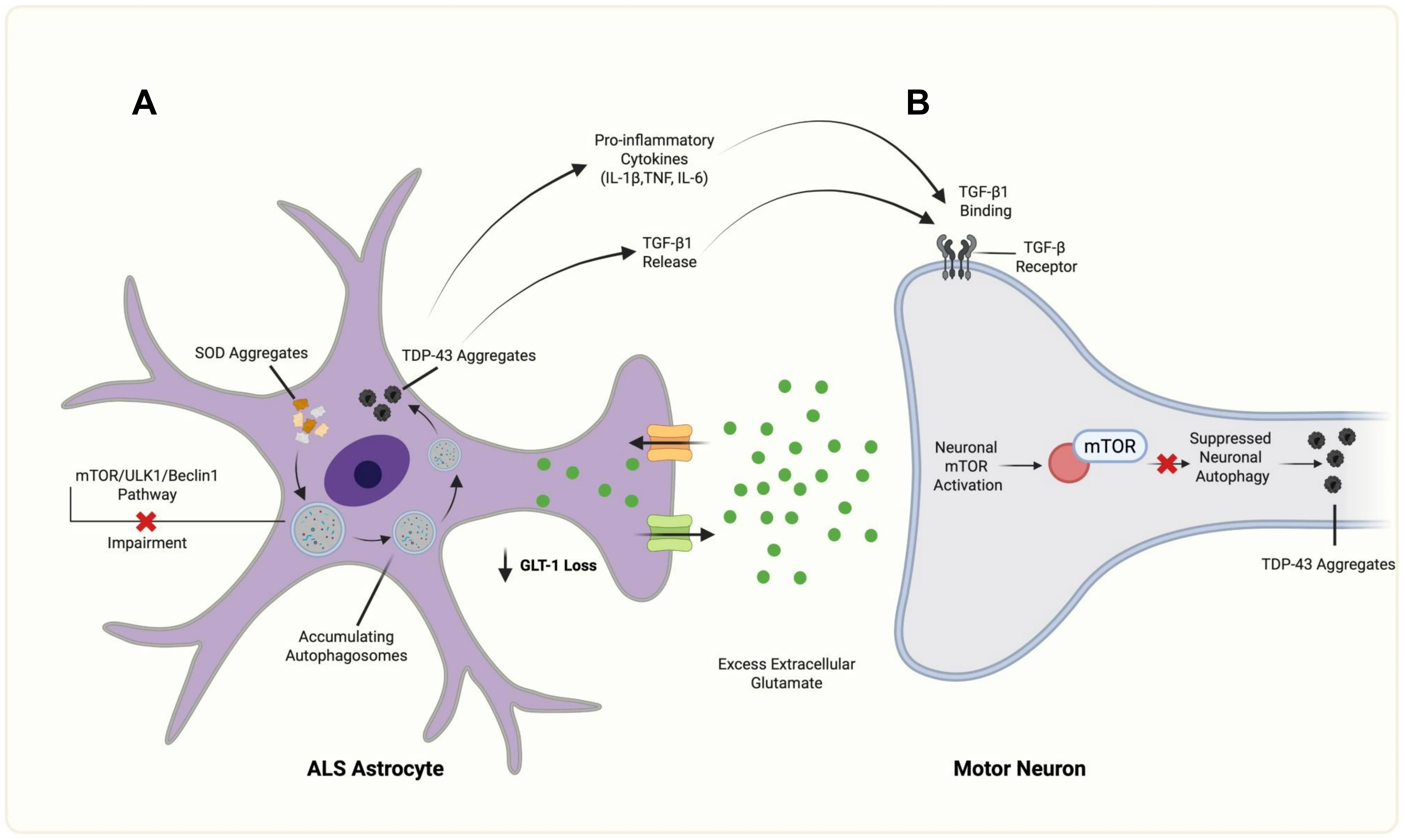
Figure 4. Astrocyte-mediated toxicity in amyotrophic lateral sclerosis. (A) Intrinsic astrocytic defects: ALS astrocytes harboring SOD1 or TDP-43 aggregates exhibit impaired mTOR/ULK1/Beclin-1 pathways. This dysfunction leads to the loss of metabolic support (e.g., GLT1 downregulation) and the secretion of pro-inflammatory cytokines such as IL-1β, TNF, and IL-6; (B) Suppression of neuronal autophagy: Astrocytes exert non-cell-autonomous toxicity by secreting TGF-β1. This factor binds to receptors on motor neurons, activating the mTOR pathway within the neuron. The resulting inhibition of neuronal autophagy compromises protein clearance and contributes to motor neuron degeneration. Created in BioRender.com. ALS: amyotrophic lateral sclerosis; SOD1: Cu/Zn superoxide dismutase; TDP-43: TAR DNA-binding protein 43; mTOR: mechanistic target of rapamycin; ULK: unc-51 like autophagy activating kinase 1; IL: interleukin; TGF: transforming growth factor.
Astrocytic autophagy has also emerged as a pivotal regulatory mechanism in this context. Impairment of the mTOR/ULK1/Beclin-1/p62/LC3B autophagy pathway has been documented in astrocytes derived from sALS patients and may drive the increased secretion of pro-inflammatory cytokines[329]. Furthermore, reduced levels of LC3B-II have been detected in astrocytes from fALS models carrying the SOD1G93A mutation[334]. These alterations suggest a compromised autophagic flux in ALS astrocytes that likely exacerbates neurotoxicity. Beyond intrinsic dysfunction, astrocytes expressing ALS-associated mutations can influence neuronal autophagy non-cell-autonomously. Conditioned media from ALS astrocytes has been shown to suppress neuronal autophagy through secreted factors such as transforming growth factor-β1 (TGF-β1), which inhibits autophagy by activating mTOR signalling within motor neurons (Figure 4B)[60,335]. This astrocyte-driven suppression underscores the importance of astrocytic autophagy in preserving motor neuron homeostasis.
Therapeutic modulation of autophagy offers potential benefits in addressing this dysfunction. The application of autophagy-inducing molecules enhances the clearance of TDP-43 in neurons and mitigates the toxicity of mutant TDP-43 in human stem cell–derived astrocytes, hinting at therapeutic utility even though direct autophagic clearance of TDP-43 in astrocytes remains to be conclusively demonstrated[309,336]. Additionally, the pathology is driven by dysregulated stress granule dynamics and impaired selective autophagic clearance of TDP-43 aggregates, particularly regarding persistent stress granules containing TDP-43[337].
In summary, these findings establish astrocytes as central drivers of ALS pathogenesis through non-cell-autonomous mechanisms involving neuroinflammatory signalling, impaired autophagy, and the loss of metabolic and trophic support. Dysfunctional astrocytic autophagy not only worsens intrinsic pathology but also accelerates motor neuron degeneration, identifying astrocytes as prime targets for therapies aimed at restoring autophagic balance. However, to the best of our knowledge, there is currently no data regarding the involvement of oligodendrocyte autophagy in ALS; therefore, detailed studies are required to clarify the role of oligodendrocyte autophagy in the course of neurodegenerative diseases, including HD and ALS.
Recent ALS research highlights a significant shift in understanding: the degeneration of motor neurons is closely tied to, and sometimes reliant on, the breakdown of autophagy in nearby support cells. Astrocytes with ALS-related mutations show a critical dysfunction in the mTOR/ULK1 autophagic pathway, which acts as a common trigger for two types of non-cell-autonomous toxicity: the reduction of essential metabolic support such as GLT1 downregulation, and the active release of neurotoxic cytokines such as TGF-β1, that directly inhibit neuronal autophagy. Although the role of astrocytes is well-established, there is a significant gap in the literature concerning the specific autophagic processes of microglia and oligodendrocytes in ALS. Filling these gaps in glial research is crucial for developing a comprehensive understanding of how non-cell-autonomous autophagic failure contributes to motor neuron death.
4. Conclusions and Future Perspectives
While the pivotal role of neuronal autophagy in neurodegeneration has been well-established, growing evidence highlights glial autophagy as a central regulator of disease progression rather than a mere compensatory response. Moving forward, the field must transition from a neurocentric view to a holistic interrogation of the glial machinery. Several critical avenues of investigation are required to translate these biological insights into effective therapeutic strategies.
A primary challenge lies in dissecting the cell-type–specific mechanisms by which degradative pathways modulate neuronal function under physiological and pathological conditions. There is a significant disparity in our understanding across glial populations; while astrocytic and microglial autophagy have been explored in AD and PD, the contributions of oligodendrocyte autophagy to demyelination and axonal degeneration in HD and ALS remain understudied. Future research must utilize advanced tools, such as human stem cell–derived models, single-cell transcriptomics, and in vivo imaging, to clarify how autophagic flux, cargo recognition, and lysosomal competence differ between neuronal and glial phenotypes across disease stages.
Understanding the seemingly contradictory roles of glial autophagy, which can have both protective and harmful effects on neurons, is critical. This dual nature indicates that the timing of intervention is vital. For example, while moderate autophagy aids in cell survival and the removal of debris, excessive or uncontrolled activation might worsen inflammatory responses or damage myelin. Additionally, aging complicates the situation; a lack of autophagy leads to SASP in microglia and precursor cells. Thus, it is important to clarify the timing of autophagy impairment and to understand the interactions between neurons and glia, as well as among glial cells, to pinpoint therapeutic opportunities that offer protection without causing unintended harm.
The conceptual framework surrounding neurodegenerative disease pathogenesis is rapidly evolving to encompass a more integrated perspective, positioning glial autophagy as a fundamental determinant of CNS homeostasis. Far from being passive bystanders, astrocytes, microglia, and oligodendrocytes utilize autophagic pathways to orchestrate critical functions, including protein quality control, inflammatory regulation, myelin maintenance, and metabolic support.
The evidence synthesized in this review underscores that disruption of glial autophagy is is not just a compensatory mechanism but a key, active factor in maintaining CNS balance. Astrocytes, microglia, and oligodendrocytes engage autophagic pathways to manage essential roles such as protein quality control, inflammation regulation, myelin upkeep, and metabolic support across major neurodegenerative disorders. In AD and PD, the failure of glial cells to degrade pathogenic aggregates, such as Aβ, tau, and α-syn, precipitates chronic inflammation and secondary neurotoxicity. Similarly, in ALS and HD, astrocytic dysfunction impairs glutamate clearance and cytokine regulation, while compromised oligodendrocyte autophagy in demyelinating conditions undermines axonal integrity. Thus, glial autophagy dysfunction creates a permissive environment for disease progression through non-cell-autonomous mechanisms.
A significant challenge is the uneven understanding of different glial populations. While the autophagic processes in astrocytes and microglia (like LAP and LANDO) are well-documented in AD and PD, there is a significant lack of understanding regarding oligodendrocyte autophagy in HD and ALS. Since oligodendrocyte dysfunction actively contributes to demyelination and axonal degeneration, not exploring their specific autophagic pathways leaves a crucial gap in understanding non-cell-autonomous disease progression. Future studies should employ advanced techniques, such as human stem cell-derived models, single-cell transcriptomics, and in vivo imaging, to explore these uncharted areas and elucidate how autophagic flux, cargo recognition, and lysosomal competence vary among all glial types throughout disease progression.
As the field progresses toward clinical implementation, it is crucial to recognize the constraints of existing experimental models. Much of our knowledge about glial autophagy comes from transgenic rodent models that overproduce mutant human proteins (such as Aβ, mHTT, α-syn) at levels that are not physiological. Additionally, the lengthy aging process typical of human neurodegeneration, along with the associated “inflamm-aging”, is challenging to replicate in the short lifespan of mouse models. Therefore, it is essential to thoroughly validate mouse findings in humanized models, like patient-derived iPSC 3D brain organoids, to ensure they are applicable to humans.
Translating these findings also involves managing intricate therapeutic considerations. For instance, using senolytics to target senescence offers a promising approach to remove SAM and decrease plaque accumulation; however, indiscriminately removing these cells could risk losing their beneficial early-stage phagocytic roles or hinder tissue repair processes. Similarly, while reducing glial neuroinflammation is a sensible objective, overly aggressive anti-inflammatory measures might unintentionally suppress the necessary immune responses needed to attract microglia to areas of active aggregate buildup. Thus, therapeutic approaches must shift from broad systemic inhibition to precise, context-specific immunomodulation.
Looking ahead, the broad pharmacological activation of autophagy carries the potential risk of disturbing cellular equilibrium or causing unintended effects in healthy cells. Therefore, creating therapeutic agents that specifically target glial cells is of utmost importance. Future treatments should focus on pathway selectivity, possibly by aiming at upstream regulators, particular lysosomal channels like TPC2 in astrocytes, or distinct cargo receptors, that are unique to astrocytes or microglia, to distinguish between beneficial metabolic support and detrimental inflammatory activation. By refining the precision of these modalities, it may be possible to restore glial homeostatic functions and delay the onset of neurodegeneration.
In conclusion, the focus on glial autophagy presents a promising yet intricate therapeutic opportunity. The difficulty is not just in boosting degradative ability but also in accurately tuning autophagic processes to balance survival-promoting roles with the reduction of neuroinflammation. By deepening our knowledge of the spatial and temporal control of these pathways, future studies may reveal new treatments that can restore the balance between neurons and glial cells, potentially slowing or halting the progression of these debilitating diseases.
Acknowledgements
We thank our colleagues at the Medical School Neurogenetics and Ageing Laboratory for their valuable feedback and insightful comments during the preparation of our manuscript. We acknowledge that not all relevant, published studies could be included in this review, and apologize to those colleagues, whose work we could not reference, due to space limitations. We, hereby, confirm that no AI models or tools were used to write the manuscript. We did use Paperpal to polish the syntax of the final draft.
Authors contribution
Çakıcı O: Conceptualization, writing-original draft, visualization.
Tavernarakis N: Conceptualization, writing-review & editing.
Conflicts of interest
Nektarios Tavernarakis is an Editorial Board Member of Geromedicine. The other author declares no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
This work in the authors’ laboratory was funded by the Hellenic Foundation for Research and Innovation (H.F.R.I.) project NeuroMitophagy (Grant No. HFRI—FM17C3-0869), the H.F.R.I. project NeuroFlame (Grant No. 15546 80145), the General Secretariat for Research and Innovation of the Greek Ministry of Development and the European Union—NextGenerationEU project BrainPrecision (Grant No. TAEDR-0535850), and the European Commission Research Executive Agency Excellence Hub “CHAngeing” (Grant No. 101087071).
Copyright
© The Author(s) 2026.
References
-
1. Vargas JNS, Hamasaki M, Kawabata T, Youle RJ, Yoshimori T. The mechanisms and roles of selective autophagy in mammals. Nat Rev Mol Cell Biol. 2023;24(3):167-185.[DOI]
-
3. Wang HD, Lv CL, Feng L, Guo JX, Zhao SY, Jiang P. The role of autophagy in brain health and disease: Insights into exosome and autophagy interactions. Heliyon. 2024;10(21):e38959.[DOI]
-
5. Steinmetz JD, Seeher KM, Schiess N, Nichols E, Cao B, Servili C, et al. Global, regional, and national burden of disorders affecting the nervous system, 1990–2021: A systematic analysis for the global burden of disease study 2021. Lancet Neurol. 2024;23(4):344-381.[DOI]
-
6. Kroemer G, Mariño G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40(2):280-293.[DOI]
-
7. Potokar M, Jorgačevski J. Targeting autophagy in astrocytes: A potential for neurodegenerative disease intervention. Front Cell Neurosci. 2025;19:1584767.[DOI]
-
8. Valori CF, Possenti A, Brambilla L, Rossi D. Challenges and opportunities of targeting astrocytes to halt neurodegenerative disorders. Cells. 2021;10(8):2019.[DOI]
-
9. Kaushik S, Cuervo AM. The coming of age of chaperone-mediated autophagy. Nat Rev Mol Cell Biol. 2018;19(6):365-381.[DOI]
-
10. Mizushima N, Komatsu M. Autophagy: Renovation of cells and tissues. Cell. 2011;147(4):728-741.[DOI]
-
11. Schuck S. Microautophagy–distinct molecular mechanisms handle cargoes of many sizes. J Cell Sci. 2020;133(17):jcs246322.[DOI]
-
12. Xie Z, Klionsky DJ. Autophagosome formation: Core machinery and adaptations. Nat Cell Biol. 2007;9(10):1102-1109.[DOI]
-
13. Yang Z, Klionsky DJ. Eaten alive: A history of macroautophagy. Nat Cell Biol. 2010;12(9):814-822.[DOI]
-
14. Nakatogawa H. Mechanisms governing autophagosome biogenesis. Nat Rev Mol Cell Biol. 2020;21(8):439-458.[DOI]
-
15. Walker SA, Ktistakis NT. Autophagosome biogenesis machinery. J Mol Biol. 2020;432(8):2449-2461.[DOI]
-
17. Johansen T, Lamark T. Selective autophagy: ATG8 family proteins, LIR motifs and cargo receptors. J Mol Biol. 2020;432(1):80-103.[DOI]
-
18. Maruyama T, Noda NN. Autophagy-regulating protease Atg4: Structure, function, regulation and inhibition. J Antibiot. 2018;71(1):72-78.[DOI]
-
19. Ohsumi Y. Historical landmarks of autophagy research. Cell Res. 2014;24(1):9-23.[DOI]
-
20. Thumm M, Egner R, Koch B, Schlumpberger M, Straub M, Veenhuis M, et al. Isolation of autophagocytosis mutants of Saccharomyces cerevisiae. FEBS Lett. 1994;349(2):275-280.[DOI]
-
21. Ktistakis NT. ER platforms mediating autophagosome generation. Biochim Biophys Acta Mol Cell Biol Lipids. 2020;1865(1):158433.[DOI]
-
22. Molino D, Zemirli N, Codogno P, Morel E. The journey of the autophagosome through mammalian cell organelles and membranes. J Mol Biol. 2017;429(4):497-514.[DOI]
-
24. Papinski D, Schuschnig M, Reiter W, Wilhelm L, Barnes CA, Maiolica A, et al. Early steps in autophagy depend on direct phosphorylation of Atg9 by the Atg1 kinase. Mol Cell. 2014;53(3):471-483.[DOI]
-
26. Henn RE, Noureldein MH, Elzinga SE, Kim B, Savelieff MG, Feldman EL. Glial-neuron crosstalk in health and disease: A focus on metabolism, obesity, and cognitive impairment. Neurobiol Dis. 2022;170:105766.[DOI]
-
27. Vainchtein ID, Molofsky AV. Astrocytes and microglia: In sickness and in health. Trends Neurosci. 2020;43(3):144-154.[DOI]
-
29. Metur SP, Klionsky DJ. Adaptive immunity at the crossroads of autophagy and metabolism. Cell Mol Immunol. 2021;18(5):1096-1105.[DOI]
-
30. Belgrad J, De Pace R, Fields RD. Autophagy in myelinating glia. J Neurosci. 2020;40(2):256-266.[DOI]
-
31. Deng Z, Zhou X, Lu JH, Yue Z. Autophagy deficiency in neurodevelopmental disorders. Cell Biosci. 2021;11(1):214.[DOI]
-
33. Lin YT, Seo J, Gao F, Feldman HM, Wen HL, Penney J, et al. APOE4 causes widespread molecular and cellular alterations associated with Alzheimer’s disease phenotypes in human iPSC-derived brain cell types. Neuron. 2018;98(6):1294.[DOI]
-
36. Pelvig DP, Pakkenberg H, Stark AK, Pakkenberg B. Neocortical glial cell numbers in human brains. Neurobiol Aging. 2008;29(11):1754-1762.[DOI]
-
37. Allen NJ, Eroglu C. Cell biology of astrocyte-synapse interactions. Neuron. 2017;96(3):697-708.[DOI]
-
38. Verkhratsky A, Nedergaard M, Hertz L. Why are astrocytes important? Neurochem Res. 2015;40(2):389-401.[DOI]
-
39. Khakh BS, Sofroniew MV. Diversity of astrocyte functions and phenotypes in neural circuits. Nat Neurosci. 2015;18(7):942-952.[DOI]
-
40. Zhang Y, Barres BA. Astrocyte heterogeneity: An underappreciated topic in neurobiology. Curr Opin Neurobiol. 2010;20(5):588-594.[DOI]
-
41. Sofroniew MV, Vinters HV. Astrocytes: Biology and pathology. Acta Neuropathol. 2010;119(1):7-35.[DOI]
-
42. Ben Haim L, Carrillo-de Sauvage MA, Ceyzériat K, Escartin C. Elusive roles for reactive astrocytes in neurodegenerative diseases. Front Cell Neurosci. 2015;9:278.[DOI]
-
43. Heneka MT, Kummer MP, Latz E. Innate immune activation in neurodegenerative disease. Nat Rev Immunol. 2014;14(7):463-477.[DOI]
-
44. Ben Haim L, Ceyzériat K, Carrillo-de Sauvage MA, Aubry F, Auregan G, Guillermier M, et al. The JAK/STAT3 pathway is a common inducer of astrocyte reactivity in Alzheimer’s and Huntington’s diseases. J Neurosci. 2015;35(6):2817-2829.[DOI]
-
46. Jänen SB, Chaachouay H, Richter-Landsberg C. Autophagy is activated by proteasomal inhibition and involved in aggresome clearance in cultured astrocytes. Glia. 2010;58(14):1766-1774.[DOI]
-
47. Hol EM, Pekny M. Glial fibrillary acidic protein (GFAP) and the astrocyte intermediate filament system in diseases of the central nervous system. Curr Opin Cell Biol. 2015;32:121-130.[DOI]
-
48. Messing A, Brenner M, Feany MB, Nedergaard M, Goldman JE. Alexander disease. J Neurosci. 2012;32(15):5017-5023.[DOI]
-
49. Tang G, Perng MD, Wilk S, Quinlan R, Goldman JE. Oligomers of mutant glial fibrillary acidic protein (GFAP) Inhibit the proteasome system in Alexander disease astrocytes, and the small heat shock protein alphaB-crystallin reverses the inhibition. J Biol Chem. 2010;285(14):10527-10537.
-
56. Komatsu M, Waguri S, Chiba T, Murata S, Iwata JI, Tanida I, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441(7095):880-884.[DOI]
-
57. Su PW, Zhai Z, Wang T, Zhang YN, Wang Y, Ma K, et al. Research progress on astrocyte autophagy in ischemic stroke. Front Neurol. 2022;13:951536.[DOI]
-
59. Nikoletopoulou V, Papandreou ME, Tavernarakis N. Autophagy in the physiology and pathology of the central nervous system. Cell Death Differ. 2015;22(3):398-407.[DOI]
-
60. Sung K, Jimenez-Sanchez M. Autophagy in astrocytes and its implications in neurodegeneration. J Mol Biol. 2020;432(8):2605-2621.[DOI]
-
63. Wang SS, Shultz JR, Burish MJ, Harrison KH, Hof PR, Towns LC, et al. Functional trade-offs in white matter axonal scaling. J Neurosci. 2008;28(15):4047-4056.[DOI]
-
64. Liu H, Yang Y, Xia Y, Zhu W, Leak RK, Wei Z, et al. Aging of cerebral white matter. Ageing Res Rev. 2017;34:64-76.[DOI]
-
65. Baumann N, Pham-Dinh D. Biology of oligodendrocyte and myelin in the mammalian central nervous system. Physiol Rev. 2001;81(2):871-927.[DOI]
-
66. Emery B. Regulation of oligodendrocyte differentiation and myelination. Science. 2010;330(6005):779-782.[DOI]
-
67. Chapman TW, Hill RA. Myelin plasticity in adulthood and aging. Neurosci Lett. 2020;715:134645.[DOI]
-
68. Sams EC. Oligodendrocytes in the aging brain. Neuronal Signal. 2021;5(3):NS20210008.[DOI]
-
73. Alam MM, Zhao XF, Liao Y, Mathur R, McCallum SE, Mazurkiewicz JE, et al. Deficiency of microglial autophagy increases the density of oligodendrocytes and susceptibility to severe forms of seizures. eNeuro. 2021;8(1):ENEURO-0183.[DOI]
-
75. Damoiseaux JS. Effects of aging on functional and structural brain connectivity. NeuroImage. 2017;160:32-40.[DOI]
-
77. Jeromin AJ, Bowser RB. Biomarkers in neurodegenerative diseases. In: Beart P, Robinson M, Rattray M, Maragakis NJ, editors. Neurodegenerative diseases. Cham: Springer; 2017. p. 491-528.[DOI]
-
78. Morrison JH, Hof PR. Life and death of neurons in the aging brain. Science. 1997;278(5337):412-419.[DOI]
-
79. Pakkenberg B, Gundersen HJG. Neocortical neuron number in humans: Effect of sex and age. J Comp Neurol. 1997;384(2):312-320.[DOI]
-
80. Sousounis K, Baddour JA, Tsonis PA. Aging and regeneration in vertebrates. Curr Top Dev Biol. 2014;108:217-246.[DOI]
-
81. Zhang X, Huang N, Xiao L, Wang F, Li T. Replenishing the aged brains: Targeting oligodendrocytes and myelination? Front Aging Neurosci. 2021;13:760200.[DOI]
-
82. Saraswat Ohri S, Bankston AN, Mullins SA, Liu Y, Andres KR, Beare JE, et al. Blocking autophagy in oligodendrocytes limits functional recovery after spinal cord injury. J Neurosci. 2018;38(26):5900-5912.[DOI]
-
83. Franco-Bocanegra DK, Gourari Y, McAuley C, Chatelet DS, Johnston DA, Nicoll JAR, et al. Microglial morphology in Alzheimer’s disease and after Aβ immunotherapy. Sci Rep. 2021;11:15955.[DOI]
-
84. Prinz M, Tay TL, Wolf Y, Jung S. Microglia: Unique and common features with other tissue macrophages. Acta Neuropathol. 2014;128(3):319-331.[DOI]
-
86. Borst K, Dumas AA, Prinz M. Microglia: Immune and non-immune functions. Immunity. 2021;54(10):2194-2208.[DOI]
-
88. Kalafatakis I, Karagogeos D. Oligodendrocytes and microglia: Key players in myelin development, damage and repair. Biomolecules. 2021;11(7):1058.[DOI]
-
90. Prinz M, Jung S, Priller J. Microglia biology: One century of evolving concepts. Cell. 2019;179(2):292-311.[DOI]
-
91. Réu P, Khosravi A, Bernard S, Mold JE, Salehpour M, Alkass K, et al. The lifespan and turnover of microglia in the human brain. Cell Rep. 2017;20(4):779-784.[DOI]
-
92. Guo S, Wang H, Yin Y. Microglia polarization from M1 to M2 in neurodegenerative diseases. Front Aging Neurosci. 2022;14:815347.[DOI]
-
93. Jordão MJC, Sankowski R, Brendecke SM, Sagar , Locatelli G, Tai YH, et al. Single-cell profiling identifies myeloid cell subsets with distinct fates during neuroinflammation. Science. 2019;363(6425):eaat7554.[DOI]
-
96. Li G, Sherchan P, Tang Z, Tang J. Autophagy & phagocytosis in neurological disorders and their possible cross-talk. Curr Neuropharmacol. 2021;19(11):1912-1924.[DOI]
-
97. Minchev D, Kazakova M, Sarafian V. Neuroinflammation and autophagy in Parkinson’s disease: Novel perspectives. Int J Mol Sci. 2022;23(23):14997.[DOI]
-
98. Münz C. Canonical and non-canonical functions of the autophagy machinery in MHC restricted antigen presentation. Front Immunol. 2022;13:868888.[DOI]
-
99. Pradel B, Robert-Hebmann V, Espert L. Regulation of innate immune responses by autophagy: A goldmine for viruses. Front Immunol. 2020;11:578038.[DOI]
-
100. Wu MY, Lu JH. Autophagy and macrophage functions: Inflammatory response and phagocytosis. Cells. 2020;9(1):70.[DOI]
-
102. Berglund R, Guerreiro-Cacais AO, Adzemovic MZ, Zeitelhofer M, Lund H, Ewing E, et al. Microglial autophagy–associated phagocytosis is essential for recovery from neuroinflammation. Sci Immunol. 2020;5(52):eabb5077.[DOI]
-
103. Jin MM, Wang F, Qi D, Liu WW, Gu C, Mao CJ, et al. A critical role of autophagy in regulating microglia polarization in neurodegeneration. Front Aging Neurosci. 2018;10:378.[DOI]
-
104. Xu Y, Propson NE, Du S, Xiong W, Zheng H. Autophagy deficiency modulates microglial lipid homeostasis and aggravates tau pathology and spreading. Proc Natl Acad Sci U S A. 2021;118(27):e2023418118.[DOI]
-
106. Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, et al. Intraneuronal β-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formation. J Neurosci. 2006;26(40):10129-10140.[DOI]
-
111. Franceschi C, Garagnani P, Vitale G, Capri M, Salvioli S. Inflammaging and ‘garb-aging’. Trends Endocrinol Metab. 2017;28(3):199-212.[DOI]
-
112. Diot A, Morten K, Poulton J. Mitophagy plays a central role in mitochondrial ageing. Mamm Genome. 2016;27:381-395.[DOI]
-
113. Vellai T, Takács-Vellai K, Sass M, Klionsky DJ. The regulation of aging: Does autophagy underlie longevity? Trends Cell Biol. 2009;19(10):487-494.[DOI]
-
115. Takahama M, Akira S, Saitoh T. Autophagy limits activation of the inflammasomes. Immunol Rev. 2018;281(1):62-73.[DOI]
-
116. Lee JH, Chang W, Min SS, Song DY, Yoo HI. Beyond support cells: Astrocytic autophagy as a central regulator of CNS homeostasis and neurodegenerative diseases. Cells. 2025;14(17):1342.[DOI]
-
117. Yang QQ, Zhou JW. Neuroinflammation in the central nervous system: Symphony of glial cells. Glia. 2019;67(6):1017-1035.[DOI]
-
118. Naffaa MM. Mechanisms of astrocytic and microglial purinergic signaling in homeostatic regulation and implications for neurological disease. Explor Neurosci. 2025;4:100676.[DOI]
-
119. Giera S, Luo R, Ying Y, Ackerman SD, Jeong SJ, Stoveken HM, et al. Microglial transglutaminase-2 drives myelination and myelin repair via GPR56/ADGRG1 in oligodendrocyte precursor cells. eLife. 2018;7:e33385.[DOI]
-
120. Hughes AN. Glial cells promote myelin formation and elimination. Front Cell Dev Biol. 2021;9:661486.[DOI]
-
121. Adamu A, Li S, Gao F, Xue G. The role of neuroinflammation in neurodegenerative diseases: Current understanding and future therapeutic targets. Front Aging Neurosci. 2024;16:1347987.[DOI]
-
122. Takata F, Nakagawa S, Matsumoto J, Dohgu S. Blood-brain barrier dysfunction amplifies the development of neuroinflammation: Understanding of cellular events in brain microvascular endothelial cells for prevention and treatment of BBB dysfunction. Front Cell Neurosci. 2021;15:661838.[DOI]
-
125. Masters CL, Bateman R, Blennow K, Rowe CC, Sperling RA, Cummings JL. Alzheimer’s disease. Nat Rev Dis Primers. 2015;1:15056.[DOI]
-
126. Selkoe DJ. Alzheimer’s disease: Genes, proteins, and therapy. Physiol Rev. 2001;81(2):741-766.[DOI]
-
127. Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015;14(4):388-405.[DOI]
-
128. Phillips EC, Croft CL, Kurbatskaya K, O’Neill MJ, Hutton ML, Hanger DP, et al. Astrocytes and neuroinflammation in Alzheimer’s disease. Biochem Soc Trans. 2014;42(5):1321-1325.[DOI]
-
129. Ingelsson M, Fukumoto H, Newell KL, Growdon JH, Hedley-Whyte ET, Frosch MP, et al. Early Aβ accumulation and progressive synaptic loss, gliosis, and tangle formation in AD brain. Neurology. 2004;62(6):925-931.[DOI]
-
130. Itagaki S, McGeer PL, Akiyama H, Zhu S, Selkoe D. Relationship of microglia and astrocytes to amyloid deposits of Alzheimer disease. J Neuroimmunol. 1989;24(3):173-182.[DOI]
-
132. Wisniewski HM, Wegiel J. Spatial relationships between astrocytes and classical plaque components. Neurobiol Aging. 1991;12(5):593-600.[DOI]
-
137. Palmer JE, Wilson N, Son SM, Obrocki P, Wrobel L, Rob M, et al. Autophagy, aging, and age-related neurodegeneration. Neuron. 2025;113(1):29-48.[DOI]
-
138. Husain MA, Laurent B, Plourde M. APOE and Alzheimer’s disease: From lipid transport to physiopathology and therapeutics. Front Neurosci. 2021;15:630502.[DOI]
-
140. Ries M, Sastre M. Mechanisms of aβ clearance and degradation by glial cells. Front Aging Neurosci. 2016;8:160.[DOI]
-
143. Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, et al. Apolipoprotein E: High-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90(5):1977-1981.
-
145. Simonovitch S, Schmukler E, Bespalko A, Iram T, Frenkel D, Holtzman DM, et al. Impaired autophagy in APOE4 astrocytes. J Alzheimers Dis. 2016;51(3):915-927.[DOI]
-
146. Nixon RA, Rubinsztein DC. Mechanisms of autophagy–lysosome dysfunction in neurodegenerative diseases. Nat Rev Mol Cell Biol. 2024;25(11):926-946.[DOI]
-
148. Xiao Q, Yan P, Ma X, Liu H, Perez R, Zhu A, et al. Enhancing astrocytic lysosome biogenesis facilitates aβ clearance and attenuates amyloid plaque pathogenesis. J Neurosci. 2014;34(29):9607-9620.[DOI]
-
149. Settembre C, Di Malta C, Polito VA, Arencibia MG, Vetrini F, Erdin S, et al. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332(6036):1429-1433.[DOI]
-
152. Abeti R, Abramov AY, Duchen MR. β-amyloid activates PARP causing astrocytic metabolic failure and neuronal death. Brain. 2011;134(6):1658-1672.[DOI]
-
154. Iram T, Trudler D, Kain D, Kanner S, Galron R, Vassar R, et al. Astrocytes from old Alzheimer’s disease mice are impaired in Aβ uptake and in neuroprotection. Neurobiol Dis. 2016;96:84-94.[DOI]
-
155. Jana A, Pahan K. Fibrillar amyloid-β-activated human astroglia kill primary human neurons via neutral sphingomyelinase: Implications for Alzheimer’s disease. J Neurosci. 2010;30(38):12676-12689.[DOI]
-
156. Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541(7638):481-487.[DOI]
-
157. Orre M, Kamphuis W, Osborn LM, Jansen AHP, Kooijman L, Bossers K, et al. Isolation of glia from Alzheimer’s mice reveals inflammation and dysfunction. Neurobiol Aging. 2014;35(12):2746-2760.[DOI]
-
158. Krüger U, Wang Y, Kumar S, Mandelkow EM. Autophagic degradation of tau in primary neurons and its enhancement by trehalose. Neurobiol Aging. 2012;33(10):2291-2305.[DOI]
-
160. Ozcelik S, Fraser G, Castets P, Schaeffer V, Skachokova Z, Breu K, et al. Rapamycin attenuates the progression of tau pathology in P301S tau transgenic mice. PLoS One. 2013;8(5):e62459.[DOI]
-
164. Bartzokis G. Alzheimer’s disease as homeostatic responses to age-related myelin breakdown. Neurobiol Aging. 2011;32(8):1341-1371.[DOI]
-
166. Depp C, Sun T, Sasmita AO, Spieth L, Berghoff SA, Nazarenko T, et al. Myelin dysfunction drives amyloid-β deposition in models of Alzheimer’s disease. Nature. 2023;618(7964):349-357.[DOI]
-
167. Vanzulli I, Papanikolaou M, De-La-Rocha IC, Pieropan F, Rivera AD, Gomez-Nicola D, et al. Disruption of oligodendrocyte progenitor cells is an early sign of pathology in the triple transgenic mouse model of Alzheimer’s disease. Neurobiol Aging. 2020;94:130-139.[DOI]
-
168. Ktena N, Kaplanis SI, Kolotuev I, Georgilis A, Kallergi E, Stavroulaki V, et al. Autophagic degradation of CNS myelin maintains axon integrity. Cell Stress. 2022;6(12):93-107.[DOI]
-
169. Wang MR, Zhang XJ, Liu HC, Ma WD, Zhang ML, Zhang Y, et al. Matrine protects oligodendrocytes by inhibiting their apoptosis and enhancing mitochondrial autophagy. Brain Res Bull. 2019;153:30-38.[DOI]
-
171. Litwiniuk A, Juszczak GR, Stankiewicz AM, Urbańska K. The role of glial autophagy in Alzheimer’s disease. Mol Psychiatry. 2023;28(11):4528-4539.[DOI]
-
173. Houtman J, Freitag K, Gimber N, Schmoranzer J, Heppner FL, Jendrach M. Beclin1-driven autophagy modulates the inflammatory response of microglia viaNLRP3. EMBO J. 2019;38(4):e99430.[DOI]
-
174. Gabandé-Rodríguez E, Keane L, Capasso M. Microglial phagocytosis in aging and Alzheimer’s disease. J Neurosci Res. 2020;98(2):284-298.[DOI]
-
175. Heckmann BL, Teubner BJW, Tummers B, Boada-Romero E, Harris L, Yang M, et al. LC3-associated endocytosis facilitates β-amyloid clearance and mitigates neurodegeneration in murine Alzheimer’s disease. Cell. 2019;178(3):536-551.[DOI]
-
177. Plaza-Zabala A, Sierra-Torre V, Sierra A. Autophagy and microglia: Novel partners in neurodegeneration and aging. Int J Mol Sci. 2017;18(3):598.[DOI]
-
181. Estfanous S, Daily KP, Eltobgy M, Deems NP, Anne MNK, Krause K, et al. Elevated expression of miR-17 in microglia of Alzheimer’s disease patients abrogates autophagy-mediated amyloid-β degradation. Front Immunol. 2021;12:705581.[DOI]
-
182. Lu J, Zhou W, Dou F, Wang C, Yu Z. TRPV1 sustains microglial metabolic reprogramming in Alzheimer’s disease. EMBO Rep. 2021;22(6):e52013.[DOI]
-
183. Fairley LH, Wong JH, Barron AM. Mitochondrial regulation of microglial immunometabolism in Alzheimer’s disease. Front Immunol. 2021;12:624538.[DOI]
-
184. Fang EF, Hou Y, Palikaras K, Adriaanse BA, Kerr JS, Yang B, et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat Neurosci. 2019;22(3):401-412.[DOI]
-
185. Poewe W, Seppi K, Tanner CM, Halliday GM, Brundin P, Volkmann J, et al. Parkinson disease. Nat Rev Dis Primers. 2017;3:17013.[DOI]
-
186. Simon DK, Tanner CM, Brundin P. Parkinson disease epidemiology, pathology, genetics, and pathophysiology. Clin Geriatr Med. 2020;36(1):1-12.[DOI]
-
187. Mulak A. Brain-gut-microbiota axis in Parkinson’s disease. World J Gastroenterol. 2015;21(37):10609.[DOI]
-
188. Chandra R, Hiniker A, Kuo YM, Nussbaum RL, Liddle RA. α-Synuclein in gut endocrine cells and its implications for Parkinson’s disease. JCI Insight. 2017;2(12):e92295.[DOI]
-
189. Li Q, Meng LB, Chen LJ, Shi X, Tu L, Zhou Q, et al. The role of the microbiota-gut-brain axis and intestinal microbiome dysregulation in Parkinson’s disease. Front Neurol. 2023;14:1185375.[DOI]
-
190. Montalbán-Rodríguez A, Abalo R, López-Gómez L. From the gut to the brain: The role of enteric glial cells and their involvement in the pathogenesis of Parkinson’s disease. Int J Mol Sci. 2024;25(2):1294.[DOI]
-
194. Shadrina M, Slominsky P. Modeling Parkinson’s disease: Not only rodents? Front Aging Neurosci. 2021;13:695718.[DOI]
-
196. Lindström V, Gustafsson G, Sanders LH, Howlett EH, Sigvardson J, Kasrayan A, et al. Extensive uptake of α-synuclein oligomers in astrocytes results in sustained intracellular deposits and mitochondrial damage. Mol Cell Neurosci. 2017;82:143-156.[DOI]
-
197. Stefanis L, Emmanouilidou E, Pantazopoulou M, Kirik D, Vekrellis K, Tofaris GK. How is alpha-synuclein cleared from the cell? J Neurochem. 2019;150(5):577-590.[DOI]
-
198. Tremblay ME, Cookson MR, Civiero L. Glial phagocytic clearance in Parkinson’s disease. Mol Neurodegener. 2019;14:16.[DOI]
-
199. Valdinocci D, Radford R, Siow S, Chung R, Pountney D. Potential modes of intercellular α-synuclein transmission. Int J Mol Sci. 2017;18(2):469.[DOI]
-
200. Angot E, Steiner JA, Lema Tomé CM, Ekström P, Mattsson B, Björklund A, et al. Alpha-synuclein cell-to-cell transfer and seeding in grafted dopaminergic neurons in vivo. PLoS One. 2012;7(6):e39465.[DOI]
-
204. Jewett M, Dickson E, Brolin K, Negrini M, Jimenez-Ferrer I, Swanberg M. Glutathione S-transferase alpha 4 prevents dopamine neurodegeneration in a rat alpha-synuclein model of Parkinson’s disease. Front Neurol. 2018;9:222.[DOI]
-
205. Lu SZ, Guo YS, Liang PZ, Zhang SZ, Yin S, Yin YQ, et al. Suppression of astrocytic autophagy by αB-crystallin contributes to α-synuclein inclusion formation. Transl Neurodegener. 2019;8:3.[DOI]
-
207. Cavaliere F, Cerf L, Dehay B, Ramos-Gonzalez P, De Giorgi F, Bourdenx M, et al. In vitro α-synuclein neurotoxicity and spreading among neurons and astrocytes using Lewy body extracts from Parkinson disease brains. Neurobiol Dis. 2017;103:101-112.[DOI]
-
208. Gu XL, Long CX, Sun L, Xie C, Lin X, Cai H. Astrocytic expression of Parkinson’s disease-related A53T α-synuclein causes neurodegeneration in mice. Mol Brain. 2010;3:12.[DOI]
-
210. Booth HDE, Hirst WD, Wade-Martins R. The role of astrocyte dysfunction in Parkinson’s disease pathogenesis. Trends Neurosci. 2017;40(6):358-370.[DOI]
-
212. Tsunemi T, Ishiguro Y, Yoroisaka A, Valdez C, Miyamoto K, Ishikawa K, et al. Astrocytes protect human dopaminergic neurons from α-synuclein accumulation and propagation. J Neurosci. 2020;40(45):8618-8628.[DOI]
-
215. Aflaki E, Stubblefield BK, McGlinchey RP, McMahon B, Ory DS, Sidransky E. A characterization of Gaucher iPS-derived astrocytes: Potential implications for Parkinson’s disease. Neurobiol Dis. 2020;134:104647.[DOI]
-
219. Hu ZL, Sun T, Lu M, Ding JH, Du RH, Hu G. Kir6.1/K-ATP channel on astrocytes protects against dopaminergic neurodegeneration in the MPTP mouse model of Parkinson’s disease via promoting mitophagy. Brain Behav Immun. 2019;81:509-522.[DOI]
-
221. Karabiyik C, Lee MJ, Rubinsztein DC. Autophagy impairment in Parkinson’s disease. Essays Biochem. 2017;61(6):711-720.[DOI]
-
222. McBean G. Cysteine, glutathione, and thiol redox balance in astrocytes. Antioxidants. 2017;6(3):62.[DOI]
-
223. Hirsch EC, Hunot S, Damier P, Brugg B, Faucheux BA, Michel PP, et al. Glial cell participation in the degeneration of dopaminergic neurons in Parkinson’s disease. Adv Neurol. 1999;80:9-18.[PubMed]
-
225. Ventura S, Pena-Díaz S. One ring is sufficient to inhibit α-synuclein aggregation. Neural Regen Res. 2022;17(3):508.[DOI]
-
226. Cardinale A, Calabrese V, de Iure A, Picconi B. Alpha-synuclein as a prominent actor in the inflammatory synaptopathy of Parkinson’s disease. Int J Mol Sci. 2021;22(12):6517.[DOI]
-
227. Kam TI, Hinkle JT, Dawson TM, Dawson VL. Microglia and astrocyte dysfunction in Parkinson’s disease. Neurobiol Dis. 2020;144:105028.[DOI]
-
228. Kim C, Ho DH, Suk JE, You S, Michael S, Kang J, et al. Neuron-released oligomeric α-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat Commun. 2013;4:1562.[DOI]
-
229. Verma DK, Seo BA, Ghosh A, Ma SX, Hernandez-Quijada K, Andersen JK, et al. Alpha-synuclein preformed fibrils induce cellular senescence in Parkinson’s disease models. Cells. 2021;10(7):1694.[DOI]
-
231. Izco M, Blesa J, Verona G, Cooper JM, Alvarez-Erviti L. Glial activation precedes alpha-synuclein pathology in a mouse model of Parkinson’s disease. Neurosci Res. 2021;170:330-340.[DOI]
-
232. Chou TW, Chang NP, Krishnagiri M, Patel AP, Lindman M, Angel JP, et al. Fibrillar α-synuclein induces neurotoxic astrocyte activation via RIP kinase signaling and NF-κB. Cell Death Dis. 2021;12(8):756.[DOI]
-
236. Zhao ZH, Chen ZT, Zhou RL, Zhang X, Ye QY, Wang YZ. Increased DJ-1 and α-synuclein in plasma neural-derived exosomes as potential markers for Parkinson’s disease. Front Aging Neurosci. 2019;10:438.[DOI]
-
237. Rostami J, Holmqvist S, Lindström V, Sigvardson J, Westermark GT, Ingelsson M, et al. Human astrocytes transfer aggregated alpha-synuclein via tunneling nanotubes. J Neurosci. 2017;37(49):11835-11853.[DOI]
-
240. Bechler ME, Byrne L, ffrench-Constant C. CNS myelin sheath lengths are an intrinsic property of oligodendrocytes. Curr Biol. 2015;25(18):2411-2416.[DOI]
-
247. Nalls MA, Blauwendraat C, Vallerga CL, Heilbron K, Bandres-Ciga S, Chang D, et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: A meta-analysis of genome-wide association studies. Lancet Neurol. 2019;18(12):1091-1102.[DOI]
-
249. Qin Y, Qiu J, Wang P, Liu J, Zhao Y, Jiang F, et al. Impaired autophagy in microglia aggravates dopaminergic neurodegeneration by regulating NLRP3 inflammasome activation in experimental models of Parkinson’s disease. Brain Behav Immun. 2021;91:324-338.[DOI]
-
256. Colonna M, Butovsky O. Microglia function in the central nervous system during health and neurodegeneration. Annu Rev Immunol. 2017;35:441-468.[DOI]
-
257. Stratoulias V, Venero JL, Tremblay M, Joseph B. Microglial subtypes: Diversity within the microglial community. EMBO J. 2019;38(17):e101997.[DOI]
-
259. Miyamoto A, Wake H, Ishikawa AW, Eto K, Shibata K, Murakoshi H, et al. Microglia contact induces synapse formation in developing somatosensory cortex. Nat Commun. 2016;7:12540.[DOI]
-
260. Tu HY, Yuan BS, Hou XO, Zhang XJ, Pei CS, Ma YT, et al. α-synuclein suppresses microglial autophagy and promotes neurodegeneration in a mouse model of Parkinson’s disease. Aging Cell. 2021;20(12):e13522.[DOI]
-
261. Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010;140(6):918-934.[DOI]
-
262. Wu Q, Zou C. Microglial dysfunction in neurodegenerative diseases via RIPK1 and ROS. Antioxidants. 2022;11(11):2201.[DOI]
-
265. Srivastava IN, Shperdheja J, Baybis M, Ferguson T, Crino PB. mTOR pathway inhibition prevents neuroinflammation and neuronal death in a mouse model of cerebral palsy. Neurobiol Dis. 2016;85:144-154.[DOI]
-
267. Mastroeni D, Nolz J, Sekar S, Delvaux E, Serrano G, Cuyugan L, et al. Laser-captured microglia in the Alzheimer’s and Parkinson’s brain reveal unique regional expression profiles and suggest a potential role for hepatitis B in the Alzheimer’s brain. Neurobiol Aging. 2018;63:12-21.[DOI]
-
268. Ghosh D, Mehra S, Sahay S, Singh PK, Maji SK. α-synuclein aggregation and its modulation. Int J Biol Macromol. 2017;100:37-54.[DOI]
-
269. George S, Rey NL, Tyson T, Esquibel C, Meyerdirk L, Schulz E, et al. Microglia affect α-synuclein cell-to-cell transfer in a mouse model of Parkinson’s disease. Mol Neurodegener. 2019;14:34.[DOI]
-
270. Tan EK, Chao YX, West A, Chan LL, Poewe W, Jankovic J. Parkinson disease and the immune system: Associations, mechanisms and therapeutics. Nat Rev Neurol. 2020;16(6):303-318.[DOI]
-
272. Xia Y, Zhang G, Han C, Ma K, Guo X, Wan F, et al. Microglia as modulators of exosomal alpha-synuclein transmission. Cell Death Dis. 2019;10:174.[DOI]
-
273. Li Y, Xia Y, Yin S, Wan F, Hu J, Kou L, et al. Targeting microglial α-synuclein/TLRs/NF-kappaB/NLRP3 inflammasome axis in Parkinson’s disease. Front Immunol. 2021;12:719807.[DOI]
-
279. Wilton DK, Stevens B. The contribution of glial cells to Huntington’s disease pathogenesis. Neurobiol Dis. 2020;143:104963.[DOI]
-
280. MacDonald M. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. 1993;72(6):971-983.[DOI]
-
281. Jimenez-Sanchez M, Licitra F, Underwood BR, Rubinsztein DC. Huntington’s disease: Mechanisms of pathogenesis and therapeutic strategies. Cold Spring Harb Perspect Med. 2017;7(7):a024240.[DOI]
-
285. Ortega Z, Lucas JJ. Ubiquitin-proteasome system involvement in Huntington’s disease. Front Mol Neurosci. 2014;7:77.[DOI]
-
286. Cortes CJ, La Spada AR. The many faces of autophagy dysfunction in Huntington’s disease: From mechanism to therapy. Drug Discov Today. 2014;19(7):963-971.[DOI]
-
288. Wong YC, Holzbaur ELF. The regulation of autophagosome dynamics by huntingtin and HAP1 is disrupted by expression of mutant huntingtin, leading to defective cargo degradation. J Neurosci. 2014;34(4):1293-1305.[DOI]
-
289. Ashkenazi A, Bento CF, Ricketts T, Vicinanza M, Siddiqi F, Pavel M, et al. Polyglutamine tracts regulate beclin 1-dependent autophagy. Nature. 2017;545(7652):108-111.[DOI]
-
291. Rocha NP, Ribeiro FM, Furr-Stimming E, Teixeira AL. Neuroimmunology of Huntington’s disease: Revisiting evidence from human studies. Mediat Inflamm. 2016;2016:8653132.[DOI]
-
292. Festa BP, Siddiqi FH, Jimenez-Sanchez M, Won H, Rob M, Djajadikerta A, et al. Microglial-to-neuronal CCR5 signaling regulates autophagy in neurodegeneration. Neuron. 2023;111(13):2021-2037.[DOI]
-
298. Liévens JC, Woodman B, Mahal A, Spasic-Boscovic O, Samuel D, Kerkerian-Le Goff L, et al. Impaired glutamate uptake in the R6 Huntington’s disease transgenic mice. Neurobiol Dis. 2001;8(5):807-821.[DOI]
-
302. Hong Y, Zhao T, Li XJ, Li S. Mutant huntingtin impairs BDNF release from astrocytes by disrupting conversion of Rab3a-GTP into Rab3a-GDP. J Neurosci. 2016;36(34):8790-8801.[DOI]
-
304. Tydlacka S, Wang CE, Wang X, Li S, Li XJ. Differential activities of the ubiquitin–proteasome system in neurons versus glia may account for the preferential accumulation of misfolded proteins in neurons. J Neurosci. 2008;28(49):13285-13295.[DOI]
-
307. Barmaki H, Nourazarian A, Shademan B, khaki-khatibi F. The autophagy paradox: A new hypothesis in neurodegenerative disorders. Neurochem Int. 2024;179:105827.[DOI]
-
309. Perucho J, Gómez A, Muñoz MP, de Yébenes JG, Mena MÁ, Casarejos MJ. Trehalose rescues glial cell dysfunction in striatal cultures from HD R6/1 mice at early postnatal development. Mol Cell Neurosci. 2016;74:128-145.[DOI]
-
310. Mandrioli J, Mediani L, Alberti S, Carra S. ALS and FTD: Where RNA metabolism meets protein quality control. Semin Cell Dev Biol. 2020;99:183-192.[DOI]
-
311. Geloso MC, Corvino V, Marchese E, Serrano A, Michetti F, D’Ambrosi N. The dual role of microglia in ALS: Mechanisms and therapeutic approaches. Front Aging Neurosci. 2017;9:242.[DOI]
-
312. Massenzio F, Peña-Altamira E, Petralla S, Virgili M, Zuccheri G, Miti A, et al. Microglial overexpression of fALS-linked mutant SOD1 induces SOD1 processing impairment, activation and neurotoxicity and is counteracted by the autophagy inducer trehalose. Biochim Biophys Acta Mol Basis Dis. 2018;1864(12):3771-3785.
-
313. Taylor JP, Brown RH Jr, Cleveland DW. Decoding ALS From genes to mechanism. Nature. 2016;539(7628):197-206.[DOI]
-
315. Fecto F. SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch Neurol. 2011;68(11):1440.[DOI]
-
317. Maruyama H, Morino H, Ito H, Izumi Y, Kato H, Watanabe Y, et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature. 2010;465(7295):223-226.[DOI]
-
318. Lino MM, Schneider C, Caroni P. Accumulation of SOD1 mutants in postnatal motoneurons does not cause motoneuron pathology or motoneuron disease. J Neurosci. 2002;22(12):4825-4832.[DOI]
-
319. Pramatarova A, Laganière J, Roussel J, Brisebois K, Rouleau GA. Neuron-specific expression of mutant superoxide dismutase 1 in transgenic mice does not lead to motor impairment. J Neurosci. 2001;21(10):3369-3374.[DOI]
-
320. Gong YH, Parsadanian AS, Andreeva A, Snider WD, Elliott JL. Restricted expression of G86R Cu/Zn superoxide dismutase in astrocytes results in astrocytosis but does not cause motoneuron degeneration. J Neurosci. 2000;20(2):660-665.[DOI]
-
325. Aebischer J, Cassina P, Otsmane B, Moumen A, Seilhean D, Meininger V, et al. IFNγ triggers a LIGHT-dependent selective death of motoneurons contributing to the non-cell-autonomous effects of mutant SOD1. Cell Death Differ. 2011;18(5):754-768.[DOI]
-
328. Nagai M, Re DB, Nagata T, Chalazonitis A, Jessell TM, Wichterle H, et al. Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat Neurosci. 2007;10(5):615-622.[DOI]
-
329. BaofengFeng , Amponsah AE, Guo R, Liu X, Zhang J, Du X, et al. Autophagy-mediated inflammatory cytokine secretion in sporadic ALS patient iPSC-derived astrocytes. Oxid Med Cell Longev. 2022;2022:6483582.[DOI]
-
332. Stewart VC, Heslegrave AJ, Brown GC, Clark JB, Heales SJR. Nitric oxide-dependent damage to neuronal mitochondria involves the NMDA receptor. Eur J Neurosci. 2002;15(3):458-464.[DOI]
-
333. Barton SK, Gregory JM, Chandran S, Turner BJ. Could an impairment in local translation of mRNAs in glia be contributing to pathogenesis in ALS? Front Mol Neurosci. 2019;12:124.[DOI]
-
337. Ryan L, Rubinsztein DC. The autophagy of stress granules. FEBS Lett. 2024;598(1):59-72.[DOI]
Copyright
© The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Publisher’s Note
Science Exploration remains a neutral stance on jurisdictional claims in published
maps
and institutional affiliations. The views expressed in this article are solely those
of
the author(s) and do not reflect the opinions of the Editors or the publisher.
Share And Cite



Science Exploration Style
Çakıcı O, Tavernarakis N. The role of glia autophagy in CNS homeostasis, ageing and disease. Geromedicine. 2026;2:202527. https://doi.org/10.70401/Geromedicine.2026.0019
Tips
Copy completed.
Submit a Manuscript
Author Instructions
Cite this Article
Article Metrics
0
View
0
Download
Cited
Article Updates

- Abstract
- Keywords
- 1. Introduction
- 2. Autophagy in Glia
- 3. Glial Autophagy in Neurodegenerative Diseases
- 4. Conclusions and Future Perspectives
- Acknowledgements
- Authors contribution
- Conflicts of interest
- Ethical approval
- Consent to participate
- Consent for publication
- Availability of data and materials
- Funding
- References
- Copyright
Science Exploration Style
Çakıcı O, Tavernarakis N. The role of glia autophagy in CNS homeostasis, ageing and disease. Geromedicine. 2026;2:202527. https://doi.org/10.70401/Geromedicine.2026.0019
copy
Share Link
copy