Is it time to rethink the single lifetime measurement of Lipoprotein(a)?
Lavalle-Cobo Augusto
1,*

,
Garcia-Zamora Sebastian
2
,
María Gabriela Matta
3
,
Schreier Laura
4
,
Corral Pablo
5
*Correspondence to:
Lavalle-Cobo Augusto, Department of Cardiology, Sanatorio Otamendi, Ciudad Autónoma de Buenos Aires C1007ABJ, Buenos Aires, Argentina.
E-mail: lavallea@otmendi.com.ar
Adv Lipoprotein(a) Res. 2026;1:202601. 10.70401/alr.2026.0007
Received: January 02, 2026Accepted: March 16, 2026Published: March 18, 2026
Abstract
Lipoprotein(a) [Lp(a)] is a genetically determined cardiovascular risk factor that has traditionally been considered stable throughout life, leading major scientific societies to recommend a single lifetime measurement in most individuals. However, emerging evidence challenges this long-standing assumption, suggesting the presence of clinically relevant intra-individual variability. While many individuals remain within the same risk category over time, a non-negligible proportion, particularly those with intermediate or borderline levels, may experience changes sufficient to alter cardiovascular risk classification or potential eligibility for emerging Lp(a)-lowering therapies. Variability appears more pronounced at lower baseline concentrations and may be influenced by demographic, metabolic, and inflammatory factors. Importantly, although long-term studies confirm that a single Lp(a) measurement is predictive of cardiovascular risk, the growing recognition of biological variability raises questions regarding risk stratification in selected patients. Standardization of variability definitions and further longitudinal research are needed to clarify the clinical implications of repeat testing. These considerations are particularly relevant in the era of targeted Lp(a)-lowering therapies, where accurate classification may influence treatment decisions. In this review, we summarize current data on Lp(a) variability across diverse populations and clinical contexts.
Keywords
Lipoprotein(a), variability, cardiovascular disease, risk stratification
1. Introduction
Elevated plasma concentrations of lipoprotein(a) [Lp(a)] are an independent risk factor for atherosclerotic cardiovascular disease (ASCVD), with more than 90% of circulating levels determined genetically[1]. For decades, it has been widely assumed that this strong genetic predisposition results in relatively stable plasma Lp(a) concentrations throughout life, with minimal influence from environmental or pharmacological factors. This understanding has led major scientific societies to recommend a single lifetime Lp(a) measurement for most individuals, in contrast to the serial assessments routinely performed for other lipid parameters in cardiovascular risk stratification[2,3].
More recent data challenge this paradigm, suggesting the existence of clinically relevant intra-individual variability in Lp(a) levels. In this review, we summarize the current evidence regarding Lp(a) variability and explore whether selected patient populations may benefit from repeated Lp(a) measurements.
2. Biological and Analytical Considerations in Lp(a) Measurement
Lp(a) is a structurally complex particle composed of an low density lipoprotein (LDL)-like moiety covalently bound to apolipoprotein(a), a highly polymorphic protein characterized by variable numbers of kringle IV type 2 repeats[1,3]. This structural heterogeneity contributes to substantial interindividual differences in particle size and mass, complicating assay calibration and interpretation[2,3].
Lp(a) can be reported in mass units (mg/dL) or molar units (nmol/L), which are not directly interchangeable due to isoform size variability[3]. Although modern assays are largely isoform-insensitive, residual analytical variation persists and must be distinguished from true biological variability[4].
Understanding these structural and analytical complexities is essential when interpreting serial Lp(a) measurements and assessing whether observed changes reflect assay imprecision or clinically meaningful biological fluctuation[5].
3. Intra-Individual Variability
Intra-individual variability is a complex and non-uniform concept that can be defined and quantified using different methodological approaches. It may reflect true biological fluctuations over time and can be influenced by physiological or pathological conditions. However, it must be clearly distinguished from analytical variability related to assay methodology.
Early evidence of intra-individual variability was reported by Marcovina et al.[4], who demonstrated that biological variation in Lp(a) levels differed according to baseline concentration, with greater relative variability observed at lower levels. Although this initial study was conducted in a small cohort of healthy individuals and with the methodology used at that time, subsequent investigations across broader populations have confirmed that Lp(a) concentrations are not entirely immutable over time. A summary of the principal studies evaluating intra-individual Lp(a) variability is provided in Table 1.
Table 1. Summary of studies evaluating intra-individual Lp(a) variability.
| Study (year) [ref] | Population Type | N | Follow-up/Interval | Definition of Variability | % Significant Variability | Risk Reclassification | Key Observations |
| Marcovina et al.[4] 1994 | Healthy individuals | 20 | Serial samples | CVb | Individual-level data only | Not reported | Greater relative variability at lower baseline Lp(a) levels |
| Hernández et al.[11] 2003 | Diabetes cohort | 70 | 4 serial measurements | CVb | 31.7% (CVb) | Yes (~20% misclassification) | Greater variability at lower baseline levels |
| Marcovina et al.[13] 2018 | IONIS-APO(a) placebo arm | 52 | Up to 192 days (7-12 samples) | Outlier if > 25% from baseline at any visit | 40.4% | 10.4% in borderline level patients | Frequent sampling increases detection of variability |
| de Boer et al.[16] 2022 | Pediatric lipid clinic (≈68% FH) | 2,740 | Longitudinal (childhood–adulthood) | Relative change ≥ 20% | 68% | Not specified | Developmental and metabolic influences likely |
| Trinder et al.[5] 2022 | UK Biobank (general population) | 16,017 | Median 4.2 years | Absolute change ≥ 25 nmol/L | 15% | Change ≥ 25 nmol/L was not association with incident CAD | Overall stability predominates; variability in minority |
| Deshotels et al.[6] 2022 | ARIC cohort (general population) | 4,734 | Visit 4–5 interval | Absolute change > 20 mg/dL | 14.7% | 58.1% in borderline group move to high Lp(a) at visit 5 | Intermediate levels most prone to reclassification |
| Harb et al.[12] 2024 | Clinical database cohort | 609 | Serial measurements | ≥ 10 mg/dL AND ≥ 25% relative change | 40.5% | 53% in the intermediate risk (20% to low risk and 33% to high risk) | Higher variability in women and Black individuals |
| Gaba et al.[14] 2024 | OCEAN(a)-DOSE placebo arm | 53 | 72 weeks | ≥ 25% vs. individual mean OR ≥ 50 nmol/L | 47% | - | Patients had baseline Lp(a) levels ≥ 150 nmol/L |
| Matta et al.[15] 2024 | GAELp(a) clinical cohort | 61 | ≥ 4 months | Relative change ≥ 25% (max discrepancy vs. baseline) | 34.4% | Yes (mainly in intermediate levels) | Real-world clinical cohort; instability concentrated in borderline range |
| Burzyńska et al.[8] 2025 | STAR-Lp(a) observational cohort | 1,263 | Two serial measurements | Relative change ≥ 20% | 44.3% | 6.2% shifted risk category (3.1% increase and 3.1% decrease) | Extremes stable; reclassification in intermediate levels |
| Joo et al.[7] 2025 | Korean multicentre cohort | 5,305 | ≥ 90 days between measures | > 10 mg/dL AND > 25% relative change | 19.9% | Yes; ~51.9% in grey zone (29.3% move to high risk and 22.5% to low risk) | Instability concentrated in intermediate range |
| Awad et al.[9] 2025 | Large academic health system | > 10,000 | Serial routine testing | Category shifts + absolute changes | Majority stable | Yes, 51.2% with borderline levels change their category. | Real-world confirmation of stability at extremes |
| Ghouse et al.[10] 2025 | Population-based cohort | 12,202 | Median 4.4 years | Absolute change + category shifts | Overall strong stability | 29.5% of patients at intermediate risk were reclassified to high risk and 9% to low risk. | High within-person correlation; limited movement at extremes |
Lp(a): lipoprotein(a); CVb: coefficient of biological variation; ARIC: Atherosclerosis Risk in Communities; FH: familial hypercholesterolemia; CAD: coronary artery disease.
Absolute thresholds (mg/dL vs. nmol/L) are not directly interchangeable due to apo(a) isoform size variability. Definitions of variability differ substantially across studies (absolute vs. relative thresholds, biological coefficient of variation (CV), single repeat vs. multiple serial measures), which likely explains part of the heterogeneity in reported rates. Reclassification rates are highest in individuals with intermediate baseline Lp(a) concentrations.
3.1 Variability in general population cohorts
Large community-based studies suggest that most individuals remain within the same Lp(a)-based risk category over time, although clinically relevant variability is observed in a subset.
In the UK Biobank, a large prospective population-based cohort (n = 16,017), variability defined as a change ≥ 25 nmol/L was observed in 15% of participants over 4.2 years, without an association with incident coronary artery disease[5]. Similarly, in the Atherosclerosis Risk in Communities (ARIC) study (n = 4,734), 14.7% of individuals exhibited changes > 20 mg/dL between visits[6].
In a Korean multicenter observational study conducted across three tertiary hospitals (n = 5,305), median intra-individual variability was 26.3%, and approximately 20% of participants were classified as having high variability[7]. Notably, individuals within the intermediate “grey zone” demonstrated greater instability, with nearly half being reclassified during follow-up[7]. In this study, increased Lp(a) variability was associated with lower baseline Lp(a) concentrations and higher follow-up levels, as well as with lower body mass index, higher hemoglobin levels, elevated white blood cell and platelet counts, increased serum glucose, lower high-density lipoprotein cholesterol levels, and the use of antihypertensive medications.
In the STAR-Lp(a) observational cohort (n = 1,263), although most individuals remained within the same risk category, 6.2% experienced shifts in risk classification[8].
More recently, Awad et al. evaluated intra-individual variability within a large academic health system population and confirmed that the majority of individuals remained stable over time, with clinically meaningful reclassification occurring predominantly among those with intermediate baseline levels[9]. Similarly, Ghouse et al. demonstrated high within-person stability of Lp(a) concentrations in a population-based cohort, further supporting the concept that Lp(a) levels remain largely stable at the extremes of distribution[10].
3.2 Variability in clinical and high-risk populations
Greater intra-individual variability has been reported in selected high-risk and clinical populations.
In patients with diabetes, Hernandez et al., in a cohort of 70 individuals with serial measurements, reported an overall within-subject biological variability of 31.7%, which was inversely correlated with serum Lp(a) levels; intra-individual variability was 42.3%, 24.1%, and 23.7% among patients with diabetes with Lp(a) concentrations < 15 mg/dL, 15-30 mg/dL, and > 30 mg/dL, respectively[11]. Harb et al., analyzing data from the Nashville Biosciences database (n = 609), observed high variability in women and black individuals, underscoring potential demographic differences in Lp(a) variability and their implications for cardiovascular risk assessment. In this analysis, variability was defined using both an absolute change of at least 10 mg/dL (approximately equivalent to the 25 nmol/L threshold applied in the UK Biobank study) and a relative change of 25%. Although one might hypothesize greater variability in women due to hormonal influences, the authors did not observe differences between women younger or older than 51 years, the average age of menopause in the United States and Europe[12].
In the GAELp(a) cohort, in a clinically selected Argentine population of stable outpatients undergoing serial Lp(a) assessment in routine clinical practice (n = 61), we observed that 34.4% had ≥ 25% intra-individual variability over a follow-up period of at least four months[15]. Importantly, reclassification occurred predominantly among individuals with borderline or intermediate baseline values, while those at the extremes of distribution remained largely stable. As a real-world clinical cohort rather than a trial population, GAELp(a) provides complementary evidence that variability is not restricted to experimental settings but is also observable in routine clinical practice.
3.3 Variability in trial placebo arms
In placebo arms of Lp(a)-lowering trials, meaningful within-person fluctuations were also observed. In the phase 2 IONIS-APO(a)Rx and IONIS-APO(a)-LRx antisense oligonucleotide trials, 40.4% of placebo-treated participants demonstrated ≥ 25% variation at least once during follow-up[13]. Similarly, the OCEAN(a)-DOSE trial evaluating olpasiran found a maximum mean percentage difference between two visits of 47%, with 25% of patients showing variability ≥ 25% in either direction[14].
Collectively, these data suggest that variability may be amplified in selected clinical settings and may meaningfully influence risk classification, particularly in patients with borderline baseline levels.
3.4 Variability in pediatric and familial hypercholesterolemia cohorts
In pediatric populations, particularly among individuals with familial hypercholesterolemia, variability may appear more pronounced. In a large lipid clinic cohort, 68% of children demonstrated ≥ 20% variation between measurements[16]. These findings may reflect developmental, metabolic, or methodological factors specific to younger populations and should be interpreted cautiously when extrapolating to adult risk stratification.
3.5 Methodological considerations
The heterogeneity observed across studies underscores the absence of a standardized definition of “clinically significant variability”. Absolute versus relative thresholds, differences in follow-up duration, assay methodologies, and population characteristics all contribute to variability in reported rates. Although analytical variation must be considered, several studies have demonstrated fluctuations exceeding expected assay imprecision[4,14], supporting the presence of true biological variability.
While the reviewed studies provide valuable insights into Lp(a) variability, they also highlight the need for greater methodological standardization. The lack of a uniform definition of “clinically significant variability”, as summarized in Table 1, complicates direct comparisons across cohorts. Moreover, the optimal number and timing of serial measurements required to establish a reliable baseline Lp(a) concentration remain uncertain. Importantly, the clinical implications of defining variability using a relative threshold (e.g., ≥ 25%) may differ substantially according to baseline Lp(a) levels. A 25% change in an individual with low baseline concentrations may have minimal clinical impact, whereas the same relative fluctuation in someone near established risk thresholds may result in meaningful reclassification.
A key consideration in interpreting reported variability is the distinction between technical assay variation and true biological fluctuation. Different assay platforms may vary in isoform sensitivity, calibration methods, and reporting units, potentially influencing measured values. However, studies evaluating serial Lp(a) measurements, including those conducted in the same laboratory within standardized assay frameworks, consistently report changes exceeding expected analytical imprecision. Marcovina et al.[4] and Gaba et al.[14], for example, observed persistent variability within the same assay platforms, suggesting that biological influences, such as inflammatory states, hormonal transitions, and metabolic conditions, contribute meaningfully to intra-individual fluctuations. While continued improvements in assay standardization remain essential, current evidence indicates that biological variability must be acknowledged when interpreting serial Lp(a) measurements in both clinical practice and research settings.
3.6 Risk reclassification and clinical implications of variability
Beyond statistical measures of intra-individual variability, the clinically relevant question is whether such fluctuations meaningfully alter cardiovascular risk classification.
Although Trinder et al.[5] did not observe an association between short-term changes in Lp(a) concentrations and incident cardiovascular events, several studies have demonstrated non-negligible rates of risk reclassification, particularly among individuals with intermediate baseline levels.
In patients with diabetes, Hernandez et al.[11] reported a misclassification rate of approximately 20% when variability was accounted for, underscoring the potential implications of biological fluctuation in higher-risk populations.
In the GAELp(a) cohort[15], where all Lp(a) determinations were performed in a single laboratory using the same analytical assay, patients with ≥ 25% intra-individual variability were stratified according to baseline Lp(a) categories (< 70 mg/dL, 70-125 mg/dL, > 125 mg/dL). Among those initially in the lowest category, 40% transitioned to the intermediate category. Of those in the intermediate range, 16.6% moved to the lowest category, while 33.3% were reclassified to the highest category. Even within the highest-risk group, 20% transitioned to the intermediate category. These findings highlight that reclassification was predominantly observed among individuals in the borderline or intermediate range.
Similarly, Harb et al.[12] categorized participants as low (< 30 mg/dL), intermediate (30-50 mg/dL), and high (≥ 50 mg/dL). In the intermediate-risk group, 20% transitioned to low risk and 33% to high risk upon repeat testing, whereas only small proportions shifted from clearly low or clearly high categories. These findings reinforce the concept that the greatest instability occurs within the “grey zone”.
Consistent patterns have been observed in large population-based cohorts. In the ARIC study[6], 58.1% of individuals with Lp(a) values between 30 and 49 mg/dL at one visit had values exceeding 50 mg/dL at the subsequent visit, further illustrating the dynamic behavior of intermediate concentrations. In the placebo arm of the IONIS trials[13], 10.4% of participants with baseline Lp(a) levels > 75 nmol/L demonstrated levels < 75 nmol/L during follow-up, predominantly among those with borderline elevations.
Joo et al.[7] similarly reported that while Lp(a)-based risk classification remained largely stable in clearly low- and high-risk categories, approximately 51.9% of individuals within the intermediate “grey-zone” were reclassified during follow-up. In the STAR-Lp(a) cohort[8], 6.2% of individuals experienced category shifts (3.1% upward and 3.1% downward), despite overall stability at the extremes.
Collectively, these data indicate that intra-individual variability does not uniformly affect all risk strata. Rather, reclassification is concentrated among individuals with intermediate baseline concentrations, whereas those in clearly low or clearly elevated categories tend to remain stable. This pattern has important implications for the selective use of repeat Lp(a) testing.
3.7 Stability and long-term risk prediction
Although increasing evidence supports the presence of intra-individual variability, a single Lp(a) measurement has consistently been shown to predict long-term cardiovascular risk in large prospective cohorts. In a 30-year follow-up study of women, Ridker et al. demonstrated that baseline Lp(a) concentrations were independently associated with incident cardiovascular events, even after adjustment for inflammatory markers and traditional lipid parameters[17].
These findings reinforce that, despite measurable variability in selected individuals, Lp(a) remains a robust long-term risk marker. Consistent with this, major population-based studies and expert consensus statements have confirmed that Lp(a) levels remain largely stable at the extremes of distribution and that a single measurement provides meaningful long-term prognostic information[2,3,5,9,10].
Importantly, current clinical practice still faces challenges in achieving universal Lp(a) screening, and many individuals have not had even a single measurement performed. Recognition of intra-individual variability should not undermine the fundamental recommendation that Lp(a) be measured at least once in all adults[2,3]. Rather, repeat testing may be considered selectively in individuals with borderline or intermediate values, or when clinical circumstances change.
It is also important to acknowledge that currently used Lp(a) cut-offs are somewhat arbitrary and represent pragmatic thresholds rather than biologically discrete categories. Nonetheless, multiple studies consistently demonstrate that individuals classified as clearly “low” or clearly “high” tend to remain within the same risk category over time, whereas the greatest degree of reclassification occurs among those with intermediate concentrations, particularly in the range of approximately 30-50 mg/dL or its equivalent in nmol/L.
In this intermediate range, repeat testing may be particularly informative, especially if clinical conditions known to influence Lp(a) concentrations develop. Secondary factors such as hypothyroidism, chronic kidney disease, nephrotic syndrome, acute inflammatory states, and major hormonal transitions, including menopause, have been associated with changes in Lp(a) levels[18].
From a particle-based perspective, Lp(a) constitutes one component of the total circulating apolipoprotein B100 (apoB) pool. Each Lp(a) particle contains one molecule of apoB; however, its per-particle atherogenicity differs from non–Lp(a) apoB-containing lipoproteins due to the presence of apolipoprotein(a) and excess oxidized phospholipids. Recent mechanistic and epidemiologic analyses have emphasized that total apoB burden reflects the number of circulating atherogenic particles and represents a central determinant of ASCVD risk[19].
Accordingly, interpretation of Lp(a) concentrations, and evaluation of Lp(a)-lowering efficacy trials, should be considered within the broader framework of total apoB burden.
3.8 Clinical implications
At present, Lp(a)-lowering therapies are not yet widely available in routine practice, which limits the immediate therapeutic consequences of serial testing in unselected populations. However, repeat measurement may be clinically relevant in selected scenarios, particularly in secondary prevention, in individuals with borderline baseline concentrations, or when values approach treatment eligibility thresholds for emerging Lp(a)-targeted therapies. Until such therapies become broadly accessible, the priority remains ensuring universal one-time Lp(a) assessment, as recommended by current guidelines, with repeat testing reserved for patients in whom reclassification would meaningfully influence management[2,3,18].
A conceptual framework summarizing the potential implications of single versus repeated Lp(a) measurement for cardiovascular risk stratification is shown in Figure 1.
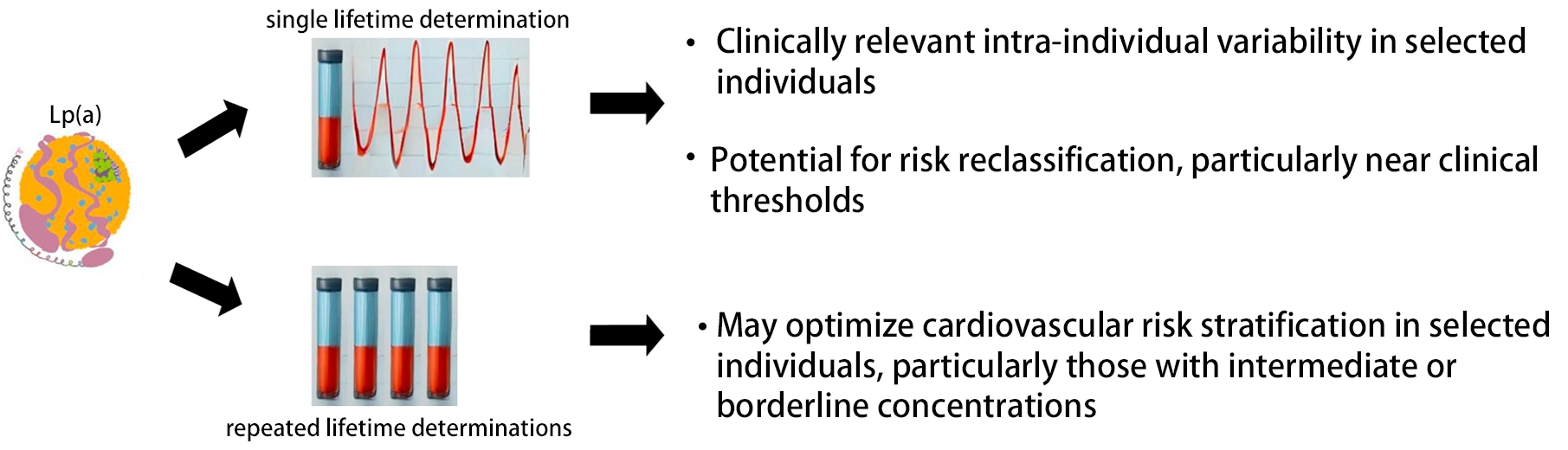
Figure 1. Conceptual illustration of potential variability in Lp(a) concentrations and its implications for cardiovascular risk assessment. While a single lifetime Lp(a) measurement is recommended by current guidelines, intra-individual variability has been reported in some longitudinal studies. In individuals with borderline concentrations, repeat testing may provide additional information that could influence clinical risk categorization in selected scenarios. Lp(a): lipoprotein(a).
4. Conclusion
Accumulating evidence suggests that Lp(a) concentrations are not uniformly stable over time. While most individuals remain within the same risk category, a meaningful subset exhibits clinically relevant intra-individual variability, particularly those with borderline or intermediate baseline levels, where reclassification is most likely. These observations do not undermine the value of a single lifetime Lp(a) measurement, which remains a robust predictor of long-term cardiovascular risk, but instead support a more nuanced strategy: universal one-time testing as the foundation of care, complemented by selective repeat measurement in individuals near clinical decision thresholds or in the presence of conditions known to influence Lp(a). As Lp(a)-targeted therapies advance, integrating an understanding of variability within the broader framework of total apoB burden will become increasingly important for accurate risk stratification and therapeutic interpretation.
Authors contribution
Pablo C, Laura S: Conceptualization, methodology, writing-review & editing.
Augusto LC: Conceptualization, formal analysis, writing-original draft, writing-review & editing.
Matta MG, Sebastian GZ: Formal analysis, writing-original draft, writing-review & editing.
Conflicts of interest
Corral Pablo serves as a Youth Editorial Board Member of the Advances in Lipoprotein(a) Research. The other authors declare no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
None.
Copyright
© The Author(s) 2026.
References
-
4. Marcovina SM, Gaur VP, Albers JJ. Biological variability of cholesterol, triglyceride, low- and high-density lipoprotein cholesterol, lipoprotein(a), and apolipoproteins A-I and B. Clin Chem. 1994;40(4):574-578.[PubMed]
-
14. Gaba P, Rosenson RS, López JAG, Watts GF, Leucker TM, Kuder JF, et al. Intraindividual variability in serial lipoprotein(a) concentrations among placebo-treated patients in the OCEAN(a)-DOSE trial. J Am Coll Cardiol. 2025;85(5):550-553.[DOI]
-
16. de Boer LM, Hof MH, Wiegman A, Stroobants AK, Kastelein JJP, Hutten BA. Lipoprotein(a) levels from childhood to adulthood: Data in nearly 3, 000 children who visited a pediatric lipid clinic. Atherosclerosis. 2022;349:227-232.[DOI]
-
18. Koschinsky ML, Bajaj A, Boffa MB, Dixon DL, Ferdinand KC, Gidding SS, et al. A focused update to the 2019 NLA scientific statement on use of lipoprotein(a) in clinical practice. J Clin Lipidol. 2024;18(3):e308-e319.[DOI]
-
19. Marston NA, Melloni GEM, Murphy SA, Morze J, Kamanu FK, Ellinor PT, et al. Per-particle cardiovascular risk of lipoprotein (a) vs non-Lp (a) apolipoprotein B-containing lipoproteins. J Am Coll Cardiol. 2024;83(3):470-472.[DOI]
Copyright
© The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Publisher’s Note
Science Exploration remains a neutral stance on jurisdictional claims in published
maps
and institutional affiliations. The views expressed in this article are solely those
of
the author(s) and do not reflect the opinions of the Editors or the publisher.
Share And Cite



Science Exploration Style
Augusto LC, Sebastian GZ, Matta MG, Laura S, Pablo C. Is it time to rethink the single lifetime measurement of Lipoprotein(a)? Adv Lipoprotein(a) Res. 2026;1:202601. https://doi.org/10.70401/alr.2026.0007
Tips
Copy completed.
Submit a Manuscript
Author Instructions
Cite this Article
Article Metrics
0
View
0
Download
Cited
Article Updates

- Abstract
- Keywords
- 1. Introduction
- 2. Biological and Analytical Considerations in Lp(a) Measurement
- 3. Intra-Individual Variability
- 4. Conclusion
- Authors contribution
- Conflicts of interest
- Ethical approval
- Consent to participate
- Consent for publication
- Availability of data and materials
- Funding
- References
- Copyright
Science Exploration Style
Augusto LC, Sebastian GZ, Matta MG, Laura S, Pablo C. Is it time to rethink the single lifetime measurement of Lipoprotein(a)? Adv Lipoprotein(a) Res. 2026;1:202601. https://doi.org/10.70401/alr.2026.0007
copy
Share Link
copy