Targeting ferroptosis pathways in cancer: Emerging molecular targets and therapeutic strategies
*Correspondence to:
Xiao-Feng Zhu, State Key Laboratory of Oncology in South China, Guangdong Provincial Clinical Research Center for Cancer, Sun Yat-sen University Cancer Center, Guangzhou 510060, Guangdong, China.
E-mail: zhuxfeng@mail.sysu.edu.cn
Ferroptosis Oxid Stress. 2026;2:202510. 10.70401/fos.2025.0011
Received: October 21, 2025Accepted: December 23, 2025Published: December 31, 2025
Abstract
Ferroptosis, a regulated form of cell death driven by iron-dependent lipid peroxidation, has emerged as a crucial tumor suppressive mechanism and a promising therapeutic target in oncology. This review synthesizes the current understanding of its core molecular machinery, encompassing lipid metabolism, iron homeostasis, and multi-layered cellular defense systems. We highlight the unique metabolic and genetic vulnerabilities that render specific cancer cell types intrinsically susceptible to ferroptosis. Furthermore, we discuss the dynamic propagation of ferroptotic signals within the tumor microenvironment and their complex immunomodulatory effects. Central to this review is a strategic framework for targeting ferroptosis, synthesizing recent advances in the development of specific ferroptosis inducers and evaluating their synergistic potential when combined with chemotherapy, radiotherapy, targeted therapy, and immunotherapy. By integrating mechanistic insight with translational perspectives, this work provides a systematic guide for rationally exploiting ferroptosis in cancer treatment.
Keywords
Ferroptosis, lipid peroxidation, iron metabolism, propagation, ferroptosis vulnerability, tumor treatment, potential targets
1. Introduction
Regulated cell death (RCD) plays crucial roles in maintaining tissue homeostasis and eliminating abnormal cells, with its dysregulation commonly observed in various diseases, including cancer[1]. Among the diverse forms of RCD, ferroptosis has gained increasing attention as a unique, iron-dependent, and lipid peroxidation-driven cell death modality. Ferroptosis is characterized by the accumulation of iron and the excessive peroxidation of polyunsaturated fatty acid-containing phospholipids (PUFA-PLs) on biological membranes, ultimately leading to membrane damage and cell demise[2,3].
In recent years, our understanding of the intricate molecular mechanisms regulating ferroptosis has advanced significantly. In this review, we summarize the latest insights into the molecular mechanisms of ferroptosis, encompassing its prerequisites (the critical roles of lipid peroxidation and iron metabolism) and its sophisticated defense systems. We then delve into a comprehensive analysis of the mechanistic basis of ferroptosis in tumor biology. Beyond intracellular events, recent evidence further indicates that ferroptosis can propagate both within and between cells, influencing the tumor microenvironment and neighboring cells.
The burgeoning interest in ferroptosis stems from its profound implications in tumor biology. Given the inherent or acquired ferroptosis vulnerabilities present in refractory cancers and within the tumor microenvironment, this process is increasingly recognized as a novel therapeutic target for cancer treatment[4,5]. We attempt to summarize potential targets that can be exploited to modulate the ferroptosis sensitivity of tumor cells and propose a conceptual framework for strategically targeting this vulnerability in cancer therapy. Furthermore, we highlight various current therapeutic strategies aimed at inducing ferroptosis in cancer cells and underscore their synergistic potential when combined with traditional cancer treatment modalities. By integrating existing knowledge, this review aims to provide researchers and clinicians with novel perspectives and insights on activating ferroptosis to combat cancer.
2. Molecular Mechanisms of Ferroptosis
2.1 Lipid peroxidation and iron metabolism
Lipid peroxidation is a process driven by iron-dependent oxidative damage to polyunsaturated fatty acids (PUFAs) in cellular membranes. Many oxidizable lipids are involved in the process, particularly PUFA-PLs, which are esterified and incorporated into membranes via Acyl-CoA synthetase long-chain family member 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3). When such peroxidation reactions exceed the buffering capacity of the ferroptosis defense system (Section 3), they induce the lethal accumulation of lipid peroxides. This ultimately results in plasma membrane rupture and cell death. Iron is an essential yet potentially toxic trace element, existing as Fe2+ and Fe3+ to enable redox reactions critical for cellular metabolism[6]. Iron homeostasis dynamically regulates ferroptosis by controlling the labile iron pool (LIP) through balanced absorption, storage, utilization, and excretion. Both lipid peroxidation and iron metabolism are prerequisites for ferroptosis (Figure 1).
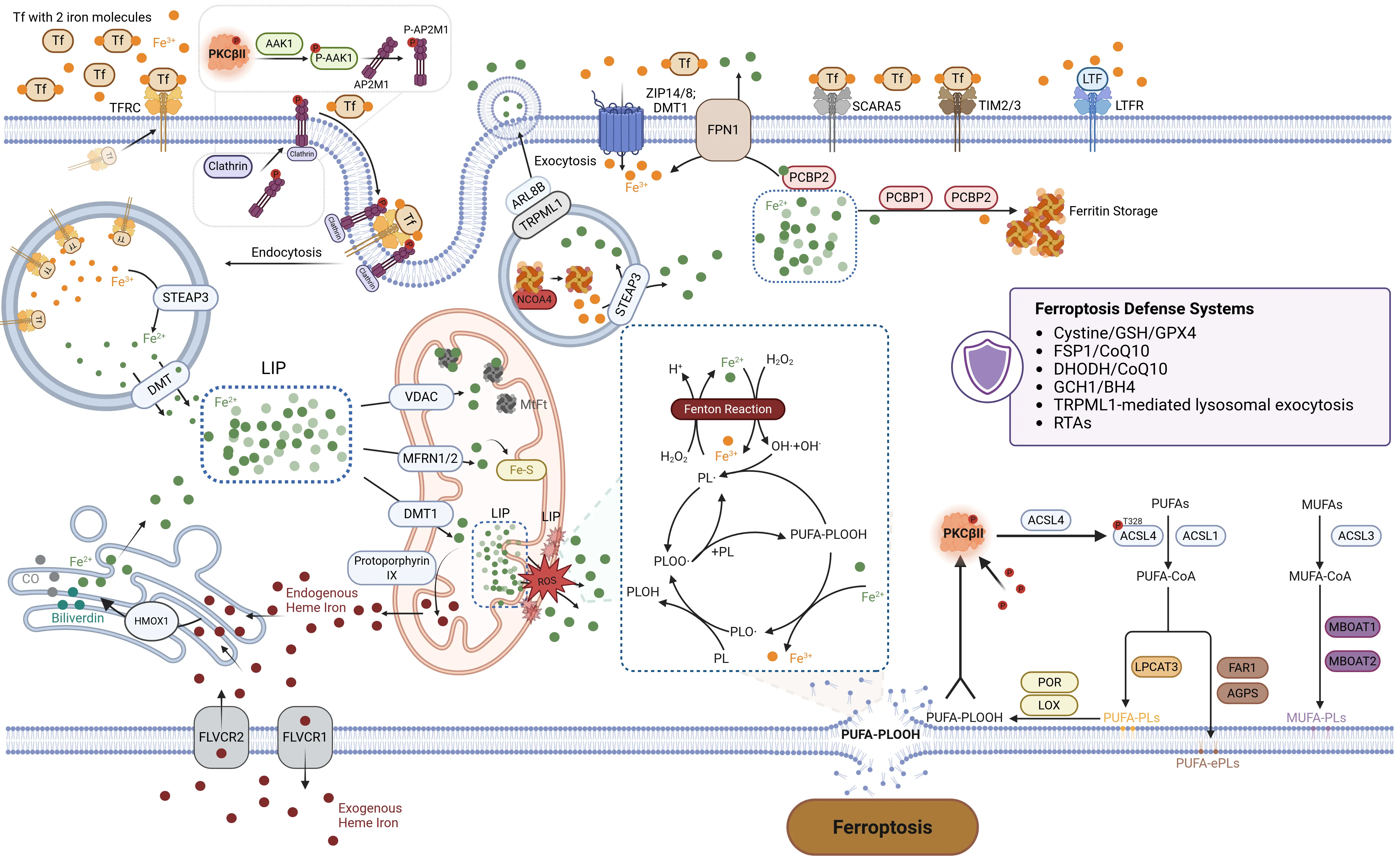
Figure 1. Lipid peroxidation and iron metabolism of ferroptosis. The hallmark of ferroptosis is the iron-dependent accumulation of ROS that drives the peroxidation of PUFAs, ultimately leading to plasma membrane rupture and cell death. Lipid peroxidation and iron metabolism are essential prerequisites for the initiation, progression, and execution of ferroptosis. The balance between iron-driven lipid peroxidation and ferroptosis defense systems determines ferroptosis occurrence. Created in BioRender.com. Tf: transferrin; TFRC: transferrin receptor; DMT: divalent metal transporter; DMT1: divalent metal transporter 1; AAK1: AP2-associated protein kinase 1; AP2M1: adaptor related protein complex 2 subunit mu 1; TRPML1: Transient Receptor Potential Mucolipin 1; ARL8B: adenosine 5′-diphosphate Ribosylation factor–like GTPase 8B;
2.1.1 Oxidizable lipids in ferroptosis
2.1.1.1 Fatty acids: the basis of lipid metabolism
Lipids are not only important energy storage molecules but also key biomolecules that regulate cellular structure, mediate intercellular communication, and coordinate genetic programs. Fatty acids (FAs), which primarily include saturated fatty acids (SFAs), monounsaturated fatty acids (MUFAs), and PUFAs, are fundamental components of cellular lipid metabolism. On one hand, mammals require dietary intake of essential PUFAs, namely α-linolenic acid (18:3 n-3) and linoleic acid (18:2 n-6). These exogenous fatty acids can enter cells through a series of transporters, such as the fatty acid transporter family, CD36, and plasma membrane fatty acid binding protein, to participate in metabolism[7]. On the other hand, SFAs and MUFAs can be synthesized de novo intracellularly. De novo synthesis of SFAs begins with acetyl-CoA in the cytoplasm. Acetyl-CoA carboxylase mediates the conversion of acetyl-CoA to malonyl-CoA, which is subsequently utilized by fatty acid synthase (FASN) to generate the long-chain SFA palmitic acid (PA, 16:0). PA can be further elongated to stearic acid (SA, 18:0) by elongases, while PA and SA can be desaturated to palmitoleic acid and oleic
2.1.1.2 PUFA-PLs: the “engine” of ferroptosis
PUFAs contain more than one double bond and are particularly prone to peroxidation due to the bisallylic groups (–CH=CH–CH2–CH=CH–). The C–H bonds at the bisallylic positions, such as C-7, C-10, and C-13 in arachidonate, are the weakest bonds in the molecules, and the hydrogen atoms at these positions are preferentially abstracted by a peroxyl radical to generate lipid radicals[9]. The composition and asymmetrical distribution of fatty acyl chains in individual phospholipids are modified after their de novo synthesis by a remodeling process known as Lands’ cycle, which results in the incorporation of PUFAs at the sn-2 position of PLs by undergoing a series of deacylation and reacylation reactions[10].
PUFAs must be activated and esterified into membrane phospholipids to function as the essential peroxidation substrates for ferroptosis. Ferroptosis is mechanistically driven by the peroxidation of specific phosphatidylethanolamine (PE)-containining PUFAs, particularly arachidonic acid (AA, C20:4 n-6) and adrenic acid (AdA, C22:4 n-6) species[11]. ACSL4 has a strong preference for AA and AdA, and catalyzes the formation of fatty acyl-CoA by inserting CoA into PUFAs[11,12]. PUFA-CoA is then incorporated into phospholipids primarily in the endoplasmic reticulum (ER) by LPCAT3, which preferentially targets acetylated AA[13]. The glycerol backbone of most phospholipids contains a SFAs chain at the sn1 position, while the sn2 position may harbor SFAs, MUFAs, or PUFAs[14].
ACSL4 is an essential component for ferroptosis execution. IFNγ induces the upregulation of ACSL4 expression via the JAK/STAT1 signaling pathway, leading to increased incorporation of PUFAs-CoA into membrane PLs and inducing immunogenic tumor ferroptosis[17]. A recent study found that ACSL4 enhances membrane fluidity and cellular invasiveness, and ACSL4/enoyl-CoA hydratase 1 co-inhibition suppress cancer metastasis[18]. In contrast, α6β4-mediated activation of Src and STAT3 suppresses expression of ACSL4, therefore protecting cells from ferroptosis[19]. Unlike ACSL4, ACSL3, another member of the ACSL family, exhibits expression patterns associated with ferroptosis resistance[20]. Mechanistically, ACSL3 contributes to intracellular lipid metabolism by facilitating the biogenesis and maturation of lipid droplets. Under LD-deficient conditions, ACSL3 predominantly localizes to the ER. Upon cellular uptake of exogenous fatty acids, ACSL3 orchestrates LD growth and maturation via ER-derived budding processes[21]. Crucially, ACSL3 is essential for activating MUFAs, and synergistically, exogenous MUFAs with ACSL3 activity confer cellular protection against ferroptosis[22].
Acylglycerol-3-phosphate O-acyltransferase 3 (AGPAT3) is also found to be a pro-ferroptotic hit. AGPAT3 exhibited acyltransferase activity with a preference for PUFA-CoA, such as arachidonoyl(20:4n-6)-CoA and docosahexanoyl(22:6n-3)-CoA, and it was shown to be involved in the formation of PUFA-containing PC[23,24]. But the AGPAT family does not appear to have a specific substrate preference toward fatty acyl-CoAs at the sn-2 position. Furthermore, AGPAT3 functions in the ER to synthesize polyunsaturated ether phospholipids (PUFA-ePLs) in the downstream of the peroxisomal pathway, thereby promoting the occurrence of ferroptosis[25]. In contrast, LPCAT1 enhances membrane phospholipid saturation through the Lands cycle, which reduces PUFA levels in membrane, protecting cells from phospholipid peroxidation-induced membrane damage, and ultimately inhibits ferroptosis[26].
2.1.1.3 MUFAs: the “brake” of ferroptosis
Contrary to PUFAs, MUFAs, activated by ACSL3, can displace PUFAs from PLs located at the plasma membrane, thus preventing the accumulation of lipid ROS in the plasma membranes, and effectively inhibit the process of iron-dependent oxidative cell death[22]. MUFAs, such as oleic acid, contain only one double bond, endowing them with potent antiperoxidative activity. The synthesis of MUFAs initiates with SCD1. By converting SFAs to MUFAs, SCD1 increases MUFA availability on one hand and reduces SFAs substrates available for the synthesis of ferroptosis-promoting PUFA-containing phospholipids on the other hand. Subsequently,
2.1.1.4 Ether phospholipids: another disruptor of membrane homeostasis
Ether PLs are a specialized class of glycerophospholipids. Unlike diacyl-containing PLs, these lipids are characterized by a unique ether linkage between the glycerol backbone and the acyl chain at the sn-1 position. Their sn-2 position, particularly in plasmalogens, tends to be esterified with long-chain PUFAs. This sn-2 position also frequently serves as a direct target for lipid peroxidation[25,59]. The unique ether linkage structure protects them from easy degradation but renders their oxidation-sensitive PUFAs “explosive”, that continuously trigger membrane damage[30].
Transmembrane protein 189 introduces vinyl-ether double bond into alkyl-ether lipids to generate plasmalogens, rendering the cells resistant to ferroptosis by inhibiting of fatty acyl CoA reductase (FAR1)[31]. Similarly, TMEM164 acts as an acyltransferase with a conserved cysteine (C123) to selectively transfer C20:4 acyl chains from PC to lyso-ePLs to produce PUFA-ePLs. Genetic deletion of TMEM164 resulted in robust decreases in C20:4 ePE lipids with minimal changes in C20:4 diacyl PEs, therefore protecting cells from ferroptosis[32].
2.1.1.5 Cholesterol and its intermediates: Sterol contributions to ferroptosis
Cholesterol and multiple intermediate metabolites generated during its biosynthetic pathway are closely associated with ferroptosis. On the one hand, cholesterol itself is susceptible to auto-oxidation, leading to the formation of cholesterol hydroperoxides, and its high abundance in the plasma membrane of eukaryotic cells renders it a potential substrate for lipid peroxidation[33]. On the other hand, aberrantly elevated cholesterol levels within the tumor microenvironment have been shown to disrupt lipid metabolic homeostasis in CD8+ T cells and induce endoplasmic reticulum stress, thereby impairing their antitumor function and altering cellular sensitivity to ferroptosis[34]. Cholesterol is generated from isoprenoid precursors produced by the mevalonate pathway, together with several essential intermediate metabolites, including isopentenyl pyrophosphate, squalene, and CoQ10. Using statins to inhibit HMG-CoA reductase, a rate limiting enzyme in the mevalonate pathway, might induce ferroptosis by inactivating glutathione peroxidase 4 (GPX4)[35]. In ALK+ anaplastic large cell lymphoma cell lines, squalene is elevated by the loss of squalene monooxygenase and prevents damage to membrane PUFAs under oxidative stress, to protect cancer cells from ferroptotic cell death[36].
2.1.2 The process of lipid peroxidation
Lipid peroxidation refers to the process by which oxidants, such as free radicals or non-radical substances, attack lipids containing carbon-carbon double bonds, particularly PUFAs.
2.1.2.1 Enzymatic lipid peroxidation
Enzymatic lipid peroxidation is mainly accomplished by lipoxygenases (LOXs), which are a family of non-heme iron-containing dioxygenases that directly catalyze the dioxygenation of PUFAs containing at least two isolated cis-double bonds. There are six isoforms that exist in humans, including ALOX5, ALOX12, ALOX12B, ALOX15, ALOX15B, and ALOXE3[39]. AA and LA are the most common substrates of LOXs. These enzymes oxidize PUFAs to their corresponding hydroperoxy derivatives, which serve as precursors for bioactive lipid mediators such as eicosanoids[40]. LOX‐catalyzed lipid hydroperoxides in cellular membranes were found to promote susceptibility to ferroptosis[41].
Inhibition or knockout of ALOX12 or ALOX15 can block the ferroptosis process in various pathological contexts, including neurodegenerative diseases and cancer[42]. ALOX12 and ALOX15 can not only synthesize 12-hydroperoxyeicosatetraenoic
2.1.2.2 Non-enzymatic lipid peroxidation
Non-enzymatic lipid peroxidation is mediated by the spontaneous generation of free radicals and catalyzed by redox active iron. Briefly, the lipid peroxidation mechanism consists of three steps: initiation, propagation, and termination. In the initial stage, free ferrous labile iron reacts with hydrogen peroxide (H2O2), known as the Fenton reaction, generating oxidative free radicals like hydroxyl radical (•OH) to initiate the oxidation of PUFAs. PUFAs lose one molecule of hydrogen at the carbon atom in the middle of the bisallylic groups and form phospholipid radical(L•). Once L• is formed, it can be further oxidized to lipid peroxide radical (LOO•) under the action of oxygen and other substances. LOO• can continue to seize a hydrogen atom on the central carbon atom of the diallyl group on the adjacent PUFAs, thereby forming a new L• and a lipid hydroperoxide (LOOH). Like H2O2, LOOH can undergo an
2.1.2.3 The products of lipid peroxidation
Lipid peroxidation produces a diverse array of oxidation byproducts such as reactive aldehydes within cells. LO• undergo
2.1.2.4 Other metabolites and associated enzymes modulating ferroptosis
Beyond lipid metabolism, alcohol- and aldehyde-related metabolites generated during lipid peroxidation, together with their associated detoxifying enzymes, have emerged as important modulators of ferroptosis in cancer. Enhanced expression of alcohol dehydrogenase exacerbates ethanol-evoked lipid peroxidation, endoplasmic reticulum stress, and ferroptotic signaling[56]. Genetic dysfunction of aldehyde dehydrogenase 2 (ALDH2), which detoxifies reactive aldehyde metabolites, leads to an “aldehyde storm” with glutathione depletion and heightened lipid peroxidation[57]. In cancer cells, ALDH2 deficiency increases ferroptosis susceptibility by promoting the accumulation of lipid-derived reactive aldehydes and weakening the antioxidant defense[58]. Purine metabolism has also been confirmed to be involved in the regulation of ferroptosis. Purine synthesis and its key metabolic enzymes in tumor cells can support the stability of mitochondria and membrane-related antioxidant systems by maintaining the supply of nucleotides and reducing equivalents, thereby indirectly suppressing lipid peroxidation to reduce ferroptosis susceptibility[59]. mTORC1-mediated purine catabolism was shown to regulate the intracellular purine pool in tumor-infiltrating CD8+ T cells, and suppression of this catabolic axis by DEPDC5 protected these T cells from ferroptosis[60].
2.1.3 Cellular sensing of lipid peroxides
Although lipid peroxidation is a well-established hallmark of ferroptosis, the precise mechanisms by which lipid peroxidation is sensed and triggers ferroptosis are not fully understood to date. In our study, we found that protein kinase C β II (PKCβII) acts as a sensor for lipid peroxidation during ferroptosis. Ferroptosis inducers promote a slight accumulation of lipid peroxides. PKCβII senses the initial lipid peroxides by directly interacting with and phosphorylating ACSL4 at Thr328, which leads to ACSL4 activation, triggering PUFA-containing lipid biosynthesis and promoting the abundant generation of lipid-peroxidation products. The
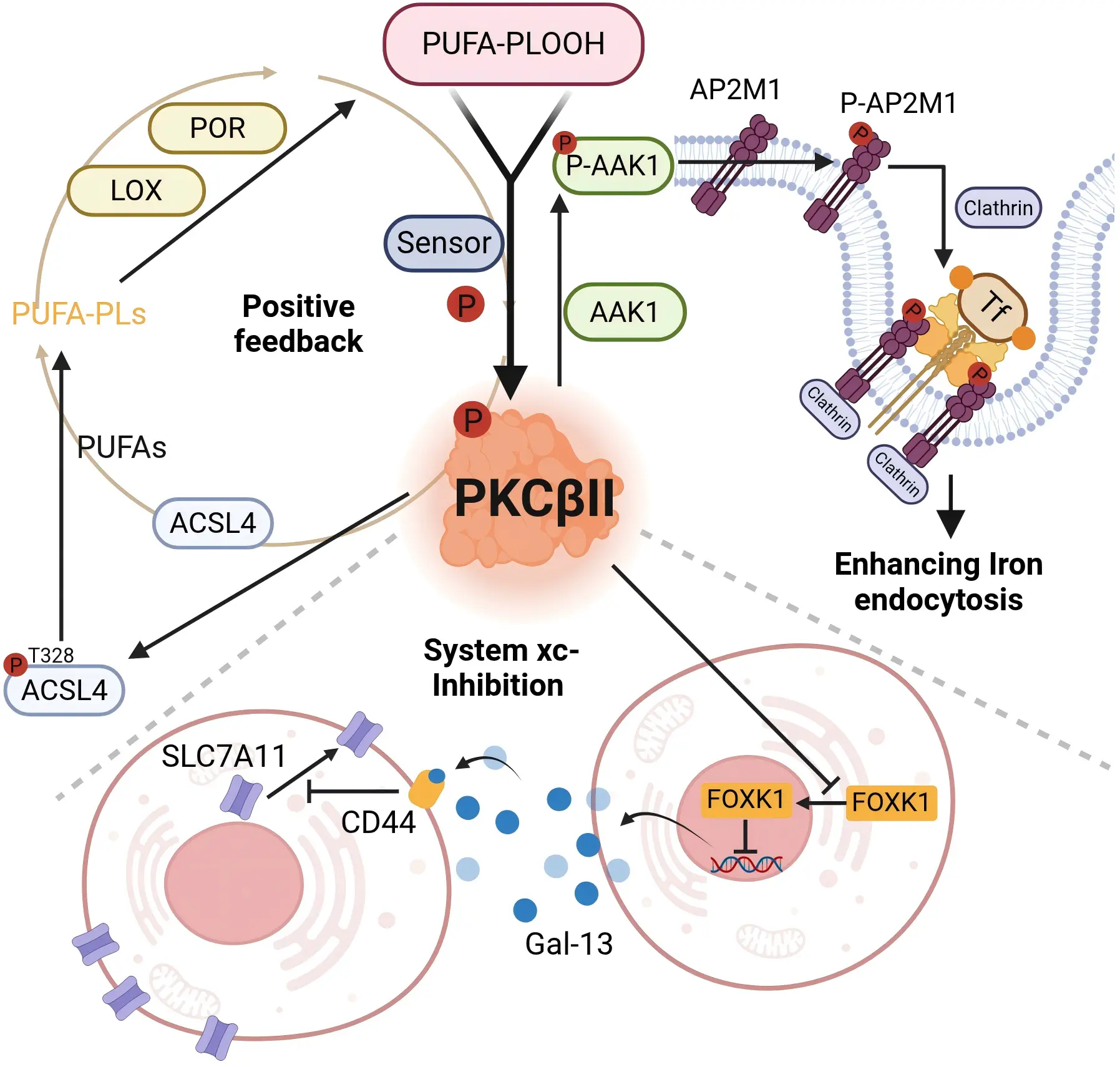
Figure 2. PKCβII as a pleiotropic lipid peroxide sensor. PKCβII functions not merely as an isolated kinase but as a pleiotropic signaling integrator, strategically positioned at the critical junctions of both lipid peroxidation and iron metabolism—two core pathways governing ferroptosis. This unique positioning enables PKCβII to coordinate death signals from distinct molecular origins, thereby playing an indispensable role in the initiation and propagation of ferroptosis. Created in BioRender.com. Tf: transferrin;
PKCβII acts as a central regulatory hub in ferroptosis. Beyond its role as a lipid peroxide sensor, it simultaneously compromises cellular defense systems (ferroptosis defense system) and amplifies offensive signals (iron metabolism), thereby precisely governing the execution of ferroptosis. Our findings demonstrate a dual mechanism: first, PKCβII impairs SLC7A11 plasma membrane localization by suppressing FOXK1 transcriptional activity, thereby disrupting intracellular glutathione synthesis and lipid peroxide clearance. Second, PKC-mediated AP2-associated protein kinase 1 (AAK1) activation promotes transferrin receptor endocytosis, consequently elevating cellular total iron and ferrous iron levels (see subsequent sections). (Figure 2).
2.1.4 Absorption, intake and export of iron
In the human diet, iron exists primarily in three forms: heme, ferritin, and ferric iron. Heme iron and non-heme iron, the latter encompassing ferritin and free iron, are absorbed through distinct physiological mechanisms[62,63].
2.1.4.1 Absorption and intake of iron
Non-heme iron is primarily absorbed as Fe2+ via divalent metal transporter 1 (DMT1) at the apical brush border of enterocytes in the small intestine. The absorption of dietary iron requires both the acidic environment provided by gastric acid and the enzymatic reduction of Fe3+ to Fe2+ by duodenal cytochrome B. After export from enterocytes, Fe2+ is oxidized to Fe3+ by enzymes such as hephaestin and ceruloplasmin, binds to transferrin (TF), and is transported to other tissues via blood circulation[64,65].
In addition to small intestinal epithelial cells, other cell types predominantly acquire iron through several pathways. For example, transferrin receptor 1 (TFR1/TFRC)-mediated iron endocytosis is considered a critical process in the progression of ferroptosis[66]. Saturated TF, which binds two Fe3+ ions in plasma, is recognized by TFR1/TFRC on target cells, forming an Fe3+-TF-TFR1/TFRC complex.
This complex is internalized via clathrin-mediated endocytosis, forming an intracellular vesicle termed a siderosome[67]. According to our study, this process is also closely associated with PKCβII. Specifically, PKCβII phosphorylates and activates AAK1, which in turn phosphorylates adaptor related protein complex 2 subunit mu 1 (AP2M1). This facilitates the recruitment of clathrin to mediate the endocytosis of TFR1, increasing the levels of both cellular total iron and ferrous iron and thereby promoting ferroptosis. We have identified the PKCβII-AAK1-AP2M1 pathway as a crucial mechanism for the regulation of cellular iron uptake during ferroptosis, which is correlated with the survival prognosis of breast cancer patients (unpublished) (Figure 1, Figure 2). Within the acidic siderosomal environment, Fe3+ dissociates from TF and is reduced to Fe2+ by the metalloreductase STEAP3 or lysosomal cytochrome B, then transported into the cytoplasmic LIP via DMT1. Subsequently, TFR1/TFRC is recycled to the cell membrane, while TF re-enters circulation[64,68]. Lactoferrin, structurally analogous to TF, serves as another critical pathway for iron uptake[69,70]. Upon binding to multiple Fe3+, the lactoferrin receptor or other receptors, form a complex that is subsequently internalized through endocytosis for cellular transport[71].
Cells also acquire iron through heme metabolism, supplementing sources beyond extracellular free iron. These sources include exogenous and recycled intracellular heme. The uptake of exogenous heme relies on membrane transporters such as the Feline Leukemia Virus Subgroup C Receptor and heme transporter HRG1/SLC48A1, coupled with cytoplasmic heme oxygenase activity[67]. FLVCR1 facilitates heme export, whereas FLVCR2 mediates heme import[72]. Both exogenous and endogenous heme are degraded in the ER, where heme oxygenase 1 (HMOX1) catalyzes heme breakdown into carbon monoxide, biliverdin, and Fe2+. HMOX1-mediated heme degradation releases Fe2+ into the LIP, fueling Fenton reaction-derived ROS. ROS accumulation exacerbates lipid peroxidation, driving ferroptosis[73]. Paradoxically, HMOX1 also suppresses ferroptosis by upregulating SLC7A11 and glutathione (GSH) levels[74,75]. Its dual role hinges on intracellular iron and ROS levels: elevated iron/ROS shifts HMOX1 from anti- to pro-ferroptotic activity[76]. Moreover, the expression of HMOX1 is also affected by erastin[74], polydopamine[77] or hypoxia[78].
2.1.4.2 Export of iron
Multiple pathways mediate extracellular iron efflux. Cytosolic poly(rC) binding protein 2 (PCBP2) acquires iron from DMT1(SLC11A2) and delivers it to ferroportin 1 (FPN1/SLC40A1) on the cell membrane[79-81]. As the primary iron efflux transporter, FPN1 oxidizes cytoplasmic Fe2+ to Fe3+ and releases it into the bloodstream, where it binds TF for systemic distribution. This oxidation process requires oxygen-dependent ferroxidases, including hephaestin, zyklopen, and ceruloplasmin, to enable Fe3+ loading onto TF[82]. Without ferroxidase activity, Fe2+ accumulates in the cytoplasm rather than being exported[83,84]. FPN1-mediated iron export is tightly regulated by hepcidin. During iron overload, hepcidin secretion increases significantly, suppressing FPN1 activity and enhancing iron import proteins such as TFRC/TFR1 and FTH1. This leads to greater intracellular iron retention and promotes ferroptosis[85-87].
Under conditions of intracellular iron overload, multivesicular bodies undergo fusion with the plasma membrane, resulting in the release of exosomes containing ferritin into the extracellular space. This mechanism serves to decrease cytoplasmic iron levels and reduce the potential for iron-induced cellular toxicity[88,89]. Conversely, when cellular iron export is impaired, the LIP accumulates within the cytoplasm. This accumulation promotes glutathione depletion, inhibits GPX4 activity, compromises the clearance of lipid peroxides, and ultimately leads to the induction of ferroptosis.
2.1.5 Iron metabolism within the mitochondria and cytoplasm
2.1.5.1 Iron metabolism within the mitochondria
The majority of free Fe2+ in the cytoplasm is transported into mitochondria via a coordinated mechanism involving DMT1[90], mitochondrial iron importers MFRN1 (SLC25A37) and MFRN2 (SLC25A28)[91], and the mitochondrial calcium uniporter[92]. In addition to these transporters, the mitochondrial membrane harbors the voltage-dependent anion channel (VDAC), a porin responsible for ion and metabolite exchange across the outer membrane. Three VDAC isoforms, VDAC1, VDAC2, and VDAC3, have been identified. The stabilization of VDAC1 to preserve mitochondrial homeostasis has been shown to inhibit ferroptosis in prostate and breast cancer cells. Conversely, erastin directly binds to VDAC2 and/or VDAC3, altering outer mitochondrial membrane permeability, which reduces NADH oxidation rates and promotes ferroptosis[93,94]. Despite these findings, the precise mechanisms underlying VDAC-mediated regulation of ferroptosis remain incompletely understood.
Within mitochondria, Fe2+ participates in critical physiological processes, including iron-sulfur (Fe-S) cluster and heme biosynthesis, as well as storage in mitochondrial ferritin (MtFt)[95].
Heme, a prosthetic group in proteins such as hemoglobin and cytochromes, serves a vital role in the electron transport chain (ETC) as a component of protein complexes embedded in the inner mitochondrial membrane. Heme biosynthesis proceeds through a conserved enzymatic pathway, wherein iron is incorporated into protoporphyrin IX to form heme, with key steps localized to the mitochondrial matrix and inner membrane[96]. Fe-S clusters act as essential cofactors for mitochondrial enzymes involved in the TCA cycle and ETC, as well as cellular processes like DNA repair. Mitochondrial Fe-S cluster assembly is mediated by specialized proteins, including FDX2, NFS1, frataxin, and ISCU. Any event that interferes with the biosynthesis of heme or Fe-S clusters can lead to mitochondrial iron overload[97-99]. For instance, doxorubicin primarily accumulates in mitochondria through integration into mtDNA, which disrupts the heme biosynthesis pathway and promotes mitochondrial iron accumulation in cardiomyocytes[100,101].
MtFt is an iron storage protein localized in mitochondria, sharing high structural and functional homology with the cytosolic ferritin H-chain. Unlike cytosolic ferritin, MtFt expression is not regulated by the canonical iron-responsive element/iron-regulatory protein system. Its primary role involves modulating ROS generation by controlling iron distribution between the cytosol and mitochondria, as well as mitochondrial iron bioavailability, thereby maintaining mitochondrial iron homeostasis[102]. Studies demonstrate that MtFt overexpression redistributes iron from cytosolic ferritin to mitochondria, thereby depleting the cytoplasmic labile iron pool. However, iron sequestered within MtFt shows reduced chelation accessibility, leading to mitochondrial iron accumulation. This pathological iron deposition disrupts mitochondrial dynamics, impairs respiratory chain complexes I and III, and ultimately enhances ROS production and ferroptosis through impaired electron transport[103,104].
In addition to regulating MtFt, Heme, and Fe-S synthesis, mitochondria also coordinate the maintenance of iron homeostasis and functional stability through other mechanisms such as mitophagy. Mitophagy, a selective degradation pathway for dysfunctional mitochondria, serves as a critical mitochondrial quality control system that preserves intracellular homeostasis[105]. During early stages of cytoplasmic LIP overload, excess free iron translocates into mitochondria, activating the PINK1/Parkin-mediated mitophagy pathway. This process sequesters free iron within mitophagosomes to restore cellular homeostasis[106,107]. However, mitochondrial free iron can participate in the Fenton reaction, oxidizing PUFAs on mitochondrial membranes and destabilizing their structure. The subsequent release of sequestered iron drives ROS and lipid peroxide overproduction, thereby promoting ferroptosis[108].
2.1.5.2 Iron metabolism within the cytoplasm
Ferritin, a critical iron storage protein primarily localized in the cytoplasm, consists of heavy (FTH1) and light (FTL) chain subunits. FTH1 uniquely contains an iron oxidase center essential for iron loading[109]. FTH1 oxidizes cytosolic LIP-associated Fe2+ to Fe3+, forming stable iron oxides/hydroxides that are stored non-toxically and mobilized as needed. This process is mediated by the cytosolic iron chaperones PCBP1 and PCBP2[110-112], alongside endophilin A2, encoded by the SH3GL1 gene[113]. PCBP1-deficient hepatocytes exhibit elevated unchelated iron and redox activity, triggering ferroptosis due to Fe2+ instability. Excess Fe2+ acts as both a substrate and a catalyst in the Fenton reaction with H2O2, generating cytotoxic ROS that induce oxidative damage to DNA, lipids, and biomolecules, culminating in cell death[110,114].
When cellular iron demand increases, NCOA4 binds to FTH1 and directs its transport to autophagosomes and subsequently to lysosomes for degradation, a process mediated by autophagy-related proteins (ATG) such as GABARAP and GABARAPL1. Within lysosomes, Fe3+ stored in ferritin is reduced to Fe2+ by STEAP3 and released into the cytoplasm as bioavailable iron. This selective degradation mechanism is termed ferritinophagy[115]. In addition to the classical ATG-dependent pathway, the NCOA4-FTH1 complex can also be tightly regulated for lysosomal degradation through the ESCRT machinery[116]. Perturbation of these regulatory pathways disrupts cytoplasmic iron homeostasis.
2.2 The propagation of ferroptosis
Ferroptosis is not a standalone process but rather a tightly regulated form of cell death that relies on coordinated interactions and precise signal transduction among specific organelles. Organelles such as lysosomes, the ER, and mitochondria play pivotal roles in modulating ferroptosis through essential biological functions, including the regulation of iron homeostasis, lipid metabolism, and redox balance. The intrinsic mechanisms within these organelles ultimately dictate whether a ferroptotic signal can be effectively initiated.
Of particular significance is the fact that these organelles not only function as central regulators of ferroptosis but also constitute key components in the signaling network that mediates this process. Upon initiation, the ferroptosis signal does not disseminate uniformly throughout the cell; instead, it progresses along a precisely orchestrated and spatially confined pathway, propagating in a directional fashion across distinct intracellular compartments and potentially extending beyond the cellular boundary to affect neighboring cells (Figure 3).
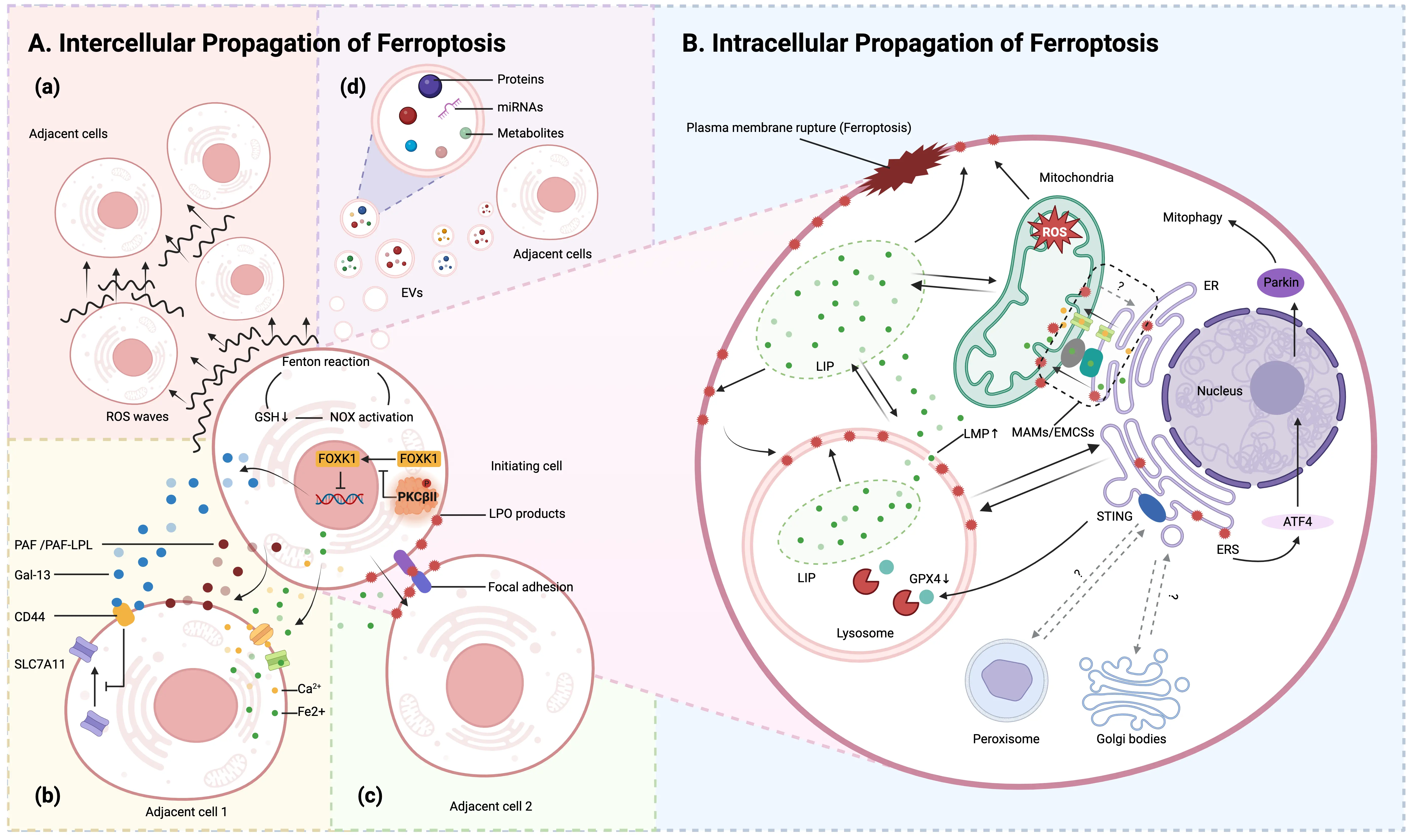
Figure 3. Intercellular and Intracellular Propagation of Ferroptosis. (A) Intercellular propagation of ferroptosis; (a) In the initiating cell, a tripartite positive feedback loop, comprising Fenton reaction induction, NOX activation, and inhibition of glutathione synthesis, is established, facilitating the dissemination of ferroptosis across the cell population via ROS waves; (b) The initiating cell promotes ferroptosis spread through the secretion of soluble mediators: PKCβII senses LPO and becomes activated, thereby inhibiting nuclear translocation of FOXK1 and suppressing transcription, resulting in upregulated expression and secretion of Gal13. Extracellular Gal13 binds to CD44, impairing plasma membrane localization of SLC7A11, reducing intracellular glutathione levels, and increasing cellular susceptibility to ferroptosis, thus promoting its propagation. Additionally, the initiating cell releases PAF/PAF-LPL, which integrates into the membranes of adjacent cells, disrupts membrane integrity, and initiate ferroptosis. Prior to plasma membrane rupture, the initiating cell also leaks low-molecular-weight species such as Fe2+ and Ca2+ into neighboring cells, contributing to ferroptotic induction; (c) Through α-catenin-mediated adherens junctions, the initiating cell facilitates the relay-like transfer of lipid peroxidation products to adjacent cells, a process potentiated by extracellular iron ions, thereby enabling coordinated propagation of ferroptosis; (d) Ferroptotic cells can communicate with surrounding cells by releasing EVs, which carry bioactive molecules such as miRNAs, proteins, or lipid metabolites; (B) Intracellular propagation of ferroptosis. The intracellular progression of ferroptosis is highly compartmentalized and fundamentally involves the directional spread of lipid peroxidation chain reactions along cellular membranes. Lysosomes serve as the initial trigger sites for this process. The ER and mitochondria function as central hubs for signal transmission and amplification. MAMs/EMCSs constitute dynamic physical and functional interfaces between the ER and mitochondria, playing a pivotal role in coordinating interorganellar crosstalk during ferroptosis regulation. Created in BioRender.com. PKCβII: protein kinase C β II; FOXK1: forkhead box protein K1; Gal13: galectin-13; SLC7A11: solute carrier family 7 member 11; PAF/PAF-LPL: platelet-activating factor or PAF-like phospholipids; ER: endoplasmic reticulum; MAMs/EMCSs: mitochondria-associated membranes or ER–mitochondria contact sites;
2.2.1 Intercellular propagation of ferroptosis
Upon crossing the single-cell boundary, ferroptotic death signals display complex population-level dynamics during inter-cellular propagation. Research has demonstrated that ferroptosis can propagate in a wave-like manner across cell populations, giving rise to a distinct spatiotemporal pattern of cell death, a phenomenon not observed in other forms of programmed cell death[117,118]. This long-range propagation mechanism fundamentally relies on reactive ROS-triggered waves. The underlying driving force of this propagation is a redox bistability switch induced by glutathione depletion. Moreover, a triple positive feedback loop comprising the Fenton reaction, NADPH oxidase activation, and inhibition of glutathione synthesis collectively enables cell populations to function as a “biological conductor” for sustained ROS propagation[119]. Within the local microenvironment, direct cell–cell contact provides an additional pathway for ferroptosis propagation: α-catenin-mediated adherens junctions, together with extracellular iron ions, facilitate the transmembrane relay transfer of lipid peroxidation products[120].
Although the ultimate outcome of ferroptosis is plasma membrane rupture, nanoscale pores can form prior to complete membrane disintegration. These pores facilitate the exchange of substances between the intracellular and extracellular environments, permitting the leakage of small molecules (e.g., Fe2+, Ca2+) while retaining large proteins. Released small molecules can infiltrate adjacent cells not yet undergoing ferroptosis, inducing intracellular calcium oscillations or elevated LIP levels. This process activates downstream signaling, initiates lipid peroxidation, and ultimately promotes ferroptosis in neighboring cells[118].
Notably, soluble factors critically mediate ferroptosis signal propagation within the tumor microenvironment. Our study reveals that during ferroptosis in initiating cells, PKCβII acts as a lipid peroxide sensor and is activated, triggering phosphorylation of FOXK1 at the Ser441 residue. This post-translational modification results in the cytoplasmic retention of FOXK1, preventing its nuclear translocation and abolishing its ability to repress Galectin-13 (Gal-13) transcription. Consequently, Gal-13 expression is upregulated, and the protein is robustly secreted into the extracellular milieu. Extracellular Gal-13 binds to CD44 on adjacent cells, impairing the plasma membrane localization of SLC7A11, leading to reduced intracellular glutathione synthesis. The depletion of this essential antioxidant compromises cellular redox homeostasis, thereby increasing susceptibility to ferroptosis and facilitating the coordinated propagation of ferroptotic cell death across the cell population[121]. Concurrently, platelet-activating factor (PAF) and PAF-like phospholipids accumulate during early ferroptosis and are actively secreted extracellularly, establishing a foundation for ferroptotic signal diffusion. These lipids embed into adjacent cell membranes, where their short acyl chains disrupt lipid bilayer organization, increasing membrane defects and water permeability. This cascade leads to ion imbalance (e.g., Ca2+ influx) and subsequent ferroptosis[122].
Ferroptotic cells can communicate with surrounding cells by releasing extracellular vesicles (EVs), which carry bioactive molecules such as miRNAs, proteins, or lipid metabolites. These molecules can be taken up by neighboring cells and induce ferroptosis or modulate ferroptosis sensitivity in recipient cells[123-126]. For instance, during the transition from acute kidney injury to chronic kidney disease, proximal tubular epithelial cells undergo ferroptosis under hypoxic conditions and release EVs containing specific miRNAs that trigger lipid peroxidation and ferroptosis in adjacent cells[123]. Another study showed that doxorubicin-stimulated breast cancer cells release small EVs delivering specific miRNAs to cardiomyocytes, thereby exacerbating ferroptosis sensitivity in the latter[124]. Furthermore, ferroptotic cells may release inflammatory mediators. For example, M1 macrophages under inflammatory conditions release mitochondrial contents via EVs, which are taken up by pancreatic β-cells and promote ferroptosis, contributing to β-cell dysfunction in acute pancreatitis[125]. Additionally, EVs derived from ferroptotic cells can carry iron ions or ferritin, which may alter cellular iron metabolism in recipient cells and promote ferroptosis[126]. This EV-mediated propagation of ferroptosis has also been explored for precise cancer therapy. For instance, an engineered EV platform has been developed to selectively target melanoma and induce YAP-dependent ferroptosis[127].
However, existing research continues to reveal critical gaps in our understanding of ferroptosis. Key unresolved issues include the potential selectivity of the propagation of ferroptosis; the impact of cellular heterogeneity, particularly differences in antioxidant capacity, on the dynamics and magnitude of ferroptotic propagation; and the involvement of or regulatory role of stromal cells in the tumor microenvironment in mediating ROS wave transmission or soluble factor diffusion (Figure 2, Figure 3A).
2.2.2 Intracellular propagation of ferroptosis
Ferroptosis follows a highly compartmentalized pattern during its intracellular propagation, characterized fundamentally by the directional diffusion of lipid peroxidation chain reactions across cellular membrane systems. Lysosomes function as the initial trigger hub. Lipid peroxidation within lysosomes increases lysosomal membrane permeability, resulting in iron release and subsequent widespread intracellular lipid peroxidation. This process not only facilitates ferroptosis but also amplifies lysosomal lipid peroxidation, thereby establishing a self-reinforcing positive feedback mechanism[128-131]. As lysosomal oxidative damage intensifies, free radical reactions transcend individual compartment boundaries and propagate to adjacent membrane structures, such as the
Subsequently, the ER functions as a central hub for the transmission and amplification of death signals. Accumulating evidence indicates that the ER not only serves as the primary site for the accumulation of peroxidized lipids, but also represents the initial subcellular compartment where lipid peroxidation markers exhibit significant accumulation, prior to their dissemination to the plasma membrane and other organelles[132]. ER stress has been shown to induce mitochondrial autophagy via ATF4-mediated transcriptional activation of Parkin, which subsequently reduces the generation of lipid peroxidation products and inhibits ferroptosis by limiting the accumulation of mitochondrial ROS[133]. Furthermore, the ER plays a central role in orchestrating an intricate inter-organelle communication network through its resident proteins, such as the stimulator of interferon genes and SLC39A7, thereby modulating the initiation and propagation of ferroptosis[134,135].
Mitochondria-associated ER membranes (MAMs) or ER-mitochondria contact sites (EMCSs), functioning as a dynamic interface between the ER and mitochondria, serve as a critical interorganellar platform for coordinating the regulation of ferroptosis[136]. Inhibiting the activity of the resident protein sigma-1 receptor on MAMs interferes with calcium transport between the ER and mitochondria, resulting in the accumulation of lipid peroxidation in both the ER and mitochondria[137,138]. Moreover, EMCSs operate as “hotspot amplifiers” for phospholipid peroxidation, enabling the targeted transfer of oxidative damage to mitochondria through specialized membrane interfaces, thereby ultimately initiating ferroptosis[139].
This sequential propagation cascade, from lysosomes → ER → MAMs/EMCSs → mitochondria, demonstrates that ferroptosis is governed by precise spatial programming at the subcellular level. However, several critical questions remain unresolved regarding the aforementioned propagation network: whether it encompasses multiple initiation points, whether its transmission follows a strictly unidirectional pattern, and whether feedback mechanisms are present that either enhance or suppress the propagation process (Figure 3B).
2.3 Ferroptosis defense systems
2.3.1 Cystine/GSH/GPX4 axis
Ferroptosis was originally described as a non-apoptotic form of cell death induced by the inhibition of cystine transport by cystine/system xc− to create a void in the antioxidant defenses of the cell, which can be rescued by Fer-1[2]. System xc– is a member of the heteromeric amino acid transporter family acting as a cystine importer and glutamate exporter, which consists of heterodimers of the catalytic subunit SLC7A11 and the partner subunit SLC3A2[140]. As the most abundant small molecule intracellular antioxidant, the synthesis of intracellular GSH depends on system xc−-mediated cystine uptake. GSH serves as the reductive co-substrate for the non-heme enzyme GPX4. Functioning as the primary enzyme catalyzing the reduction of PLOOHs in mammalian cells, GPX4 reduces PL-OOH to alcohols. This reaction is accompanied by the spontaneous oxidation of its selenocysteine residue and the oxidation of GSH to glutathione disulfide (GSSG), thereby establishing GPX4 as a central regulator of ferroptosis. Subsequently, GSSG is regenerated to GSH by glutathione reductase utilizing the two electrons provided by NADPH, completing the redox cycle[141-143]. In this process, cysteine serves as the rate-limiting substrate for GSH synthesis, being synthesized from serine and homocysteine mainly via the trans-sulfuration pathway. Furthermore, cysteine facilitates the protein synthesis of GPX4 by promoting selenium uptake and utilization.
2.3.2 Ferroptosis suppressor protein 1 (FSP1)/CoQ10 system
FSP1 (used to be known as apoptosis-inducing factor mitochondria-associated 2, AIFM2) confers protection against ferroptosis elicited by GPX4 deletion. Physiologically, FSP1 is a lipid droplet-associated protein highly enriched in brown adipose tissue and contains a flavoprotein NADH oxidoreductase domain[144]. The reduced form of ubiquinone (also known as coenzyme Q10, CoQ10), ubiquinol (CoQH2), traps lipid peroxyl radicals while FSP1 catalyzes the regeneration of ubiquinone using NAD(P)H. CoQH2 can additionally regenerate α-tocopherol and therefore enabling sustained radical scavenging[145]. This process requires the
2.3.3 Dihydroorotate dehydrogenase (quinone) (DHODH)/CoQ10 system
DHODH, a mitochondrial enzyme involved in pyrimidine biosynthesis, inhibits ferroptosis in the mitochondrial inner membrane through reducing CoQ to CoQH2[150]. DHODH shares a function analogous to FSP1, as both proteins reduce CoQ10 to prevent lipid peroxidation. The studies exploiting the effects of DHODH inhibitors, such as brequinar, revealed that these inhibitors surprisingly also suppressed FSP1. This indicates that the ferroptosis-sensitizing effect of DHODH inhibitors may be mediated through the inhibition of FSP1, rather than being directly attributable to DHODH itself[151].
2.3.4 GTP cyclohydrolase 1 (GCH1)/tetrahydrobiopterin (BH4) system
BH4 shows great radical-trapping antioxidant activity by increasing ubiquinol and protecting cells from ferroptosis[152]. Within the GCH1/BH4 pathway, GCH1 is recognized as the rate-limiting enzyme for BH4 synthesis, catalyzing the conversion of dihydrobiopterin (BH2) to BH4 upon NAD(P)H consumption. GCH1 and its metabolic derivatives BH4/BH2 drive lipid remodeling in GCH1-expressing cells alone or in synergy with α-tocopherol (vitamin E), suppressing ferroptosis through selective prevention of phospholipid depletion with two PUFA tails[152-154].
2.3.5 Transient Receptor Potential Mucolipin 1 (TRPML1)-mediated lysosomal exocytosis
Through a CRISPR-Cas9 activation screen to identify factors conferring resistance to ferroptosis, we discovered an enrichment of genes associated with lysosomal exocytosis. Among these, TRPML1 emerged as a potent suppressor of ferroptosis. Further investigation revealed that AKT directly phosphorylates TRPML1 at serine 343, which in turn inhibits K552 ubiquitination and subsequent proteasomal degradation of TRPML1. This post-translational stabilization promotes the interaction between TRPML1 and ribosylation factor–like GTPase 8B (ARL8B), triggering lysosomal exocytosis. This pathway suppresses ferroptosis via two primary mechanisms. On the one hand, it can expel intracellular Fe2+ to diminish Fenton reaction-driven lipid peroxidation. On the other hand, it will secrete acid sphingomyelinase to bolster plasma membrane repair capacity, thereby countering oxidative damage[155].
2. 3.6. Radical-trapping antioxidants (RTAs)
In addition to the enzymatic defense systems, a diverse array of endogenous RTAs suppresses ferroptosis by directly donating hydrogen atoms to lipid peroxyl radicals, thereby halting the propagation of phospholipid peroxidation. Beyond the previously discussed BH4, CoQ10, and vitamin K, other crucial RTAs include vitamin E, which quenches lipid peroxyl radicals within membranes, albeit with activity modulated by its interaction with phospholipid head groups[42,156]. Key intermediates of the cholesterol biosynthetic pathway, such as squalene[36] and 7-DHC[37,38], also function as potent lipophilic RTAs. Furthermore, hydropersulfides inhibit ferroptosis through radical scavenging and autocatalytic regeneration, a pathway dependent on cysteine availability but distinct from the canonical GPX4 axis[157,158]. Finally, several tryptophan metabolites, including serotonin, 3-hydroxyanthranilic acid, trans-3-indoleacrylic acid, and kynurenine, act as effective RTAs or modulate ferroptosis sensitivity through direct radical trapping or indirect metabolic regulation[159-161], highlighting the breadth of endogenous antioxidant mechanisms that can counteract lipid peroxidation.
3. Ferroptosis-Associated Targets in Cancer
3.1 Ferroptosis vulnerability in certain cancers
Inducing ferroptosis has emerged as a novel strategy for cancer therapy. Numerous studies have demonstrated that various oncoproteins, tumor suppressor genes, and oncogenic signal transduction pathways can regulate ferroptosis. To cope with survival pressures and meet their increasing nutritional demands, cancer cells remodel their metabolic networks and adjust the tumor microenvironment to maintain survival, proliferation, metastasis, and therapeutic resistance. Such adaptations often endow specific cancer types with new vulnerabilities, such as heightened sensitivity to ferroptosis. In-depth research to identify the types, characteristics, and biomarkers of these tumor cells is therefore crucial for treating these “refractory” tumors[162]. For instance, clear cell renal cell carcinoma (ccRCC), clear cell ovarian carcinoma, triple-negative breast cancer (TNBC), and diffuse large B-cell lymphoma, all exhibit inherently high sensitivity to ferroptosis. In this section, we systematically elaborate on several characteristic alterations in these tumor cells that confer significant susceptibility to ferroptosis (Figure 4).
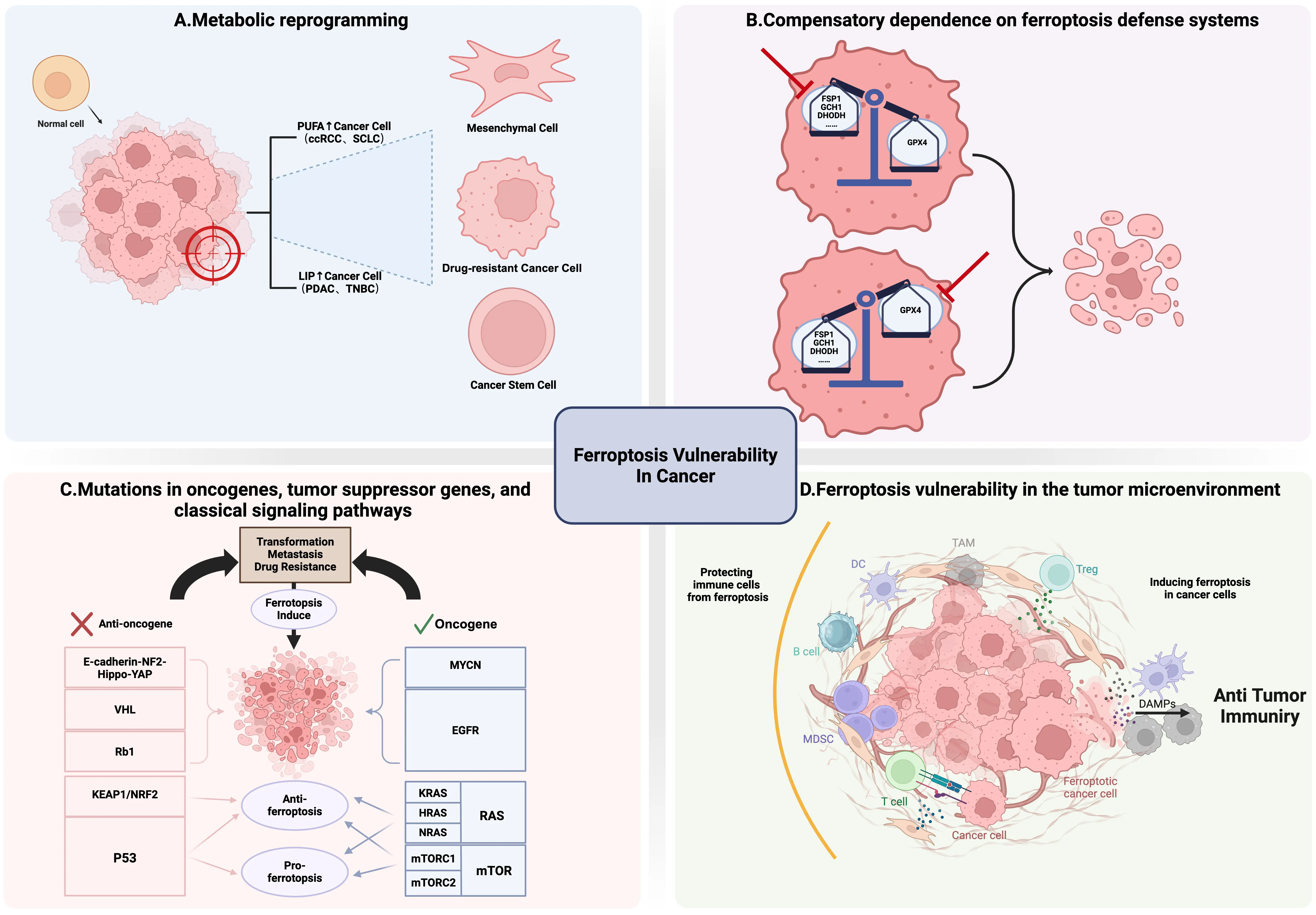
Figure 4. Characteristic Alterations in Cancer leading to Ferroptosis Vulnerability. (A) Metabolic reprogramming. Some cancer cells possess unique lipid metabolic features, such as high PUFA content or elevated levels of labile iron pools, rendering them more susceptible to ferroptosis; (B) Compensatory dependence on ferroptosis defense systems. Ferroptosis defense systems mainly include GPX4-dependent and GPX4-independent systems. Specific inhibition of the remaining major defense pathways that cancer cells depend on will trigger intense ferroptosis due to the lack of effective compensatory mechanisms; (C) Mutations in oncogenes, tumor suppressor genes, and classical signaling pathways. Mutations in oncogenes and anti-oncogenes often help tumor cells cope with survival pressure but may also lead to their vulnerability to ferroptosis, providing potential intervention windows for targeted therapy. Meanwhile, cancer cells may have differential responses to ferroptosis in the face of the same gene or pathway mutation; (D) Ferroptosis vulnerability in the tumor microenvironment. Ferroptosis and the TME, especially the immune system, interact in complex ways. On the one hand, mediators released by ferroptotic tumor cells can be transmitted to immune cells, triggering anti-tumor immune responses. On the other hand, mediators released by immune cells can regulate the sensitivity of tumor cells to ferroptosis, and the sensitivity of immune cells themselves to ferroptosis can also affect their functions. Created in BioRender.com. PUFA: polyunsaturated fatty acid; LIP: labile iron pool; ccRCC: clear cell renal cell carcinoma; SCLC: small cell lung cancer; PDAC: pancreatic ductal adenocarcinoma; TNBC: triple-negative breast cancer; GPX4: glutathione peroxidase 4; FSP1: ferroptosis suppressor protein 1; DHODH: dihydroorotate dehydrogenase (quinone); GCH1: GTP cyclohydrolase 1; DAMPs: damage-associated molecular patterns; MDSC: myeloid-derived suppressor cell; TAM: tumor-associated macrophage;
3.1.1 Metabolic reprogramming
Certain tumor cells often possess unique lipid metabolic features, such as high PUFA content or elevated levels of labile iron pools. Their overall metabolism is more active, and ROS load is higher, rendering them more susceptible to ferroptosis (Figure 4A).
As substrates for lipid peroxides, the abundance of PUFAs (e.g., AA or LA) is one of the key factors determining ferroptosis sensitivity. In ccRCC, HIF-2α has been found to selectively enrich polyunsaturated lipids by activating the expression of hypoxia-inducible lipid droplet-associated protein[163]. Meanwhile, this type of cell highly expresses alkylglycerone phosphate synthase, a key enzyme involved in the synthesis of PUFA-ePLs, leading to the accumulation of large amounts of PUFA-ePLs and subsequent increase in lipid peroxidation[25]. Chromophobe renal cell carcinoma primarily maintains extremely high intracellular levels of GSH and GSSG through the XC system. These tumor cells are highly sensitive to cystine uptake and catabolism. Compared with clear cell renal cell carcinoma, they are more sensitive to ferroptosis[164]. Metastatic tumors, particularly metastatic ovarian cancer cells, also exhibit higher PUFA levels and lower MUFA levels. Moreover, metastatic tumor cells can utilize high ACSL4 and abundant PUFAs to better promote cancer metastasis and in vivo survival[18].
The level of intracellular labile iron pools is another key factor determining ferroptosis sensitivity. Compared with normal cells, cancer cells rely strongly on iron for growth and are therefore more prone to ferroptosis occurrence. In pancreatic ductal adenocarcinoma (PDAC), the BACH1 gene regulates EMT and ferroptosis sensitivity through multiple mechanisms. Specifically, it inhibits the NRF2 pathway by binding to NRF2, promoting an increase in labile iron within tumor cells and leading to ferroptosis. BACH1 links EMT to ferroptosis, providing new insights for the treatment of these malignant tumors[165]. TNBC cells have long been found to be highly sensitive to ferroptosis, which stems from their unique metabolic features, including abundant PUFAs, elevated labile iron pool levels, and impairment of the GPX4-GSH defense system[12,166]. Notably, there is heterogeneity in ferroptosis-related metabolites and metabolic pathways among different TNBC subtypes. The luminal androgen receptor subtype of TNBC is particularly susceptible to ferroptosis induced by GPX4 inhibition[167,168].
Mesenchymal cancer cells typically refer to tumor cells that have undergone EMT and possess high invasiveness, drug resistance, and stem cell-like properties. However, these tumor cells often have more abundant phospholipids containing PUFAs and higher levels of labile iron pools. Increased synthesis of phospholipids containing PUFAs is closely associated with the high expression of zinc finger E-box binding homeobox 1 (ZEB1). ZEB1 is a transcription factor associated with EMT and a driver of lipid biosynthesis. It promotes the accumulation of PUFA-PLs by directly activating peroxisome proliferator-activated receptor γ, thereby rendering cells highly dependent on GPX4[169]. In mesenchymal cancer cells, enhanced CD44-mediated chondroitin sulfate-dependent iron endocytosis and iron accumulation caused by decreased FPN expression also contribute to ferroptosis susceptibility[170,171]. Similarly, mesenchymal gastric cancer shows elevated levels of amino acids and adenylates due to high expression of ELOVL5 and FADS1 involved in PUFA synthesis[172].
Drug-tolerant persister (DTP) cells with similar mesenchymal characteristics also exhibit inherent sensitivity to ferroptosis and high dependence on GPX4 due to their unique and analogous metabolic features[173]. These cells develop tolerance to traditional drugs through metabolic reprogramming and epigenetic modifications, therefore entering a state of suspended proliferation with enhanced survival capacity. Therapy-resistant dedifferentiated subpopulations in melanoma cells exhibit their susceptibility to ferroptosis, which may be due to the accumulation of PUFAs and decreased glutathione levels[174]. Gefitinib-resistant lung cancer cells can also increase susceptibility to ferroptosis by downregulating apoptosis-associated tyrosine kinase, which promotes endosomal recycling and iron accumulation[175]. Additionally, most DTP cells exhibit upregulation of CD44 to promote the uptake of redox-active iron to meet metabolic demands. The newly developed Fento-1 can effectively target these CD44-highly expressing DTP cells to induce ferroptosis[130]. Through epigenetic regulation, DTP cells, on one hand, resist external survival pressure, and on the other hand, indirectly induce susceptibility to ferroptosis. For example, erlotinib-resistant DTP cells can inhibit glutaminolysis and retain their mesenchymal characteristics through histone lysine demethylase 5A-mediated inhibition of mitochondrial pyruvate carrier 1, resulting in enhanced susceptibility to ferroptosis both in vitro and in vivo[176].
Cancer stem cells (CSCs) are a group of cells with self-renewal ability, differentiation potential, and unlimited proliferative capacity, capable of generating heterogeneous tumor cells. CSCs lead to the failure of traditional radiotherapy and chemotherapy as well as tumor metastasis and recurrence. Dysregulated iron homeostasis is a hallmark feature of CSCs. To meet the demands of their tumor microenvironment and the energy requirements for self-renewal, CSCs typically exhibit stronger iron absorption capacity[177,178]. Numerous studies have found a significant association between CSCs of different cancer types and abnormal iron metabolism. For example, glioblastoma and breast cancer CSCs show significantly increased expression of transferrin receptor 1 and ferritin[179,180]. This alteration in iron metabolism is crucial for maintaining CSC stemness and increases their susceptibility to ferroptosis inducers, providing a new approach for targeted elimination. Studies have also found that enhancing stem cell characteristics of tumor cells can increase their sensitivity to ferroptosis, which may be due to their lack of antioxidant capacity and abundant iron storage. However, although CSCs have increased levels of labile iron pools due to massive iron uptake, they still employ various ways to resist potentially induced ferroptosis, including maintaining low ROS levels[181], inhibiting ACSL4[182], or enhancing SLC7A11 expression[183] and so on. Future studies need to further investigate issues such as heterogeneity of CSCs and the complexity of the tumor microenvironment to better induce ferroptosis in CSCs in vivo.
In summary, these changes in metabolic reprogramming render the aforementioned tumor cells susceptible to ferroptosis and provide promising therapeutic strategies for addressing these challenging malignant tumors.
3.1.2 Compensatory dependence on ferroptosis defense systems
As mentioned earlier, ferroptosis defense systems mainly include GPX4-dependent and GPX4-independent systems. Tumor cells usually possess multiple ferroptosis defenses, but when one of them (such as FSP1) is congenitally deficient, has low expression, or is functionally impaired, cells are forced to over-rely on the remaining major pathways (such as GPX4) to maintain survival. This “putting all eggs in one basket” state creates fatal metabolic vulnerabilities in certain tumor cells. Specific inhibition of the remaining major defense pathways that cancer cells depend on will trigger intense ferroptosis due to the lack of effective compensatory mechanisms (Figure 4B).
In some cancer cells, if GPX4 expression is low or partially inactive, cells are highly dependent on GPX4-independent defense systems to resist ferroptosis. For example, studies have found that inhibition of DHODH in NCI-H226 cells (with low GPX4 expression) results in a more significant lipid peroxidation and ferroptosis compared to cells with normal GPX4 expression. Additionally, the use of DHODH inhibitors can better suppress the growth and proliferation of GPX4-low-expressing xenograft tumors[150]. Similarly, tumor cells with low GPX4 expression also exhibit high sensitivity to FSP1 inhibitors[145]. Germinal center B-cell-like diffuse large B-cell lymphoma with low GPX4 expression is more sensitive to ferroptosis induced by dimethyl fumarate, which primarily functions by rapidly depleting GSH by succination[184].
It is worth noting that there may be a therapeutic time window for acquired inactivation of a ferroptosis defense pathway. For example, after continuous inhibition of FSP1, tumor cells can develop resistance to ferroptosis through cholesterol synthesis[37]. If GPX4-independent defense systems are lowly expressed or inactive, cells will also be highly dependent on the GPX4 system to resist ferroptosis. For example, studies have found that certain tumor cells with low FSP1 expression or FSP1 knockout are more sensitive to ferroptosis induced by GPX4 inhibitors (such as RSL3). Additionally, GPX4 knockout significantly inhibits tumor growth more in FSP1-knockout xenografts than in FSP1 wild-type tumors, and this reduction in tumor growth is attributed to more intense ferroptosis[145,146].
GCH1 is an important component of the ferroptosis defense system. Cancer cells with low GCH1 expression are also more sensitive to GPX4 inhibitors (such as RSL3) and FSP1 inhibitors (such as iFSP1)[153]. Therefore, a deep understanding of which ferroptosis defense pathways are active, absent, or weakened in specific tumor cells is crucial for selecting the most effective single-agent therapy and designing rational combination therapy strategies.
3.1.3 Mutations in oncogenes, tumor suppressor genes, and classical signaling pathways
A growing body of research supports the view that ferroptosis acts as a natural anti-tumor mechanism. In general, the interaction of multiple tumor suppressor genes with ferroptosis (such as KEAP1[185], BAP1[186]) can exert their tumor-suppressive effects, while activation of specific genes can endow cancer cells with the ability to evade ferroptosis (such as OTUB1[187], CKB[188]) to promote tumor growth. Specific targeted activation of these tumor suppressor genes or inhibition of oncogene expression is an important
Mutations in tumor suppressor genes are often associated with malignant transformation, metastasis, drug resistance, poor prognosis, and ferroptosis resistance in cancer. However, cancer cells with some tumor suppressor gene mutations are also more sensitive to ferroptosis, exposing unusual weaknesses. The E-cadherin-NF2-Hippo-YAP pathway is one of the most notable examples. E-cadherin (ECAD), mainly responsible for maintaining cell polarity, acts as an upstream regulator of the Hippo pathway. Its mediated cell-cell interactions activate the Hippo tumor suppressor signaling pathway in an NF2-dependent manner, inhibiting the transcriptional activity of YAP and transcriptional co-activator with PDZ-binding motif (TAZ), thereby regulating tumor cell proliferation[189]. Antagonizing this signaling pathway allows YAP to promote ferroptosis by upregulating various ferroptosis regulators, including ACSL4 and TFRC[190]. Meanwhile, loss-of-function mutations in ECAD, NF2, and Hippo pathway components LATS1/2, or overexpression of YAP, often occur in cancers such as gastric cancer, breast cancer, and mesothelioma. These mutations make it possible to treat them with ferroptosis inducers. ccRCC, the most common type of kidney cancer, is inherently highly sensitive to ferroptosis. ccRCC is mainly caused by the deletion of the tumor suppressor gene VHL, which in turn leads to the selective accumulation of PUFAs and impairment of fatty acid degradation, making tumor cells particularly sensitive to cystine deprivation or GPX4 inhibition[191,192]. The RB1 tumor suppressor gene inactivation is common in various therapy-resistant cancers. Studies have found that RB1 deletion and E2F activation can sensitize tumor cells to ferroptosis by upregulating ACSL4 levels and the content of phospholipids containing PUFAs. The use of the GPX4 inhibitor JKE-1674 can efficiently block the growth and metastasis of
The transcription factor p53 has long been a star molecule in the field of cancer treatment. p53 is one of the most frequently mutated genes in cancer, and its mutation predicts poor prognosis in various cancer types. p53 itself can regulate the key substrates of lipid peroxidation, executors of lipid peroxidation, and anti-ferroptosis systems, which have been elaborated in relevant reviews[194-196]. Interestingly, p53 activation plays a dual role in ferroptosis: p53 can sensitize tumor cells to ferroptosis by promoting ROS production, catalyzing lipid peroxidation, and inhibiting SLC7A11 transcription, while also resisting ferroptosis through transactivation of iPLA2β and restriction of membrane-bound dipeptidyl peptidase 4 (DPP4). This implies that p53 regulation of ferroptosis is a complex environment-dependent and pathway-specific network. As a guardian of cell fate and responder to stress, it regulates the delicate balance between death and survival. Similarly, p53 mutations in cancer also have dual effects on ferroptosis regulation. On the one hand, p53 mutations can promote certain antioxidant stress proteins and ferroptosis defense systems
Beyond the classical tumor suppressor genes, mutations in the core cellular antioxidant stress-response pathway, the KEAP1/NRF2/CUL3 axis, are also intimately linked to tumor ferroptosis sensitivity and therapy resistance. Under physiological conditions, KEAP1 acts as a sensor for oxidative stress. By binding to the CUL3 ubiquitin ligase complex, it facilitates the continuous ubiquitination and degradation of the transcription factor NRF2, thereby restricting its activity[202]. However, in various cancers
Activation of some proto-oncogenes in tumor cells may also expose sensitivity to ferroptosis by enhancing their dependence on the GPX4 pathway. For example, MYCN-amplified neuroblastomas are highly invasive. Due to the regulation of cell proliferation, iron uptake, and other cellular activities by MYCN, high iron content in these tumor cells enhances their dependence on cysteine/cystine. These cells mainly rely on trans-sulfuration-mediated cysteine supply and LRP8-mediated organic selenium absorption to resist inherent ferroptosis susceptibility[211]. Triple therapy with co-inhibition of GPX4, cystine uptake, and trans-sulfuration pathways has been confirmed to effectively inhibit the growth of MYCN-amplified neuroblastomas[212,213]. The epidermal growth factor receptor (EGFR) is a key driver gene in solid tumors such as lung cancer. The EGFR signaling pathway significantly upregulates SLC7A11 expression by activating key downstream effectors including PI3K/AKT and RAS/MAPK[214]. This creates an abnormal dependency of mutant EGFR cells on extracellular cystine uptake to support their rapid proliferation and antioxidant stress response, concurrently rendering them highly susceptible to SLC7A11 inhibitors and cystine deprivation-induced ferroptosis[214].
The RAS family is one of the most frequently mutated oncogenes in human cancers and the first oncogene found to be closely associated with ferroptosis[141]. RAS mainly includes three mutants, including KRAS, HRAS, and NRAS, and different RAS mutation types have different effects on ferroptosis[215]. For example, HRAS-mutant cells show increased clearance of lysophospholipids and resistance to SCD1 inhibition[216]. The KRAS gene plays a pivotal role in regulating diverse intracellular signaling pathways and cellular activities, including the activation of the MAPK and PI3K-AKT pathways[141]. Tumors harboring KRAS mutations manifest a distinct metabolic profile. On one hand, their accelerated metabolism generates substantial ferroptosis pressure; on the other hand, this is counterbalanced by the activation of robust compensatory defense networks[141]. KRAS mutations drive the Warburg effect and glutaminolysis, leading to persistently elevated intracellular ROS levels, thereby creating a pro-oxidative stress microenvironment[217]. Concurrently, KRAS-mutant cells have evolved multi-layered, redundant ferroptosis defense systems. KRAS mutations can evade ferroptosis through ACSL3-dependent FASN-mediated increased synthesis of saturated and monounsaturated fatty acids in the Lands cycle[218]. Downstream of FASN, ACSL3 is crucial for tumorigenesis in KRAS-mutant lung cancer[219]. In addition, KRAS-mutant cells can also exhibit increased GSH synthesis and FSP1 expression driven by NRF2 and MAPK signaling[217,220], and can protect tumor cells from ferroptosis by upregulating the NRF2/SLC7A1 axis[217]. Combined use of SLC7A11 inhibitors or FSP1 inhibitors with traditional treatments is an effective strategy for treating KRAS-mutant tumors. KRAS can also resist ferroptosis by affecting lactate levels in the tumor microenvironment. Specifically, KRAS mutation-induced lactate promotes acylation of the glutamate-cysteine ligase (GCL) modifier subunit mediated by acetyl-CoA acyltransferase 2 (ACAT2). This modification reduces GCL enzyme activity, inhibits GSH synthesis through targeting ACAT2, and thereby overcomes tumor ferroptosis resistance[221]. Recent studies have also found that KRAS-mutant pancreatic cancer can develop ferroptosis resistance by inducing ALOX15B depalmitoylation and membrane translocation to promote its degradation[222].
Mammalian target of rapamycin (mTOR), a serine/threonine protein kinase, is involved in the PI3K/AKT/mTOR signaling pathway and plays an important role in regulating cell growth, proliferation, and death[223]. mTOR can form two functional complexes: mTORC1 and mTORC2, and their effects on ferroptosis depend not only on the complex type but also on the tissue microenvironment. mTORC2 can promote ferroptosis in tumor cells by phosphorylating serine 26 of SLC7A11 to inhibit the activity of the system xc−[224]. Studies using a mouse model of acute lymphocytic choriomeningitis virus (LCMV) infection found that mTORC2 can also activate the AKT/GSK3β/NRF2 signaling axis to resist ferroptosis in memory CD4+ T cells and prolong the survival of antigen-specific memory T cells[225]. In most tumor cells, mTORC1 inhibits ferroptosis mainly through three ways: inhibiting autophagy, promoting GPX4 protein synthesis, and promoting MUFA synthesis. mTORC1 can phosphorylate and inhibit ULK1 of ATG complexes, thereby effectively suppressing autophagy. For example, knockdown of glucose-6-phosphate dehydrogenase can promote redox homeostasis, trigger phosphorylation activation of AMPK, and reduce mTORC1 activity, thereby effectively activating the autophagy pathway[226]. Under high cell density, the Hippo pathway is activated, leading to phosphorylation activation of mTORC1, which in turn inhibits
Mutations in oncogenes, tumor suppressor genes, and certain signaling pathways often help tumor cells cope with survival pressure but may also lead to their vulnerability to ferroptosis, providing potential intervention windows for targeted therapy of specific tumor types. Meanwhile, the above studies also imply that cancer cells may have differential responses to ferroptosis in the face of the same gene or pathway mutation. Future studies need to further clarify the impact of cell-specific differences on ferroptosis sensitivity.
3.1.4 Ferroptosis vulnerability in the tumor microenvironment
The tumor microenvironment (TME) is a dynamic and complex system composed of tumor cells, immune cells, stromal cells, vascular systems, and other non-cellular components. In the TME, these components coexist and interact with each other, playing an important role in tumor growth and proliferation[229]. Ferroptosis and the TME, especially the immune system, interact in complex ways. On the one hand, mediators released by dying tumor cells can be transmitted to immune cells, triggering anti-tumor immune responses. On the other hand, mediators released by immune cells can regulate the sensitivity of tumor cells to ferroptosis in the TME, and the sensitivity of immune cells themselves to ferroptosis can also affect their functions, thereby reshaping tumor development in the TME (Figure 4D).
3.1.4.1 Immunogenicity of ferroptosis
ICD refers to the process by which cells activate the immune system during death, triggering a specific immune response in the body against antigens related to dying cells[230]. Exploring methods to induce immunogenic cell death is a new approach to cancer treatment, because the treatment itself kills tumor cells, and the immunostimulatory signals released by dead cells can further activate the adaptive immune system, thereby synergistically inhibiting tumors. Due to the lack of observation of specific, effective, and orderly release of danger signals to strongly activate adaptive immune responses in early studies, ferroptosis was generally regarded as a low-immunogenic death mode. In recent years, increasing evidence has shown that ferroptosis does not necessarily lead to immune silence, especially in the TME. Efimova et al. first demonstrated that ferroptosis is immunogenic in vitro and in vivo, and found that early ferroptotic cells with intact membrane structures can promote the phenotypic maturation of bone
It should be noted that the immunogenicity of ferroptosis is highly environment-dependent, affected by multiple factors such as cell type, ferroptosis induction method, and tumor microenvironment. For example, studies have shown that ferroptotic cancer cells can impair the maturation of dendritic cells, and their derived damage-associated molecular patterns may have negative effects on adaptive immune responses[241]. Moreover, ferroptosis induced by RSL3 or erastin cannot expose calreticulin before complete rupture of the plasma membrane, indicating that certain specific ferroptosis induction methods and environments affect the immunogenicity of ferroptosis[241]. Ferroptosis of various immune cells is also associated with immune dysfunction in mouse cancer models[242]. Meanwhile, ferroptosis-induced immune responses focus more on strong innate immune activation, and more exploration is needed on how to initiate effective adaptive immunity. Future studies need to further understand and optimize how to utilize the immunostimulatory potential of ferroptosis and overcome its potential immunosuppressive effects.
3.1.4.2 Ferroptotic immune cells and immune cells on cancer cells ferroptosis
In addition to specifically inducing ferroptosis in tumor cells to generate adaptive immune responses, protecting immune cells from ferroptosis is also crucial. Ferroptosis inducers show good tumor-killing effects in in vitro experiments but are relatively less effective in animal models, except in immunodeficient mice. Ferroptosis of immune cells affects not only their survival but also their functions. Their sensitivity to ferroptosis varies significantly with the environment.
T cells, especially CD8+ T cells, are the main executors of anti-tumor immunity in the TME. IFNγ derived from immune-activated CD8+ T cells can combine with AA in the TME to form endogenous ferroptosis inducers and synergistically induce ferroptosis in tumor cell through ACSL4[17]. Activated CD8+ T cells can also deplete intracellular GSH in tumor cells by inhibiting SLC7A11 mediated by TNFγ to promote ferroptosis[243]. CD8+ T cells appear to be more susceptible to GPX4 inhibitor-induced ferroptosis than ordinary cancer
The characteristic high dependence on GPX4 also extends to CD4+ T cells. Follicular helper T cells (TFH) are a specialized subset of CD4+ T cells, mainly supporting germinal center responses. Deletion of GPX4 in T cells selectively eliminates TFH numbers and germinal center responses in mouse models, suggesting that TFH are highly dependent on GPX4[249]. Regulatory T (Treg) cells are a subset with immunosuppressive activity and an important barrier to prevent autoimmunity. Treg cells have inherently high GPX4 expression, so GPX4-deficient Treg cells show high sensitivity to ferroptosis and promote the production of IL-1β and the response of helper T cell 17, thereby enhancing the body’s anti-tumor immune capacity[250]. Notably, T cell populations are more dependent on GPX4 than SLC7A11. Deletion of SLC7A11 in mouse models does not affect the growth and anti-tumor immunity of T cells in vivo and even enhances the efficacy of ICIs[251]. This may be due to the low and non-essential expression of SLC7A11 in T cells[252]. Recent studies on the lipid profile of immune system cells have shown differences in PUFA-PLs abundance, which explain and support the susceptibility of T cells to ferroptosis[253].
B cells are another pillar of anti-tumor immunity and exhibit significant heterogeneity. Different B cells subset exert complex and interrelated effects in the TME. Innate-like B cells (including B1 cells and marginal zone B cells) exhibit more active lipid metabolic characteristics compared with follicular B2 cells and are more susceptible to GPX4 inhibition-induced ferroptosis. These studies highlight the importance of ferroptosis in B cell survival and function, but the impact of ferroptosis on tumor-infiltrating B cells and B cell-mediated tumor immunity remains to be further studied[254]. B cells and T cells have very similar PUFA-PLs compositions, but B cells are more resistant to ferroptosis than T cells, possibly due to differences in peroxidized PLs components or undiscovered protective mechanisms[253]. However, studies on the impact of ferroptosis on tumor-infiltrating B cells and B cell-mediated humoral immunity are relatively few and still need further exploration.
The innate immune system also plays a role in regulating ferroptosis in tumor cells, and its own functions are affected by both ferroptosis and secretions from related cells. For example, tumor-associated macrophages (TAMs) can differentiate into immunostimulatory M1 phenotypes and immunosuppressive M2 phenotypes. During treatment, we tend to eliminate M2 phenotype cells or promote their conversion to M1 phenotype. M1 TAMs show significantly enhanced resistance to ferroptosis compared with M2 TAMs. On the one hand, M1 TAMs have higher expression of nitric oxide synthase and production of nitric oxide radicals (NO·), which can neutralize lipid radicals produced by 15-LOX[255]. On the other hand, M1 TAMs have increased ferritin heavy chain 1 expression and decreased iron exporter expression, which may lead to relatively stable intracellular iron content and resistance to ferroptosis[256]. Immunosuppressive M2 TAMs are relatively sensitive to ferroptosis. Moreover, multiple studies have confirmed that applying ferroptosis stimulation can reprogram M2 TAMs into M1 phenotype, thereby further inhibiting tumor progression[257-259]. Targeting ferroptosis in TAMs is expected to specifically eliminate M2 TAMs that promote tumor growth or promote the production of M1 TAMs, providing a promising strategy for immunotherapy. Pathologically activated neutrophils, also known as myeloid-derived suppressor cells (PMN-MDSCs), are another relevant immune cell type in the TME, with significant immunosuppressive capacity and heterogeneity. PMN-MDSCs are prone to ferroptosis and can even undergo spontaneous ferroptosis in the TME. Meanwhile, accumulated peroxidized lipids and release of PEG2 affect the activity of tumor-infiltrating CD8+ T cells and tumor-associated macrophages, making them more immunosuppressive[260]. Studies based on mouse hepatocyte models have also found that GPX4 deficiency leads to increased CXCL10-dependent CD8+ T cell infiltration, elevated PD-L1 expression, and promotes PMN-MDSC infiltration into the TME. Here, PMN-MDSCs do not show susceptibility to ferroptosis, and the strategy of combining GPX4 blockade with blocking MDSC recruitment significantly enhances the efficacy of immune checkpoint therapy and improves the survival of
These studies collectively reveal the complex interrelationship between ferroptosis and both adaptive and innate immunity in the TME. To improve cancer immunotherapy strategies, it is crucial to further clarify the heterogeneity of these immune cells in the TME and their connection with ferroptosis.
3.1.4.3 Metabolic competition in the TME shapes ferroptosis sensitivity
Despite its diverse immune effects, ferroptosis in the TME does not operate in isolation, and increasing evidence suggests that its immune consequences are not only dictated by cell-intrinsic programs, but are further shaped by metabolic interactions and nutrient competition among tumor and immune cells[262]. For instance, tumor cells exhibiting high SLC7A11 expression can preferentially take up cystine, depriving tumor-infiltrating CD8+ T cells of this essential amino acid. This cystine starvation disrupts glutathione synthesis and promotes CD36-mediated lipid uptake and lipid peroxidation, triggering ferroptosis and dysfunction in T cells[263]. Similarly, competition for glutamine selectively compromises immune cells with limited metabolic flexibility. Glutamine deprivation disrupts TCA cycle anaplerosis and NADPH production in T cells and dendritic cells, weakening lipid peroxide detoxification and lowering the ferroptotic threshold, whereas tumor cells maintain redox homeostasis through metabolic
3.2 Application of FINs in tumor treatment
In the process of tumorigenesis and development, tumor cells adopt multiple mechanisms to evade death. Currently, widely used clinical methods such as chemotherapy and radiotherapy can eliminate tumors by inducing various forms of regulated cell death in tumor cells, including necrosis, apoptosis, and ferroptosis. However, these approaches lack tumor specificity and are associated with multiple toxic side effects. As mentioned above, ferroptosis is a weakness of certain cancer types. Inducing ferroptosis or combining ferroptosis with traditional treatments provides new opportunities for cancer treatment. Identifying susceptible tumor cell types, delineating the specific mechanisms through which ferroptosis is induced, determining whether these mechanisms concurrently trigger death in non-malignant cells, and developing targeted therapies against key ferroptosis regulators are crucial for achieving precision medicine in oncology. The following section highlights ferroptosis-associated targets in cancer, alongside the developments in the application of diverse FINs in combination with chemotherapy, radiotherapy, immunotherapy, and other therapeutic modalities.
Our toolkit for manipulating ferroptosis in humans remains limited. Although no drugs specifically designed to induce ferroptosis have yet been approved for cancer therapy, several existing marketed drugs, such as metformin[268], lovastatin[269], and
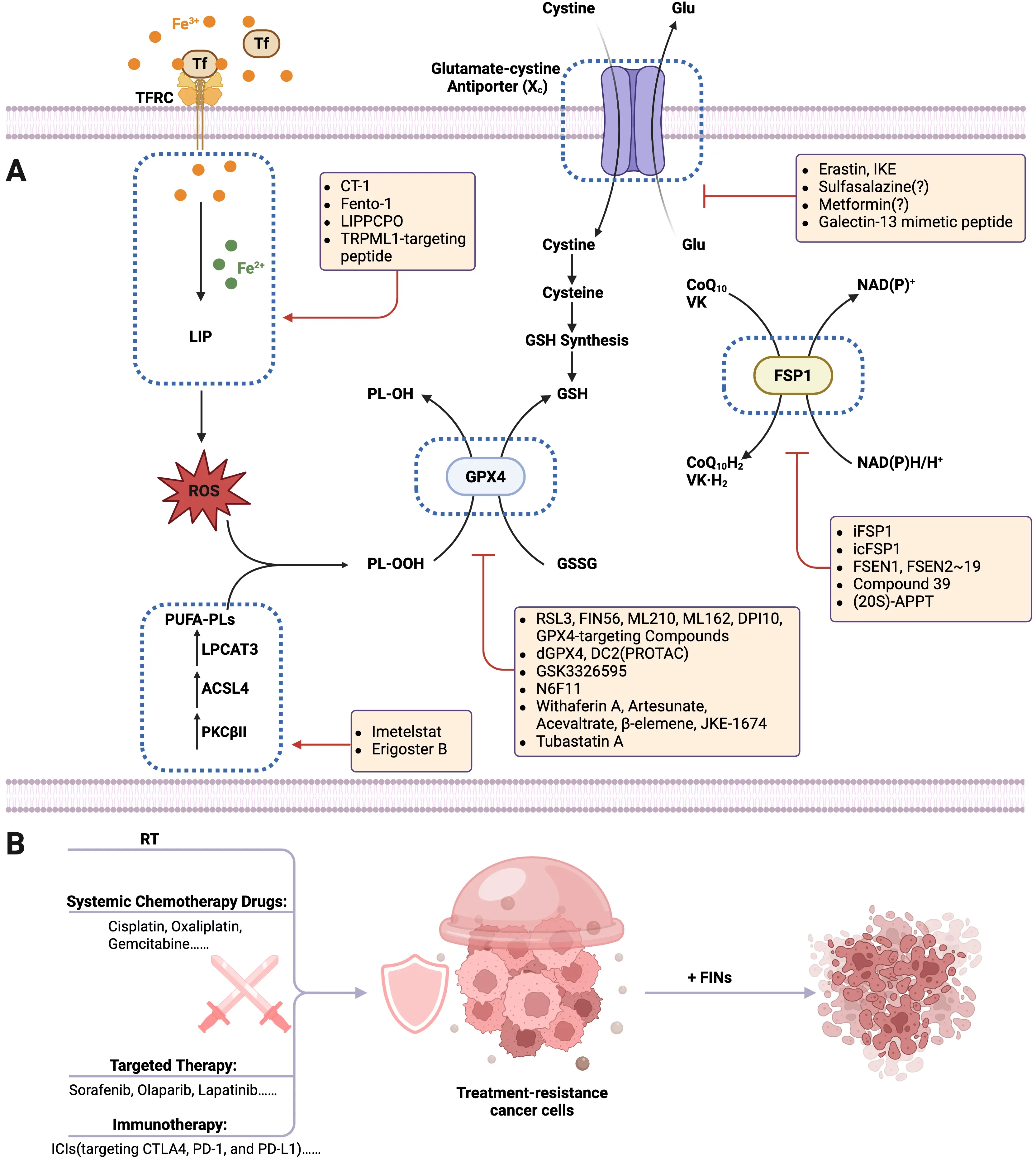
Figure 5. Application of FINs in tumor treatment. (A) Preclinical or clinical antitumor agents that target key points within the ferroptotic cascade. A growing number of FINs are being discovered and developed, actively advancing toward clinical application globally. Common targets include, for example, GPX4, FSP1, system xc–, as well as targets lipid peroxidation and iron metabolism in cancer cells; (B) Application of FINs in combination with common tumor treatments. Cancer cells exhibiting resistance to conventional radiotherapy, systemic chemotherapy, targeted therapy, or immunotherapy demonstrate marked sensitivity and vulnerability to FINs under specific conditions. The strategic combination of FINs with these established therapeutic modalities may provide an effective approach for managing certain refractory tumors. Created in BioRender.com. IKE: imidazolone erastin; (20S)-APPT: (20S)-protopanaxatriol; LIPPCPO: phosphatidylcholine peroxide-decorating liposomes; IR: ionizing radiation;
3.2.1 Anti-tumor drugs targeting GPX4
GPX4 is the core of ferroptosis regulation. Pharmacological inhibition of GPX4 induces uncontrolled, massive lipid peroxidation and triggers intense ferroptosis in both in vitro and in vivo environments. In in vitro experiments, we often use small molecule inducers such as RSL3, FIN56, ML210, ML162, and DPI10 to inhibit the classical GPX4-regulated ferroptosis defense pathway, thereby promoting ferroptosis[271]. Studies have attempted to develop GPX4 degraders, dGPX4 and DC2, based on PROTAC. The former covalently links ML162 with pomalidomide, while the latter uses the active ingredient of ML210, both showing stronger ferroptosis-inducing ability and anti-cancer activity[272,273]. The PRMT5 antagonist GSK3326595 can also promote ubiquitination and degradation of GPX4 by preventing its methylation, and is currently in phase I and II clinical trials[274]. It is worth noting that systemic inhibition of GPX4 will inevitably damage other healthy cells. Therefore, through drug screening, the first cell type-specific GPX4 degrader, N6F11, was discovered. It acts as a direct activator of E3 ubiquitin ligase tripartite motif containing 25, inducing tumor-specific GPX4 ubiquitination and proteasomal degradation. In a mouse model of pancreatic cancer, N6F11 enhanced the effect of immune checkpoint inhibitors and improved the viability of cytotoxic CD8+ T cells[275]. Withaferin A is a natural ferroptosis inducer that both promotes the release of Fe2+ from heme and inhibits GPX4 activity, and is even more effective than traditional etoposide or cisplatin in the treatment of neuroblastoma[276]. Artesunate is a water-soluble derivative of artemisinin, and its newly synthesized compound A7 can target and degrade GPX4 to induce ferroptosis in cancer cells, showing strong tumor-suppressive activity in the T24 xenograft model[277]. The recently screened natural product acevaltrate, as the first small molecule inhibitor targeting both iron chaperones PCBP1/2 and GPX4, has strong ferroptosis-inducing potential and shows good efficacy in cancer cells and cancer organoid
3.2.2 Anti-tumor drugs targeting system xc–
Inhibiting system xc– is an effective mechanism to induce ferroptosis. Various structurally different inhibitors of this antiporter, such as erastin, imidazolone erastin (IKE), and sulfasalazine, are widely used for their efficient induction of ferroptosis. In addition, cystine deficiency depletes the extracellular substrate of system xc– and induces ferroptosis. In a B-cell lymphoma xenograft model, IKE can induce cellular ferroptosis and inhibit tumor growth[284]. Targeting cystine and system xc– has also shown good tumor-suppressive effects in pancreatic ductal carcinoma and melanoma models[285,286]. Sulfasalazine, a commonly used oral anti-inflammatory drug, can also suppress the growth of lymphoma and other cancer cells in preclinical models by inhibiting system xc– to induce ferroptosis. However, the specific mechanisms underlying its antitumor effects remain poorly understood. The oral hypoglycemic drug metformin is also proven to reduce the protein stability of SLC7A11, thereby increasing intracellular Fe2+ content and lipid ROS levels, inducing ferroptosis[268].These inhibitors have off-target effects during treatment[287], and safer drugs for in vivo use are still under development. Based on the finding that Galectin-13 is a key factor mediating the spread of cellular ferroptosis, we developed a Galectin-13 mimetic peptide, which binds to CD44 to reduce SLC7A11 membrane localization, showing high efficiency and low toxicity in both in in vitro and in vivo tumor models. In particular, cancer stem cells were more vulnerable to the combination of Galectin-13 mimetic peptide and FINs[121] (Figure 5A).
3.2.3 Anti-tumor drugs targeting the FSP1-CoQ10-NAD(P)H axis
FSP1 is a highly promising anti-tumor target, but similar to system xc–, targeting FSP1 alone is usually insufficient to induce intense ferroptosis. However, FSP1 is highly expressed in various cancer cells and is closely related to GPX4 inhibitor resistance. Cancer cells lacking GPX4 can be effectively killed by the FSP1-specific inhibitor iFSP1, while in cancer cells with normal GPX4, iFSP1 has a synergistic effect with RSL3[145]. Unlike iFSP1, which competitively inhibits FSP1 enzymatic activity, icFSP1, identified through small molecule library screening, triggers FSP1 phase separation, significantly sensitizing GPX4 inhibitors and reducing off-target
3.2.4 Anti-tumor drugs targeting peroxides, iron, or polyunsaturated fatty acids
Inducing lipid peroxidation by targeting peroxides, iron, or PUFA overload is another main mechanism to induce ferroptosis. The telomerase inhibitor imetelstat promotes the formation of PUFA-PLs, leading to increased lipid peroxidation and oxidative stress levels, providing a new perspective for the treatment of acute myeloid leukemia[291]. Similarly, the DECR1 inhibitor Erigoster B can enhance phosphatidylcholine metabolism, accompanied by activation of PLA2G12A and massive accumulation of AA, showing good therapeutic effects in the 4T1 xenograft model[292]. CT-1, a derivative of cryptotanshinone, can target FTH1 and promote the interaction between NCOA4 and ferritin, thereby triggering ferritinophagy-mediated ferroptosis and significantly inhibiting the growth and proliferation of TNBC[293]. Lysosomal active iron may be the trigger of ferroptosis. The new small molecule compound Fento-1 can selectively kill CD44-highly expressed metastatic tumor cells by activating lysosomal iron and inducing phospholipid peroxidation[130]. Newly synthesized phosphatidylcholine peroxide-decorating liposomes promote ferrous iron efflux by inducing cysteinylation of lysosomal DMT1. Notably, when loaded with other ferroptosis inducers such as artesunate, these liposomes significantly suppress tumor growth and metastasis[294]. Similarly, our previous studies demonstrated that
Notably, although numerous FINs have been developed against the aforementioned targets, a key challenge in their clinical translation remains achieving cancer cell-specific killing while maximally sparing normal cells. This selectivity relies on both the intrinsic differences between cancer and normal cells in metabolism, redox homeostasis, and microenvironmental adaptation, as well as the continuous innovation in drug discovery strategies. As mentioned, to sustain rapid proliferation, tumor cells often exhibit a unique lipid metabolism profile and disrupted iron homeostasis—a state characterized by elevated PUFA and high iron levels—which renders them more susceptible to lipid peroxidation and leads to an over-reliance on ferroptosis defense systems. Concurrently, specific genetic mutations (e.g., inactivation of tumor suppressor genes or activation of oncogenes) can further sensitize cancer cells to ferroptosis. On the other hand, factors within the TME, such as hypoxia, chronic oxidative stress, and immune interactions, compel cancer cells to depend heavily on certain compensatory survival pathways, thereby amplifying their metabolic vulnerabilities. Building on these biological distinctions, current research is developing selective ferroptosis-targeting strategies through multiple dimensions. These include precise interventions based on biomarkers such as GPX4 and FSP1, as well as the application of novel technological approaches, such as natural product screening, PROTAC degraders, gene editing, nanodelivery systems, peptidomimetics, and biologics, to enhance anticancer specificity and reduce damage to normal tissues.
3.3 Combination of FINs with radiotherapy in tumor treatment
Radiation therapy (RT) is an efficient tumor treatment method that selectively targets and eliminates tumor cells through precise administration of ionizing radiation (IR). Previous studies have suggested that IR can directly eliminate cancer by inducing DNA damage, generating free radicals, and causing tumor cells to undergo different forms of RCD, such as apoptosis, necrosis, ferroptosis, and cuproptosis[295,296]. Radiotherapy itself can induce ferroptosis through multiple mechanisms. Beyond directly generating ROS,
As previously mentioned, for tumors harboring KEAP1 mutations with constitutively high NRF2 activity, combining SLC7A11 inhibitors with RT can effectively block their primary compensatory defense mechanism[147,209]. Other mechanisms that inhibit ferroptosis can also contribute to tumor resistance to radiotherapy. OA in radioresistant tumor cells can protect cells from ferroptosis caused by the accumulation of phospholipids containing PUFAs in an ACSL3-dependent manner[305]. In esophageal squamous cell carcinoma, stanniocalcin 2 can upregulate SLC7A11 in a PRMT5-dependent manner, leading to radioresistance. Similarly, IR induces decreased expression of copper metabolism MURR1 domain 10 in HCC, inhibiting the ubiquitin-mediated degradation of HIF1α and promoting SLC7A11 transcription, thereby easily causing radioresistance. Recent studies have identified that intracellular O-GlcNAc transferase (OGT) can act as a ROS sensor in tumor cells. It enhances O-GlcNAc signaling and activates the ROS–OGT–FOXK2–SLC7A11 signaling axis, thereby suppressing ferroptosis and promoting RT resistance in cancer cells[306].
Given these complex mechanisms, a future direction lies in tailoring precise combination therapies based on a tumor’s molecular profile to reverse RT resistance. For example, in some RT-resistant NSCLC with high GPX4 expression, inhibiting FSP1 has been shown to effectively induce ferroptosis and restore radiosensitivity[307]. The timing of intervention is also crucial. Preclinical models suggest that administering ferroptosis inducers within a specific post-RT window (e.g., 24-48 hours) may maximize the exploitation of the metabolic vulnerability induced by radiotherapy[308]. Our previous work found that RT can not only activate NRF2-mediated GPX4 transcription in tumor cells but also inhibit lysosome-mediated GPX4 degradation, thereby generating radioresistance. Our screening identified Tubastatin A as a direct enzymatic inhibitor of GPX4. When combined with radiotherapy, this compound potentiated tumor suppression in mouse xenograft models through ferroptosis sensitization[280]. High-level clinical evidence (NCT01730937) indicates that a sequential combination strategy of stereotactic body radiotherapy (SBRT) followed by sorafenib, a regimen that synergistically induces ferroptosis, significantly prolonged the survival of patients with hepatocellular carcinoma, further validating the feasibility of such combined approaches. Furthermore, the combination of RT, ferroptosis induction, and immunotherapy is currently one of the most promising therapeutic avenues. RT promotes tumor antigen release and presentation, while ferroptosis, as an ICD modality, further activates the immune system. The triple combination not only directly kills tumor cells but also can convert “cold” tumors into “hot” tumors, synergizing with ICIs to achieve durable systemic antitumor immunity[309] (Figure 5B).
3.3.1 Combination of FINs with systemic chemotherapy drugs in tumor treatment
Systemic chemotherapy remains one of the cornerstones of cancer treatment, occupying a central position in multi-tumor and
Cisplatin is a classic platinum-based chemotherapeutic drug. Since its approval in 1978, it has become a basic drug for chemotherapy of various solid tumors. Cisplatin resistance is often accompanied by compensatory enhancement of intracellular antioxidant capacity, driven predominantly by aberrant activation of the NRF2/SLC7A11 antioxidant axis or increased translocation of SLC7A11 to the cell membrane[311], implying its strong resistance to ferroptosis. Adding SLC7A11-targeted FINs can significantly eliminate cisplatin resistance in head and neck squamous cell carcinoma, oral squamous cell carcinoma, and gastric cancer cells[312-314]. In addition, in NSCLC cells resistant to cisplatin and AZD9291, the natural NQO1 substrate 2-methoxy-6-acetyl-7-methyljuglone is found to target NQO1 to induce ferroptosis. The finding regarding NQO1 provides a new option for overcoming cisplatin resistance[315]. Meanwhile, studies have also identified HMOX1 as another key gene mediating ferroptosis resistance in NSCLC to develop cisplatin resistance. Small molecule inhibitors targeting HMOX1 can effectively reverse drug resistance[316].
The resistance mechanism to oxaliplatin involves the disruption of ferroptosis execution. A prominent feature is the reduction in lipid peroxidation substrates. Studies in CRC have shown that oxaliplatin-resistant cells can exert their effect through CDK1-mediated ubiquitination and degradation of the ACSL4 protein. Combining a CDK1 inhibitor stabilizes ACSL4 protein levels, restores PUFA-PL synthesis, and thereby resensitizes cancer cells to oxaliplatin-induced ferroptosis[317]. Furthermore, the regulation of GPX4 stability by the KIF20A/NUAK1/PP1β signaling pathway contributes an additional layer of antioxidant defense[318]. Similarly, directly targeting GPX4 defense using GPX4 inhibitors or DHODH inhibitors (e.g., terpopitide) has also been demonstrated to be effective. For instance, in a hepatocellular carcinoma model, DHODH inhibitors can synergize with oxaliplatin by promoting GPX4 ubiquitination and degradation[319].
Gemcitabine is a deoxycytidine analog antimetabolite chemotherapeutic drug and is a cornerstone drug for pancreatic cancer, bladder cancer, and breast cancer. Numerous studies have reported that the combined use of FINs and gemcitabine has better killing effects on pancreatic cancer, lung adenocarcinoma, and other cells[320,321]. Gemcitabine resistance frequently involves intrinsic metabolic reprogramming of tumor cells and extrinsic modulation by the TME. For instance, gemcitabine has limited clinical benefits in intrahepatic cholangiocarcinoma due to drug resistance. Specifically, the long non-coding RNA PAX8-AS1 promotes GPX4 transcription by activating NRF2 and stabilizes the mRNA structure of GPX4. In preclinical models, the combination of the GPX4 inhibitor JKE-1674 with gemcitabine and cisplatin showed better anti-tumor effects[322]. Gemcitabine-resistant PDAC cells are often accompanied by significantly increased expression of Gli1, a key transcription factor downstream of the Hedgehog signaling pathway. Gli1-driven pentose phosphate cycle can resist gemcitabine-induced DNA damage by promoting pyrimidine synthesis and resist gemcitabine-induced ferroptosis by scavenging lipid reactive oxygen species. When Gli1 inhibitors (e.g., GANT21) are used in combination with gemcitabine, they can significantly enhance DNA damage and ferroptosis, showing synergistic tumor-inhibiting effects[323]. Meanwhile, CAF-derived exosomal miR-3173-5p in PDAC can adsorb ACSL4 and inhibit ferroptosis after being taken up by cancer cells, thereby promoting gemcitabine resistance. The miR-3173-5p/ACSL4 pathway is a promising therapeutic target for gemcitabine-resistant pancreatic cancer[324]. Similarly, CAF-derived exosomal DACT3-AS1 is also an inhibitory regulator of gastric cancer oxaliplatin resistance, which mediates tumor cell resistance to ferroptosis through the miR-181a-5p/SIRT1 axis[325].
Beyond the aforementioned conventional chemotherapeutic agents, substantial evidence indicates that resistance to ferroptosis represents a common core mechanism underlying the acquisition of resistance to diverse chemotherapeutic drugs. By activating ferroptosis defense pathways, tumor cells establish a universal survival barrier that transcends the specific molecular targets of individual drugs. For example, TMZ is a standard chemotherapy drug for neuroendocrine tumors. The curcumin analog ALZ003 can induce ubiquitination of the androgen receptor and inhibit GPX4, opening up new ideas for the treatment strategy of
3.3.2 Combination of FINs with targeted therapy in tumor treatment
Targeted therapy is a core pillar of modern cancer treatment, reshaping the tumor treatment landscape to a certain extent with its precise mechanism of action and its high efficiency and low toxicity. Targeted therapy enhances antitumor efficacy by modulating key ferroptosis pathways, yet tumor cells can acquire targeted resistance through ferroptosis evasion mechanisms. Acquired targeted therapy resistance is very common, and emerging evidence indicates that inducing ferroptosis has surprisingly beneficial effects in overcoming standard treatment resistance.
Sorafenib, an oral multi-tyrosine kinase inhibitor, reshaped treatment paradigms through dual-pathway antiproliferative and antiangiogenic suppression together with ferroptosis induction. However, hepatocellular carcinoma, advanced renal carcinoma, and some other cancers are resistant to sorafenib, limiting its application and leading to a poor prognosis of patients. Sorafenib resistance is caused by ferroptosis resistance through multiple mechanisms. One possibility is that it is due to the upregulation of intracellular metallothionein 1G and activation of the NRF2-MTG1 pathway, which limits GSH depletion and lipid peroxidation. The use of MTG1 inhibitors enhances the sensitivity of resistant HCC to sorafenib[331]. Similarly, studies have identified phosphoseryl-tRNA kinase (PSTK) as a key mediator of sorafenib resistance in hepatocellular carcinoma cells. PSTK inhibitors can enhance chemotherapy efficacy by disrupting redox homeostasis and inactivating GPX4[332]. In addition, studies have found that overexpression of YAP/TAZ and DPP9 in HCC and ccRCC cells causes sorafenib resistance by stabilizing the NRF2 protein and upregulating SLC7A11. Therefore, targeting YAP/TAZ and DPP9, including their downstream SLC7A11, is a new option to combat sorafenib resistance[210,333]. Stromal interaction molecule 1 can also affect the transcriptional activity of SLC7A11 by manipulating store-operated calcium entry (SOCE), leading to sorafenib resistance in HCC. The SOCE inhibitor SKF96365 can significantly enhance the sensitivity of resistant strains to sorafenib both in vivo and in vitro[334]. Recent studies have found that in sorafenib-resistant HCC cells, the deubiquitinase ubiquitin-specific protease 18 can catalyze the deubiquitination and degradation of NCOA4, thereby forming a positive feedback loop of ferroptosis resistance and drug resistance[335].
The balance of iron homeostasis and activation of other ferroptosis inhibitory molecules can also mediate drug resistance. LIFR is often downregulated in hepatocellular carcinoma. Loss of LIFR can lead to upregulation of the iron-chelating cytokine LCN2, thereby depleting iron and making it insensitive to FINs. The use of LCN2-neutralizing antibodies can enhance the effect of sorafenib in
Concurrently, precision combination strategies targeting specific driver genes represent another critical research direction. Although several small-molecule inhibitors targeting KRAS have been developed and entered clinical trials[339], the emergence of drug resistance necessitates the exploration of alternative therapeutic approaches. As discussed previously, KRAS mutations create a unique metabolic paradox, enabling rapid tumor cell proliferation while simultaneously conferring a heightened dependence on multiple ferroptosis defense networks, such as NRF2. Promoting ferroptosis is expected to enhance the efficacy of KRAS-targeted therapies. FINs, including sorafenib, lapatinib, and sulfasalazine, exhibit significant growth inhibitory effects against G12Ci-resistant KRASG12C-mutant cancer cell lines[340]. β-ElE, an active natural compound, demonstrates favorable therapeutic efficacy against
In other targeted therapy contexts, combining FINs also demonstrates significant therapeutic value. Olaparib, a well-known poly (ADP-ribose) polymerase inhibitor, can selectively kill tumors with homologous recombination repair defects (e.g., BRCA mutations). Olaparib has good therapeutic effects on ovarian cancer with BRCA1/2 defects but poor effects on tumors with normal BRCA1/2. By combining with FINs, olaparib can induce ferroptosis in ovarian cancer with normal BRCA gene expression by activating p53, reversing the original drug resistance of the tumor[347]. Lapatinib, trastuzumab, and neratinib are commonly used baseline drugs for patients with HER2-positive breast cancer. Studies have found that fibroblast growth factor receptor 4 (FGFR4) is an essential gene for acquired resistance after anti-HER2 therapy, and its high expression is often associated with poor prognosis. Inhibiting FGFR4 can enhance tumor cell sensitivity to ferroptosis through the β-catenin/TCF4-SLC7A11/FPN1 axis, reducing intrinsic and acquired resistance to anti-HER2 therapy[348] (Figure 5B).
3.3.3 Combination of FINs with immunotherapy in tumor treatment
Immunotherapy has gradually transformed from the last option to a core strategy throughout the entire course of cancer treatment. Immune checkpoint inhibitors (represented by targeting CTLA4, PD-1, and its ligand PD-L1), as representatives of immunotherapy, mark a shift in cancer treatment from directly killing tumor cells to activating the host’s own immune system, achieving unprecedented survival benefits in the treatment of various malignant tumors. However, many patients still develop resistance or non-response to ICI treatment[349]. Since ferroptosis has immunomodulatory effects and is involved in the anti-tumor effect of ICIs, inducing ferroptosis is expected to be an anti-tumor strategy to enhance the efficacy of ICIs.
On the one hand, anti-PD-L1 antibodies can stimulate CD8+ T cells to secrete IFNγ, activate the JAK–STAT1 pathway, and downregulate the expression of SLC7A11 and SLC3A2, thereby sensitizing cancer cells to ferroptosis[243]. Anti-PD-L1 antibodies and ferroptosis activators (e.g., erastin, RSL3, and cysteine proteases) synergistically induce tumor growth inhibition both in vivo and
Tumor cell resistance to ferroptosis is often related to their resistance to ICIs, and restoring their sensitivity to ferroptosis can enhance the efficacy of immunotherapy. For example, tumors with high TYRO3 expression that are resistant to anti-PD-1/PD-L1 can be sensitized to anti-PD-1 therapy by inhibiting the TYRO3-mediated NRF2 pathway to promote ferroptosis[354]. In addition, tumor cells take up itaconic acid produced by macrophages through SLC13A3, thereby escaping immune-mediated ferroptosis killing in the TME and reducing ICIs efficacy. Genetic knockout of SLC13A3 in tumors or treatment with SLC13A3 inhibitors can sensitize tumors to ferroptosis and improve ICI effectiveness[355]. Our investigation of the PKCβII-ACSL4 axis revealed its role in governing a
As mentioned earlier, the complex impact of ferroptosis on the TME varies due to ferroptosis-triggered immunosuppressive or immunostimulatory activities, making it difficult to translate single-target or single-inhibition strategies (e.g., GPX4) into clinical practice. For example, although hepatocyte-specific GPX4 inhibition-induced ferroptosis in HCC increases CD8+ T cell infiltration, this effect is offset by upregulation of PD-L1 in tumor cells and significant infiltration of HMGB1-mediated MDSCs. The triple combination strategy of FINs, ICIs, and MDSC inhibitors can effectively inhibit the growth and metastasis of liver cancer[236]. The immune-related ferroptosis induced by IFNγ released by immune checkpoint blockade (ICB) therapy can also be resisted by phosphoserine aminotransferase 1 (PSAT1), a key enzyme in the serine synthesis pathway. The CAMK2-PSAT1-GPX4 axis in tumor cells can sense cytokine stimulation in the TME and enhance GPX4-dependent ferroptosis defense capacity. PSAT1 blocking peptides and GPX4 inhibition can enhance tumor sensitivity to ferroptosis and the efficacy of ICB in TNBC[356] (Figure 5B).
4. Challenge and Future Direction
Ferroptosis is an iron-dependent form of cell death driven by lipid peroxidation, and is tightly regulated at multiple levels including epigenetic, transcriptional, post-transcriptional, and post-translational stages. Despite the rapid progress in ferroptosis research, numerous challenges in molecular biology and clinical applications remain to be addressed.
5. Mechanisms of Cell Death Execution
First, the existence of specific execution molecules responsible for terminal events such as plasma membrane rupture during ferroptosis remains unclear. Although significant advances have been made in understanding the initiation, accumulation,
Recent studies have also confirmed that NINJ1 acts as an active executor of death by mediating plasma membrane rupture in pyroptosis and necroptosis. While previous research generally regarded PUFA-PLs synthesis and peroxidation-related proteins
6. Clinical Applications
Although numerous preclinical models have demonstrated synergistic effects and good tolerability of FINs in combination with conventional cancer therapies, the clinical translation and application of FINs have stagnated. To fully realize the potential of ferroptosis-inducing strategies in cancer treatment, future research must address several additional challenges.
First, identifying and defining the criteria for selecting ideal candidates for ferroptosis-related cancer therapies is essential, as this forms the basis for further clinical trials. We have preliminarily reviewed characteristic alterations in epigenetics, metabolic reprogramming, gene expression, and microenvironmental features of tumor cells more sensitive to ferroptosis. However, additional factors influence ferroptosis vulnerability, such as cell density[190] and oxygen concentration[361]. Future studies should integrate existing research to comprehensively assess ferroptosis vulnerability. Moreover, the sensitivity of different cancer types to ferroptosis varies based on tumor origin and individual genotypes. In this context, integrating genetic information from human cancer genomes will help predict tumor responsiveness or non-responsiveness to specific FINs.
Second, there is a lack of highly targeted, safe, effective, and clinically applicable ferroptosis-inducing drugs. On the one hand, although a series of FINs targeting different molecules in the ferroptosis pathway (e.g., GPX4, SLC7A11) have been developed, most remain in preclinical stages. In vivo, their potential is limited by poor bioavailability and insufficient targeting. Emerging strategies such as PROTACs, nanoparticles, and mimetic peptides may offer promising solutions. For example, since GPX4 is critical for cell survival, circumventing the toxicity of GPX4 inhibition and specifically targeting tumor cells with high GPX4 expression are attractive for clinical translation. On the other hand, multiple approaches demonstrate robust pro-ferroptotic activity in preclinical models. These include immunotherapy, radiotherapy, chemotherapy (e.g., cisplatin), targeted therapy (e.g., sorafenib), and FDA-approved compounds such as statins. Nevertheless, comprehensive and rigorously controlled clinical trials remain essential to validate their therapeutic efficacy as ferroptosis-inducing agents in patients. We anticipate that such ferroptosis-promoting drugs, when combined with conventional cancer treatments, will yield enhanced tumor-suppressive effects.
Beyond specifically inducing ferroptosis in cancer cells, the complexity of ferroptosis among different cell types within the TME requires further clarification. As discussed earlier, ferroptosis induction may simultaneously affect immune cells, potentially impairing anti-tumor immunity. Future work must dissect how ferroptosis operates in malignant cells, anti-tumor immune effectors, and immunosuppressive populations. The mechanisms by which ferroptotic cancer cells foster immunosuppression need prompt neutralization, and cell type selective agents should be engineered. Additionally, determining optimal dosages and administration strategies for novel FINs, as well as designing therapeutic interventions to minimize adverse effects, are critical.
In the end, robust biomarkers that reliably report ferroptosis induction in vivo remain elusive. To overcome this limitation, multidimensional integration is required, including deconvoluting cellular heterogeneity, mapping the spatial distribution of key molecules, charting functional protein networks, and tracking metabolic reprogramming. To advance the clinical translation of ferroptosis, key priorities include identifying novel biomarkers of sensitivity and resistance, developing strategies to induce ferroptosis selectively in cancer cells, and elucidating its role within the tumor microenvironment and its impact on antitumor immunity.
Acknowledgements
The authors used AI-assisted tools exclusively for language polishing. No AI tools were used to generate content, data, interpretations, or figures. The authors take full responsibility for the integrity and originality of the work.
Authors contribution
Zhu XF: Conceptualization, supervision, writing-review & editing.
Zhan B, Lin XM, Chen P: Writing-original draft, writing-review & editing.
Zhang JB, Guo YQ, Deng R, Zhang HL: Writing-review & editing.
Conflicts of interest
The authors declare no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Funding
This work was supported by the National Natural Science Foundation of China (Grant No.82130079, 82321003, 82372808).
Copyright
© The Author(s) 2025
References
-
1. Tang D, Chen X, Kang R, Kroemer G. Ferroptosis: Molecular mechanisms and health implications. Cell Res. 2021;31(2):107-125.[DOI]
-
2. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060-1072.[DOI]
-
3. Jiang X, Stockwell BR, Conrad M. Ferroptosis: Mechanisms, biology and role in disease. Nat Rev Mol Cell Biol. 2021;22(4):266-282.[DOI]
-
4. Zheng J, Conrad M. Ferroptosis: When metabolism meets cell death. Physiol Rev. 2025;105(2):651-706.[DOI]
-
5. Lei G, Zhuang L, Gan B. Targeting ferroptosis as a vulnerability in cancer. Nat Rev Cancer. 2022;22(7):381-396.[DOI]
-
6. Silvestri L, Pettinato M, Furiosi V, Bavuso Volpe L, Nai A, Pagani A. Managing the dual nature of iron to preserve health. Int J Mol Sci. 2023;24(4):3995.[DOI]
-
7. Su X, Abumrad NA. Cellular fatty acid uptake: A pathway under construction. Trends Endocrinol Metab. 2009;20(2):72-77.[DOI]
-
8. Harayama T, Riezman H. Understanding the diversity of membrane lipid composition. Nat Rev Mol Cell Biol. 2018;19(5):281-296.[DOI]
-
9. Liang D, Minikes AM, Jiang X. Ferroptosis at the intersection of lipid metabolism and cellular signaling. Mol Cell. 2022;82(12):2215-2227.[DOI]
-
10. Wang B, Tontonoz P. Phospholipid remodeling in physiology and disease. Annu Rev Physiol. 2019;81:165-188.[DOI]
-
11. Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol. 2017;13(1):81-90.[DOI]
-
12. Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 2017;13(1):91-98.[DOI]
-
13. Hishikawa D, Shindou H, Kobayashi S, Nakanishi H, Taguchi R, Shimizu T. Discovery of a lysophospholipid acyltransferase family essential for membrane asymmetry and diversity. Proc Natl Acad Sci. 2008;105(8):2830-2835.[DOI]
-
14. Hishikawa D, Hashidate T, Shimizu T, Shindou H. Diversity and function of membrane glycerophospholipids generated by the remodeling pathway in mammalian cells. J Lipid Res. 2014;55(5):799-807.[DOI]
-
15. Chen D, Chu B, Yang X, Liu Z, Jin Y, Kon N, et al. IPLA2β-mediated lipid detoxification controls p53-driven ferroptosis independent of GPX4. Nat Commun. 2021;12(1):3644.[DOI]
-
16. Qiu B, Zandkarimi F, Bezjian CT, Reznik E, Soni RK, Gu W, et al. Phospholipids with two polyunsaturated fatty acyl tails promote ferroptosis. Cell. 2024;187(5):1177-1190.[DOI]
-
17. Liao P, Wang W, Wang W, Kryczek I, Li X, Bian Y, et al. CD8+ T cells and fatty acids orchestrate tumor ferroptosis and immunity via ACSL4. Cancer Cell. 2022;40(4):365-378.[DOI]
-
18. Wang Y, Hu M, Cao J, Wang F, Han JR, Wu TW, et al. ACSL4 and polyunsaturated lipids support metastatic extravasation and colonization. Cell. 2025;188(2):412-429.[DOI]
-
19. Brown CW, Amante JJ, Goel HL, Mercurio AM. The α6β4 integrin promotes resistance to ferroptosis. J Cell Biol. 2017;216(12):4287-4297.[DOI]
-
20. Li D, Li Y. The interaction between ferroptosis and lipid metabolism in cancer. Signal Transduct Target Ther. 2020;5(1):108.[DOI]
-
21. Poppelreuther M, Rudolph B, Du C, Großmann R, Becker M, Thiele C, et al. The N-terminal region of acyl-CoA synthetase 3 is essential for both the localization on lipid droplets and the function in fatty acid uptake. J Lipid Res. 2012;53(5):888-900.[DOI]
-
22. Magtanong L, Ko PJ, To M, Cao JY, Forcina GC, Tarangelo A, et al. Exogenous monounsaturated fatty acids promote a ferroptosis-resistant cell state. Cell Chem Biol. 2019;26(3):420-432.[DOI]
-
23. Yamashita A, Hayashi Y, Matsumoto N, Nemoto-Sasaki Y, Oka S, Tanikawa T, et al. Glycerophosphate/Acylglycerophosphate acyltransferases. Biology. 2014;3(4):801-830.[DOI]
-
24. Yuki K, Shindou H, Hishikawa D, Shimizu T. Characterization of mouse lysophosphatidic acid acyltransferase 3: An enzyme with dual functions in the testis. J Lipid Res. 2009;50(5):860-869.[DOI]
-
25. Zou Y, Henry WS, Ricq EL, Graham ET, Phadnis VV, Maretich P, et al. Plasticity of ether lipids promotes ferroptosis susceptibility and evasion. Nature. 2020;585(7826):603-608.[DOI]
-
26. Li Z, Hu Y, Zheng H, Li M, Liu Y, Feng R, et al. LPCAT1-mediated membrane phospholipid remodelling promotes ferroptosis evasion and tumour growth. Nat Cell Biol. 2024;26(5):811-824.[DOI]
-
27. Liang D, Feng Y, Zandkarimi F, Wang H, Zhang Z, Kim J, et al. Ferroptosis surveillance independent of GPX4 and differentially regulated by sex hormones. Cell. 2023;186(13):2748-2764.[DOI]
-
28. Ackerman D, Tumanov S, Qiu B, Michalopoulou E, Spata M, Azzam A, et al. Triglycerides promote lipid homeostasis during hypoxic stress by balancing fatty acid saturation. Cell Rep. 2018;24(10):2596-2605.[DOI]
-
29. Broniec A, Klosinski R, Pawlak A, Wrona-Krol M, Thompson D, Sarna T. Interactions of plasmalogens and their diacyl analogs with singlet oxygen in selected model systems. Free Radic Biol Med. 2011;50(7):892-898.[DOI]
-
30. Perez MA, Clostio AJ, Houston IR, Ruiz J, Magtanong L, Dixon SJ, et al. Ether lipid deficiency disrupts lipid homeostasis leading to ferroptosis sensitivity. PLoS Genet. 2022;18(9):e1010436.[DOI]
-
31. Cui W, Liu D, Gu W, Chu B. Peroxisome-driven ether-linked phospholipids biosynthesis is essential for ferroptosis. Cell Death Differ. 2021;28(8):2536-2551.[DOI]
-
32. Reed A, Ware T, Li H, Fernando Bazan J, Cravatt BF. TMEM164 is an acyltransferase that forms ferroptotic C20:4 ether phospholipids. Nat Chem Biol. 2023;19(3):378-388.[DOI]
-
33. Zielinski ZA, Pratt DA. Cholesterol autoxidation revisited: Debunking the dogma associated with the most vilified of lipids. J Am Chem Soc. 2016;138(22):6932-6935.[DOI]
-
34. Ma X, Bi E, Lu Y, Su P, Huang C, Liu L, et al. Cholesterol induces CD8+ T cell exhaustion in the tumor microenvironment. Cell Metab. 2019;30(1):143-156.[DOI]
-
35. Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore-Ludlow B, et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature. 2017;547(7664):453-457.[DOI]
-
36. Garcia-Bermudez J, Baudrier L, Bayraktar EC, Shen Y, La K, Guarecuco R, et al. Squalene accumulation in cholesterol auxotrophic lymphomas prevents oxidative cell death. Nature. 2019;567(7746):118-122.[DOI]
-
37. Li Y, Ran Q, Duan Q, Jin J, Wang Y, Yu L, et al. 7-Dehydrocholesterol dictates ferroptosis sensitivity. Nature. 2024;626(7998):411-418.[DOI]
-
38. Freitas FP, Alborzinia H, Dos Santos AF, Nepachalovich P, Pedrera L, Zilka O, et al. 7-Dehydrocholesterol is an endogenous suppressor of ferroptosis. Nature. 2024;626(7998):401-410.[DOI]
-
39. Haeggström JZ, Funk CD. Lipoxygenase and leukotriene pathways: Biochemistry, biology, and roles in disease. Chem Rev. 2011;111(10):5866-5898.[DOI]
-
40. Kuhn H, Banthiya S, van Leyen K. Mammalian lipoxygenases and their biological relevance. Biochim Biophys Acta. 2015;1851(4):308-330.[DOI]
-
41. Shintoku R, Takigawa Y, Yamada K, Kubota C, Yoshimoto Y, Takeuchi T, et al. Lipoxygenase-mediated generation of lipid peroxides enhances ferroptosis induced by erastin and RSL3. Cancer Sci. 2017;108(11):2187-2194.[DOI]
-
42. Shah R, Shchepinov MS, Pratt DA. Resolving the role of lipoxygenases in the initiation and execution of ferroptosis. ACS Cent Sci. 2018;4(3):387-396.[DOI]
-
43. Dobrian AD, Lieb DC, Cole BK, Taylor-Fishwick DA, Chakrabarti SK, Nadler JL. Functional and pathological roles of the 12- and 15-lipoxygenases. Prog Lipid Res. 2011;50(1):115-131.[DOI]
-
44. Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci. 2016;113(34):E4966-E4975.[DOI]
-
45. Wenzel SE, Tyurina YY, Zhao J, St Croix CM, Dar HH, Mao G, et al. PEBP1 wardens ferroptosis by enabling lipoxygenase generation of lipid death signals. Cell. 2017;171(3):628-641.[DOI]
-
46. Chu B, Kon N, Chen D, Li T, Liu T, Jiang L, et al. ALOX12 is required for p53-mediated tumour suppression through a distinct ferroptosis pathway. Nat Cell Biol. 2019;21(5):579-591.[DOI]
-
47. Yin H, Xu L, Porter NA. Free radical lipid peroxidation: Mechanisms and analysis. Chem Rev. 2011;111(10):5944-5972.[DOI]
-
48. Saraev DD, Pratt DA. Reactions of lipid hydroperoxides and how they may contribute to ferroptosis sensitivity. Curr Opin Chem Biol. 2024;81:102478.[DOI]
-
49. Esterbauer H, Eckl P, Ortner A. Possible mutagens derived from lipids and lipid precursors. Mutat Res. 1990;238(3):223-233.[DOI]
-
50. Huang Y, Sarkhel S, Roy A, Mohan A. Interrelationship of lipid aldehydes (MDA, 4-HNE, and 4-ONE) mediated protein oxidation in muscle foods. Crit Rev Food Sci Nutr. 2024;64(32):11809-11825.[DOI]
-
51. Dalleau S, Baradat M, Guéraud F, Huc L. Cell death and diseases related to oxidative stress: 4-hydroxynonenal (HNE) in the balance. Cell Death Differ. 2013;20(12):1615-1630.[DOI]
-
52. Zhong H, Yin H. Role of lipid peroxidation derived 4-hydroxynonenal (4-HNE) in cancer: Focusing on mitochondria. Redox Biol. 2015;4:193-199.[DOI]
-
53. Liu L, Pang J, Qin D, Li R, Zou D, Chi K, et al. Deubiquitinase OTUD5 as a novel protector against 4-HNE-triggered ferroptosis in myocardial ischemia/reperfusion injury. Adv Sci. 2023;10(28):e2301852.[DOI]
-
54. Chen X, Huang J, Yu C, Liu J, Gao W, Li J, et al. A noncanonical function of EIF4E limits ALDH1B1 activity and increases susceptibility to ferroptosis. Nat Commun. 2022;13(1):6318.[DOI]
-
55. Zhong T, Li Y, Jin M, Liu J, Wu Z, Zhu F, et al. Downregulation of 4-HNE and FOXO4 collaboratively promotes NSCLC cell migration and tumor growth. Cell Death Dis. 2024;15(7):546.[DOI]
-
56. Lu Q, Qin X, Chen C, Yu W, Lin J, Liu X, et al. Elevated levels of alcohol dehydrogenase aggravate ethanol-evoked cardiac remodeling and contractile anomalies through FKBP5-yap-mediated regulation of ferroptosis and er stress. Life Sci. 2024;343:122508.[DOI]
-
57. Takami Y, Nakamura J, Katahira J, Maeda Y, Tanaka M, Kuwamura M, et al. Systemic aldehyde storm induced by allyl alcohol exposure results in extensive hepatic ferroptosis in Aldh2∗2 knock-in mice. Free Radic Biol Med. 2025;239:177-188.[DOI]
-
58. Shan G, Bian Y, Yao G, Liang J, Shi H, Hu Z, et al. Targeting ALDH2 to augment platinum-based chemosensitivity through ferroptosis in lung adenocarcinoma. Free Radic Biol Med. 2024;224:310-324.[DOI]
-
59. Pusapati RV, Daemen A, Wilson C, Sandoval W, Gao M, Haley B, et al. mTORC1-dependent metabolic reprogramming underlies escape from glycolysis addiction in cancer cells. Cancer Cell. 2016;29(4):548-562.[DOI]
-
60. Li S, Ouyang X, Sun H, Jin J, Chen Y, Li L, et al. DEPDC5 protects CD8+ T cells from ferroptosis by limiting mTORC1-mediated purine catabolism. Cell Discov. 2024;10(1):53.[DOI]
-
61. Zhang HL, Hu BX, Li ZL, Du T, Shan JL, Ye ZP, et al. PKCβII phosphorylates ACSL4 to amplify lipid peroxidation to induce ferroptosis. Nat Cell Biol. 2022;24(1):88-98.[DOI]
-
62. Carpenter CE, Mahoney AW. Contributions of heme and nonheme iron to human nutrition. Crit Rev Food Sci Nutr. 1992;31(4):333-367.[DOI]
-
63. Piskin E, Cianciosi D, Gulec S, Tomas M, Capanoglu E. Iron absorption: Factors, limitations, and improvement methods. ACS Omega. 2022;7(24):20441-20456.[DOI]
-
64. Galy B, Conrad M, Muckenthaler M. Mechanisms controlling cellular and systemic iron homeostasis. Nat Rev Mol Cell Biol. 2024;25(2):133-155.[DOI]
-
65. Theil EC. Iron, ferritin, and nutrition. Annu Rev Nutr. 2004;24:327-343.[DOI]
-
66. Liu X, Zhao Z, Bian Z, Benthani FA, Hu Y, Liang D, et al. Endocytosis is essential for cysteine-deprivation-induced ferroptosis. Mol Cell. 2025;85(17):3333-3342.[DOI]
-
67. Dutt S, Hamza I, Bartnikas TB. Molecular mechanisms of iron and heme metabolism. Annu Rev Nutr. 2022;42:311-335.[DOI]
-
68. Catapano A, Cimmino F, Petrella L, Pizzella A, D’Angelo M, Ambrosio K, et al. Iron metabolism and ferroptosis in health and diseases: The crucial role of mitochondria in metabolically active tissues. J Nutr Biochem. 2025;140:109888.[DOI]
-
69. Kell DB, Heyden EL, Pretorius E. The biology of lactoferrin, an iron-binding protein that can help defend against viruses and bacteria. Front Immunol. 2020;11:1221.[DOI]
-
70. Wang B, Timilsena YP, Blanch E, Adhikari B. Lactoferrin: Structure, function, denaturation and digestion. Crit Rev Food Sci Nutr. 2019;59(4):580-596.[DOI]
-
71. Kruzel ML, Zimecki M, Actor JK. Lactoferrin in a context of inflammation-induced pathology. Front Immunol. 2017;8:1438.[DOI]
-
72. Khan AA, Quigley JG. Heme and FLVCR-related transporter families SLC48 and SLC49. Mol Aspects Med. 2013;34:669-682.[DOI]
-
73. Huang L, Wang X, Zheng Y, Lang D, Wang J, Yan S, et al. EGCG-NPs inhibition HO-1-mediated reprogram iron metabolism against ferroptosis after subarachnoid hemorrhage. Redox Biol. 2024;70:103075.[DOI]
-
74. Chang LC, Chiang SK, Chen SE, Yu YL, Chou RH, Chang WC. Heme oxygenase-1 mediates BAY 11-7085 induced ferroptosis. Cancer Lett. 2018;416:124-137.[DOI]
-
75. Machado SE, Spangler D, Stacks DA, Darley-Usmar V, Benavides GA, Xie M, et al. Counteraction of myocardial ferritin heavy chain deficiency by heme oxygenase-1. Int J Mol Sci. 2022;23(15):8300.[DOI]
-
76. Chiang SK, Chen SE, Chang LC. A dual role of heme oxygenase-1 in cancer cells. Int J Mol Sci. 2018;20(1):39.[DOI]
-
77. Wang X, Zhang T, Wang S, Shi H, Dong H, Huang Y, et al. Bio-nanocomplexes impair iron homeostasis to induce non-canonical ferroptosis in cancer cells. J Nanobiotechnol. 2025;23(1):121.[DOI]
-
78. Guo Z, Zhang W, Gao H, Li Y, Li X, Yang X, et al. High expression levels of haem oxygenase-1 promote ferroptosis in macrophage-derived foam cells and exacerbate plaque instability. Redox Biol. 2024;76:103345.[DOI]
-
79. Fenaroli F, Valerio A, Ingrassia R. Ischemic neuroprotection by insulin with down-regulation of divalent metal transporter 1 (DMT1) expression and ferrous iron-dependent cell death. Biomolecules. 2024;14(7):856.[DOI]
-
80. Yanatori I, Richardson DR, Imada K, Kishi F. Iron export through the transporter ferroportin 1 is modulated by the iron chaperone PCBP2. J Biol Chem. 2016;291(33):17303-17318.[DOI]
-
81. Barra J, Crosbourne I, Roberge CL, Bossardi-Ramos R, Warren JSA, Matteson K, et al. DMT1-dependent endosome-mitochondria interactions regulate mitochondrial iron translocation and metastatic outgrowth. Oncogene. 2024;43(9):650-667.[DOI]
-
82. Shang Y, Luo M, Yao F, Wang S, Yuan Z, Yang Y. Ceruloplasmin suppresses ferroptosis by regulating iron homeostasis in hepatocellular carcinoma cells. Cell Signal. 2020;72:109633.[DOI]
-
83. D’Andrea P, Giampieri F, Battino M. Nutritional modulation of hepcidin in the treatment of various anemic states. Nutrients. 2023;15(24):5081.[DOI]
-
84. Zhang H, Ostrowski R, Jiang D, Zhao Q, Liang Y, Che X, et al. Hepcidin promoted ferroptosis through iron metabolism which is associated with DMT1 signaling activation in early brain injury following subarachnoid hemorrhage. Oxid Med Cell Longev. 2021;2021:9800794.[DOI]
-
85. Wang S, Ren H, Fan C, Lin Q, Liu M, Tian J. Ochratoxin A induces renal cell ferroptosis by disrupting iron homeostasis and increasing ROS. J Agric Food Chem. 2024;72(3):1734-1744.[DOI]
-
86. Fisher AL, Phillips S, Wang CY, Paulo JA, Xiao X, Xu Y, et al. The hepcidin-ferroportin axis modulates liver endothelial cell BMP expression to influence iron homeostasis in mice. Blood. 2025;145(6):625-634.[DOI]
-
87. Donovan A, Lima CA, Pinkus JL, Pinkus GS, Zon LI, Robine S, et al. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab. 2005;1(3):191-200.[DOI]
-
88. Luo W, Wang J, Xu W, Ma C, Wan F, Huang Y, et al. LncRNA RP11-89 facilitates tumorigenesis and ferroptosis resistance through PROM2-activated iron export by sponging miR-129-5p in bladder cancer. Cell Death Dis. 2021;12(11):1043.[DOI]
-
89. Brown CW, Amante JJ, Chhoy P, Elaimy AL, Liu H, Zhu LJ, et al. Prominin2 drives ferroptosis resistance by stimulating iron export. Dev Cell. 2019;51(5):575-586.[DOI]
-
90. Wolff NA, Garrick MD, Zhao L, Garrick LM, Ghio AJ, Thévenod F. A role for divalent metal transporter (DMT1) in mitochondrial uptake of iron and manganese. Sci Rep. 2018;8(1):211.[DOI]
-
91. Seguin A, Jia X, Earl AM, Li L, Wallace J, Qiu A, et al. The mitochondrial metal transporters mitoferrin1 and mitoferrin2 are required for liver regeneration and cell proliferation in mice. J Biol Chem. 2020;295(32):11002-11020.[DOI]
-
92. Arcos M, Goodla L, Kim H, Desai SP, Liu R, Yin K, et al. PINK1-deficiency facilitates mitochondrial iron accumulation and colon tumorigenesis. Autophagy. 2025;21(4):737-753.[DOI]
-
93. Huang L, Wei B, Zhao Y, Gong X, Chen L. DYNLT1 promotes mitochondrial metabolism to fuel breast cancer development by inhibiting ubiquitination degradation of VDAC1. Mol Med. 2023;29(1):72.[DOI]
-
94. Yang J, Lu X, Hao JL, Li L, Ruan YT, An XN, et al. VSTM2L protects prostate cancer cells against ferroptosis via inhibiting VDAC1 oligomerization and maintaining mitochondria homeostasis. Nat Commun. 2025;16(1):1160.[DOI]
-
95. Ni S, Kuang Y, Yuan Y, Yu B. Mitochondrion-mediated iron accumulation promotes carcinogenesis and Warburg effect through reactive oxygen species in osteosarcoma. Cancer Cell Int. 2020;20:399.[DOI]
-
96. Schulz V, Basu S, Freibert SA, Webert H, Boss L, Mühlenhoff U, et al. Functional spectrum and specificity of mitochondrial ferredoxins FDX1 and FDX2. Nat Chem Biol. 2023;19(2):206-217.[DOI]
-
97. Gervason S, Larkem D, Mansour AB, Botzanowski T, Müller CS, Pecqueur L, et al. Physiologically relevant reconstitution of iron-sulfur cluster biosynthesis uncovers persulfide-processing functions of ferredoxin-2 and frataxin. Nat Commun. 2019;10(1):3566.[DOI]
-
98. Fox NG, Yu X, Feng X, Bailey HJ, Martelli A, Nabhan JF, et al. Structure of the human frataxin-bound iron-sulfur cluster assembly complex provides insight into its activation mechanism. Nat Commun. 2019;10(1):2210.[DOI]
-
99. Lill R, Freibert SA. Mechanisms of mitochondrial iron-sulfur protein biogenesis. Annu Rev Biochem. 2020;89:471-499.[DOI]
-
100. Abe K, Ikeda M, Ide T, Tadokoro T, Miyamoto HD, Furusawa S, et al. Doxorubicin causes ferroptosis and cardiotoxicity by intercalating into mitochondrial DNA and disrupting Alas1-dependent heme synthesis. Sci Signal. 2022;15(758):eabn8017.[DOI]
-
101. Huang J, Xie H, Li J, Huang X, Cai Y, Yang R, et al. Histone lactylation drives liver cancer metastasis by facilitating NSF1-mediated ferroptosis resistance after microwave ablation. Redox Biol. 2025;81:103553.[DOI]
-
102. Levi S, Ripamonti M, Dardi M, Cozzi A, Santambrogio P. Mitochondrial ferritin: Its role in physiological and pathological conditions. Cells. 2021;10(8):1969.[DOI]
-
103. Nie G, Sheftel AD, Kim SF, Ponka P. Overexpression of mitochondrial ferritin causes cytosolic iron depletion and changes cellular iron homeostasis. Blood. 2005;105(5):2161-2167.[DOI]
-
104. Nie G, Chen G, Sheftel AD, Pantopoulos K, Ponka P. In vivo tumor growth is inhibited by cytosolic iron deprivation caused by the expression of mitochondrial ferritin. Blood. 2006;108(7):2428-2434.[DOI]
-
105. Wang XX, Li M, Xu XW, Zhao WB, Jin YM, Li LL, et al. BNIP3-mediated mitophagy attenuates hypoxic-ischemic brain damage in neonatal rats by inhibiting ferroptosis through P62-KEAP1-NRF2 pathway activation to maintain iron and redox homeostasis. Acta Pharmacol Sin. 2025;46(1):33-51.[DOI]
-
106. Clague MJ, Urbé S. Diverse routes to mitophagy governed by ubiquitylation and mitochondrial import. Trends Cell Biol. 2025;35(6):527-538.[DOI]
-
107. Martinez A, Sanchez-Martinez A, Pickering JT, Twyning MJ, Terriente-Felix A, Chen PL, et al. Mitochondrial CISD1/Cisd accumulation blocks mitophagy and genetic or pharmacological inhibition rescues neurodegenerative phenotypes in Pink1/parkin models. Mol Neurodegener. 2024;19(1):12.[DOI]
-
108. Alvarez SW, Sviderskiy VO, Terzi EM, Papagiannakopoulos T, Moreira AL, Adams S, et al. NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nature. 2017;551:639-643.[DOI]
-
109. Mumbauer S, Pascual J, Kolotuev I, Hamaratoglu F. Ferritin heavy chain protects the developing wing from reactive oxygen species and ferroptosis. PLoS Genet. 2019;15(9):e1008396.[DOI]
-
110. Shi H, Bencze KZ, Stemmler TL, Philpott CC. A cytosolic iron chaperone that delivers iron to ferritin. Science. 2008;320(5880):1207-1210.[DOI]
-
111. Wang Y, Protchenko O, Huber KD, Shakoury-Elizeh M, Ghosh MC, Philpott CC. The iron chaperone poly(rC)-binding protein 1 regulates iron efflux through intestinal ferroportin in mice. Blood. 2023;142(19):1658-1671.[DOI]
-
112. Ryu MS, Zhang D, Protchenko O, Shakoury-Elizeh M, Philpott CC. PCBP1 and NCOA4 regulate erythroid iron storage and heme biosynthesis. J Clin Invest. 2017;127(5):1786-1797.[DOI]
-
113. Duan ZW, Wang WT, Wang Y, Wang R, Hua W, Shang CY, et al. SH3GL1-activated FTH1 inhibits ferroptosis and confers doxorubicin resistance in diffuse large B-cell lymphoma. Clin Transl Med. 2025;15(3):e70246.[DOI]
-
114. Protchenko O, Baratz E, Jadhav S, Li F, Shakoury-Elizeh M, Gavrilova O, et al. Iron chaperone poly rC binding protein 1 protects mouse liver from lipid peroxidation and steatosis. Hepatology. 2021;73(3):1176-1193.[DOI]
-
115. Luo C, Liang H, Ji M, Ye C, Lin Y, Guo Y, et al. Autophagy induced by mechanical stress sensitizes cells to ferroptosis by NCOA4-FTH1 axis. Autophagy. 2025;21(6):1263-1282.[DOI]
-
116. Goodwin JM, Dowdle WE, DeJesus R, Wang Z, Bergman P, Kobylarz M, et al. Autophagy-independent lysosomal targeting regulated by ULK1/2-FIP200 and ATG9. Cell Rep. 2017;20(10):2341-2356.[DOI]
-
117. Kim SE, Zhang L, Ma K, Riegman M, Chen F, Ingold I, et al. Ultrasmall nanoparticles induce ferroptosis in nutrient-deprived cancer cells and suppress tumour growth. Nat Nanotechnol. 2016;11(11):977-985.[DOI]
-
118. Riegman M, Sagie L, Galed C, Levin T, Steinberg N, Dixon SJ, et al. Ferroptosis occurs through an osmotic mechanism and propagates independently of cell rupture. Nat Cell Biol. 2020;22(9):1042-1048.[DOI]
-
119. Co HKC, Wu CC, Lee YC, Chen SH. Emergence of large-scale cell death through ferroptotic trigger waves. Nature. 2024;631(8021):654-662.[DOI]
-
120. Roeck BF, Lotfipour Nasudivar S, Vorndran MRH, Schueller L, Yapici FI, Rübsam M, et al. Ferroptosis spreads to neighboring cells via plasma membrane contacts. Nat Commun. 2025;16(1):2951.[DOI]
-
121. Zhang HL, Guo YQ, Liu S, Ye ZP, Li LC, Hu BX, et al. Galectin-13 reduces membrane localization of SLC7A11 for ferroptosis propagation. Nat Chem Biol. 2025;21(10):1531-1543.[DOI]
-
122. Zhang Q, Sun T, Yu F, Liu W, Gao J, Chen J, et al. PAFAH2 suppresses synchronized ferroptosis to ameliorate acute kidney injury. Nat Chem Biol. 2024;20(7):835-846.[DOI]
-
123. Wang X, Kim CS, Adams BC, Wilkinson R, Hill MM, Shah AK, et al. Human proximal tubular epithelial cell-derived small extracellular vesicles mediate synchronized tubular ferroptosis in hypoxic kidney injury. Redox Biol. 2024;70:103042.[DOI]
-
124. Han D, Wang T, Li X, Qin C, Zhang Y, Zhou T, et al. Small extracellular vesicles orchestrated pathological communications between breast cancer cells and cardiomyocytes as a novel mechanism exacerbating anthracycline cardiotoxicity by fueling ferroptosis. Redox Biol. 2025;86:103843.[DOI]
-
125. Gao Y, Mi N, Wu W, Zhao Y, Fan F, Liao W, et al. Transfer of inflammatory mitochondria via extracellular vesicles from M1 macrophages induces ferroptosis of pancreatic beta cells in acute pancreatitis. J Extracell Vesicles. 2024;13(2):e12410.[DOI]
-
126. Pei Z, Qin Y, Fu X, Yang F, Huo F, Liang X, et al. Inhibition of ferroptosis and iron accumulation alleviates pulmonary fibrosis in a bleomycin model. Redox Biol. 2022;57:102509.[DOI]
-
127. Wang J, Zhao Z, Yang H, Wang R, Wang S, Yu J, et al. Engineered nanovesicle platform simultaneously triggers YAP-dependent ferroptosis and reprograms T-cell immunity through miR-150-3p codelivery in melanoma microenvironment. Theranostics. 2025;15(16):8377-8403.[DOI]
-
128. Saimoto Y, Kusakabe D, Morimoto K, Matsuoka Y, Kozakura E, Kato N, et al. Lysosomal lipid peroxidation contributes to ferroptosis induction via lysosomal membrane permeabilization. Nat Commun. 2025;16(1):3554.[DOI]
-
129. Zhu L, Hu J, Wu X, Zhang J, Xu X, Huang X, et al. Programmed enhancement of endogenous iron-mediated lysosomal membrane permeabilization for tumor ferroptosis/pyroptosis dual-induction. Nat Commun. 2025;16(1):3017.[DOI]
-
130. Cañeque T, Baron L, Müller S, Carmona A, Colombeau L, Versini A, et al. Activation of lysosomal iron triggers ferroptosis in cancer. Nature. 2025;642(8067):492-500.[DOI]
-
131. Li S, Liao Z, Yin H, Liu O, Hua W, Wu X, et al. G3BP1 coordinates lysophagy activity to protect against compression-induced cell ferroptosis during intervertebral disc degeneration. Cell Prolif. 2023;56(3):e13368.[DOI]
-
132. von Krusenstiern AN, Robson RN, Qian N, Qiu B, Hu F, Reznik E, et al. Identification of essential sites of lipid peroxidation in ferroptosis. Nat Chem Biol. 2023;19(6):719-730.[DOI]
-
133. Liu S, Chen JH, Li LC, Ye ZP, Liu JN, Chen YH, et al. Susceptibility of mitophagy-deficient tumors to ferroptosis induction by relieving the suppression of lipid peroxidation. Adv Sci. 2025;12(6):e2412593.[DOI]
-
134. Wang X, Chen T, Chen S, Zhang J, Cai L, Liu C, et al. STING aggravates ferroptosis-dependent myocardial ischemia-reperfusion injury by targeting GPX4 for autophagic degradation. Signal Transduct Target Ther. 2025;10(1):136.[DOI]
-
135. Liang P, Tian K, Yang W, Feng R, Li Y, Hu L, et al. ACSL4-mediated ZIP7-VDAC3 interaction regulates endoplasmic reticulum-mitochondria iron transfer in hepatocytes under PFOs exposure. Sci Total Environ. 2024;957:177679.[DOI]
-
136. Liu H, Zheng S, Hou G, Dai J, Zhao Y, Yang F, et al. AKAP1/PKA-mediated GRP75 phosphorylation at mitochondria-associated endoplasmic reticulum membranes protects cancer cells against ferroptosis. Cell Death Differ. 2025;32(3):488-505.[DOI]
-
137. Nguyen NT, Jaramillo-Martinez V, Mathew M, Suresh VV, Sivaprakasam S, Bhutia YD, et al. Sigma receptors: Novel regulators of iron/heme homeostasis and ferroptosis. Int J Mol Sci. 2023;24(19):14672.[DOI]
-
138. Barwick SR, Siddiq MS, Wang J, Xiao H, Marshall B, Perry E, et al. Sigma 1 receptor co-localizes with NRF2 in retinal photoreceptor cells. Antioxidants. 2021;10(6):981.[DOI]
-
139. Sassano ML, Tyurina YY, Diokmetzidou A, Vervoort E, Tyurin VA, More S, et al. Endoplasmic reticulum-mitochondria contacts are prime hotspots of phospholipid peroxidation driving ferroptosis. Nat Cell Biol. 2025;27(6):902-917.[DOI]
-
140. Lewerenz J, Hewett SJ, Huang Y, Lambros M, Gout PW, Kalivas PW, et al. The cystine/glutamate antiporter system xc- in health and disease: From molecular mechanisms to novel therapeutic opportunities. Antioxid Redox Signal. 2013;18(5):522-555.[DOI]
-
141. Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156(1):317-331.[DOI]
-
142. Ursini F, Maiorino M. Lipid peroxidation and ferroptosis: The role of GSH and GPX4. Free Radic Biol Med. 2020;152:175-185.[DOI]
-
143. Cozza G, Rossetto M, Bosello-Travain V, Maiorino M, Roveri A, Toppo S, et al. Glutathione peroxidase 4-catalyzed reduction of lipid hydroperoxides in membranes: The polar head of membrane phospholipids binds the enzyme and addresses the fatty acid hydroperoxide group toward the redox center. Free Radic Biol Med. 2017;112:1-11.[DOI]
-
144. Nguyen HP, Yi D, Lin F, Viscarra JA, Tabuchi C, Ngo K, et al. Aifm2, a NADH oxidase, supports robust glycolysis and is required for cold- and diet-induced thermogenesis. Mol Cell. 2020;77(3):600-617.[DOI]
-
145. Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature. 2019;575(7784):693-698.[DOI]
-
146. Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. 2019;575(7784):688-692.[DOI]
-
147. Koppula P, Lei G, Zhang Y, Yan Y, Mao C, Kondiparthi L, et al. A targetable CoQ-FSP1 axis drives ferroptosis- and radiation-resistance in KEAP1 inactive lung cancers. Nat Commun. 2022;13(1):2206.[DOI]
-
148. Mishima E, Ito J, Wu Z, Nakamura T, Wahida A, Doll S, et al. A non-canonical vitamin K cycle is a potent ferroptosis suppressor. Nature. 2022;608(7924):778-783.[DOI]
-
149. Dai E, Zhang W, Cong D, Kang R, Wang J, Tang D. AIFM2 blocks ferroptosis independent of ubiquinol metabolism. Biochem Biophys Res Commun. 2020;523(4):966-971.[DOI]
-
150. Mao C, Liu X, Zhang Y, Lei G, Yan Y, Lee H, et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature. 2021;593(7860):586-590.[DOI]
-
151. Mishima E, Nakamura T, Zheng J, Zhang W, Mourão ASD, Sennhenn P, et al. DHODH inhibitors sensitize to ferroptosis by FSP1 inhibition. Nature. 2023;619(7968):E9-E18.[DOI]
-
152. Soula M, Weber RA, Zilka O, Alwaseem H, La K, Yen F, et al. Metabolic determinants of cancer cell sensitivity to canonical ferroptosis inducers. Nat Chem Biol. 2020;16(12):1351-1360.[DOI]
-
153. Kraft VAN, Bezjian CT, Pfeiffer S, Ringelstetter L, Müller C, Zandkarimi F, et al. GTP cyclohydrolase 1/tetrahydrobiopterin counteract ferroptosis through lipid remodeling. ACS Cent Sci. 2020;6(1):41-53.[DOI]
-
154. Hu Q, Wei W, Wu D, Huang F, Li M, Li W, et al. Blockade of GCH1/BH4 axis activates ferritinophagy to mitigate the resistance of colorectal cancer to erastin-induced ferroptosis. Front Cell Dev Biol. 2022;10:810327.[DOI]
-
155. Zhang HL, Hu BX, Ye ZP, Li ZL, Liu S, Zhong WQ, et al. TRPML1 triggers ferroptosis defense and is a potential therapeutic target in AKT-hyperactivated cancer. Sci Transl Med. 2024;16(753):eadk0330.[DOI]
-
156. Traber MG, Head B. Vitamin E: How much is enough, too much and why! Free Radic Biol Med. 2021;177:212-225.[DOI]
-
157. Wu Z, Khodade VS, Chauvin JR, Rodriguez D, Toscano JP, Pratt DA. Hydropersulfides inhibit lipid peroxidation and protect cells from ferroptosis. J Am Chem Soc. 2022;144(34):15825-15837.[DOI]
-
158. Barayeu U, Schilling D, Eid M, Xavier da Silva TN, Schlicker L, Mitreska N, et al. Hydropersulfides inhibit lipid peroxidation and ferroptosis by scavenging radicals. Nat Chem Biol. 2023;19(1):28-37.[DOI]
-
159. Liu D, Liang CH, Huang B, Zhuang X, Cui W, Yang L, et al. Tryptophan metabolism acts as a new anti-ferroptotic pathway to mediate tumor growth. Adv Sci. 2023;10(6):e2204006.[DOI]
-
160. Cui W, Guo M, Liu D, Xiao P, Yang C, Huang H, et al. Gut microbial metabolite facilitates colorectal cancer development via ferroptosis inhibition. Nat Cell Biol. 2024;26(1):124-137.[DOI]
-
161. Fiore A, Zeitler L, Russier M, Groß A, Hiller MK, Parker JL, et al. Kynurenine importation by SLC7A11 propagates anti-ferroptotic signaling. Mol Cell. 2022;82(5):920-932.[DOI]
-
162. Martinez-Outschoorn UE, Peiris-Pagés M, Pestell RG, Sotgia F, Lisanti MP. Cancer metabolism: A therapeutic perspective. Nat Rev Clin Oncol. 2017;14(1):11-31.[DOI]
-
163. Zou Y, Palte MJ, Deik AA, Li H, Eaton JK, Wang W, et al. A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers sensitivity to ferroptosis. Nat Commun. 2019;10(1):1617.[DOI]
-
164. Zhang L, Hobeika CS, Khabibullin D, Yu D, Filippakis H, Alchoueiry M, et al. Hypersensitivity to ferroptosis in chromophobe RCC is mediated by a glutathione metabolic dependency and cystine import via solute carrier family 7 member 11. Proc Natl Acad Sci. 2022;119(28):e2122840119.[DOI]
-
165. Sato M, Matsumoto M, Saiki Y, Alam M, Nishizawa H, Rokugo M, et al. BACH1 promotes pancreatic cancer metastasis by repressing epithelial genes and enhancing epithelial-mesenchymal transition. Cancer Res. 2020;80(6):1279-1292.[DOI]
-
166. Verma N, Vinik Y, Saroha A, Nair NU, Ruppin E, Mills G, et al. Synthetic lethal combination targeting BET uncovered intrinsic susceptibility of TNBC to ferroptosis. Sci Adv. 2020;6(34):eaba8968.[DOI]
-
167. Yang F, Xiao Y, Ding JH, Jin X, Ma D, Li DQ, et al. Ferroptosis heterogeneity in triple-negative breast cancer reveals an innovative immunotherapy combination strategy. Cell Metab. 2023;35(1):84-100.[DOI]
-
168. Ge A, Xiang W, Li Y, Zhao D, Chen J, Daga P, et al. Broadening horizons: The multifaceted role of ferroptosis in breast cancer. Front Immunol. 2024;15:1455741.[DOI]
-
169. Krebs AM, Mitschke J, Lasierra Losada M, Schmalhofer O, Boerries M, Busch H, et al. The EMT-activator Zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nat Cell Biol. 2017;19(5):518-529.[DOI]
-
170. Müller S, Sindikubwabo F, Cañeque T, Lafon A, Versini A, Lombard B, et al. CD44 regulates epigenetic plasticity by mediating iron endocytosis. Nat Chem. 2020;12(10):929-938.[DOI]
-
171. Oliveira T, Hermann E, Lin D, Chowanadisai W, Hull E, Montgomery M. HDAC inhibition induces EMT and alterations in cellular iron homeostasis to augment ferroptosis sensitivity in SW13 cells. Redox Biol. 2021;47:102149.[DOI]
-
172. Lee JY, Nam M, Son HY, Hyun K, Jang SY, Kim JW, et al. Polyunsaturated fatty acid biosynthesis pathway determines ferroptosis sensitivity in gastric cancer. Proc Natl Acad Sci. 2020;117(51):32433-32442.[DOI]
-
173. Hangauer MJ, Viswanathan VS, Ryan MJ, Bole D, Eaton JK, Matov A, et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature. 2017;551(7679):247-250.[DOI]
-
174. Tsoi J, Robert L, Paraiso K, Galvan C, Sheu KM, Lay J, et al. Multi-stage differentiation defines melanoma subtypes with differential vulnerability to drug-induced iron-dependent oxidative stress. Cancer Cell. 2018;33(5):890-904.e895.[DOI]
-
175. Lee WC, Moi SH, Yang SF, Tseng HH, Liu YP. Downregulation of AATK enhances susceptibility to ferroptosis by promoting endosome recycling in gefitinib-resistant lung cancer cells. J Pathol. 2025;265(4):422-436.[DOI]
-
176. You JH, Lee J, Roh JL. Mitochondrial pyruvate carrier 1 regulates ferroptosis in drug-tolerant persister head and neck cancer cells via epithelial-mesenchymal transition. Cancer Lett. 2021;507:40-54.[DOI]
-
177. Long H, Zhu W, Wei L, Zhao J. Iron homeostasis imbalance and ferroptosis in brain diseases. MedComm. 2023;4(4):e298.[DOI]
-
178. Mai TT, Hamaï A, Hienzsch A, Cañeque T, Müller S, Wicinski J, et al. Salinomycin kills cancer stem cells by sequestering iron in lysosomes. Nat Chem. 2017;9(10):1025-1033.[DOI]
-
179. Wang D, Wu H, Yang J, Li M, Ling C, Gao Z, et al. Loss of SLC46A1 decreases tumor iron content in hepatocellular carcinoma. Hepatol Commun. 2022;6(10):2914-2924.[DOI]
-
180. Schonberg DL, Miller TE, Wu Q, Flavahan WA, Das NK, Hale JS, et al. Preferential iron trafficking characterizes glioblastoma stem-like cells. Cancer Cell. 2015;28(4):441-455.[DOI]
-
181. Luo M, Bao L, Xue Y, Zhu M, Kumar A, Xing C, et al. ZMYND8 protects breast cancer stem cells against oxidative stress and ferroptosis through activation of NRF2. J Clin Invest. 2024;134(6):e171166.[DOI]
-
182. Li Z, Xu ZM, Chen WP, Du XJ, Ou CX, Luo ZK, et al. Tumor-repopulating cells evade ferroptosis via PCK2-dependent phospholipid remodeling. Nat Chem Biol. 2024;20:1341-1352.[DOI]
-
183. Wu M, Zhang X, Zhang W, Chiou YS, Qian W, Liu X, et al. Cancer stem cell regulated phenotypic plasticity protects metastasized cancer cells from ferroptosis. Nat Commun. 2022;13:1371.[DOI]
-
184. Schmitt A, Xu W, Bucher P, Grimm M, Konantz M, Horn H, et al. Dimethyl fumarate induces ferroptosis and impairs NF-kappaB/STAT3 signaling in DLBCL. Blood. 2021;138:871-884.[DOI]
-
185. Zhang Y, Shi J, Liu X, Feng L, Gong Z, Koppula P, et al. BAP1 links metabolic regulation of ferroptosis to tumour suppression. Nat Cell Biol. 2018;20(10):1181-1192.[DOI]
-
186. Dodson M, Castro-Portuguez R, Zhang DD. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 2019;23:101107.[DOI]
-
187. Liu T, Jiang L, Tavana O, Gu W. The deubiquitylase OTUB1 mediates ferroptosis via stabilization of SLC7A11. Cancer Res. 2019;79(8):1913-1924.[DOI]
-
188. Wu K, Yan M, Liu T, Wang Z, Duan Y, Xia Y, et al. Creatine kinase B suppresses ferroptosis by phosphorylating GPX4 through a moonlighting function. Nat Cell Biol. 2023;25(5):714-725.[DOI]
-
189. Kim NG, Koh E, Chen X, Gumbiner BM. E-cadherin mediates contact inhibition of proliferation through Hippo signaling-pathway components. Proc Natl Acad Sci U.S.A. 2011;108(29):11930-11935.[DOI]
-
190. Wu J, Minikes AM, Gao M, Bian H, Li Y, Stockwell BR, et al. Intercellular interaction dictates cancer cell ferroptosis via NF2-YAP signalling. Nature. 2019;572(7769):402-406.[DOI]
-
191. Zou Y, Li H, Graham ET, Deik AA, Eaton JK, Wang W, et al. Cytochrome P450 oxidoreductase contributes to phospholipid peroxidation in ferroptosis. Nat Chem Biol. 2020;16(3):302-309.[DOI]
-
192. Miess H, Dankworth B, Gouw AM, Rosenfeldt M, Schmitz W, Jiang M, et al. The glutathione redox system is essential to prevent ferroptosis caused by impaired lipid metabolism in clear cell renal cell carcinoma. Oncogene. 2018;37(40):5435-5450.[DOI]
-
193. Wang ME, Chen J, Lu Y, Bawcom AR, Wu J, Ou J, et al. RB1-deficient prostate tumor growth and metastasis are vulnerable to ferroptosis induction via the E2F/ACSL4 axis. J Clin Invest. 2023;133(10):e166647.[DOI]
-
194. Zhou Q, Meng Y, Li D, Yao L, Le J, Liu Y, et al. Ferroptosis in cancer: From molecular mechanisms to therapeutic strategies. Signal Transduct Target Ther. 2024;9(1):55.[DOI]
-
195. Liu Y, Stockwell BR, Jiang X, Gu W. p53-regulated non-apoptotic cell death pathways and their relevance in cancer and other diseases. Nat Rev Mol Cell Biol. 2025;26:600-614.[DOI]
-
196. Chung JY, Knutson BA. Bypassing the guardian: Regulated cell death pathways in p53-mutant cancers. Cell Mol Biol Lett. 2025;30:68.[DOI]
-
197. Peng M, Hu Q, Wu Z, Wang B, Wang C, Yu F. Mutation of TP53 confers ferroptosis resistance in lung cancer through the FOXM1/MEF2C axis. Am J Pathol. 2023;193(10):1587-1602.[DOI]
-
198. Dibra D, Xiong S, Moyer SM, El-Naggar AK, Qi Y, Su X, et al. Mutant p53 protects triple-negative breast adenocarcinomas from ferroptosis in vivo. Sci Adv. 2024;10(7):eadk1835.[DOI]
-
199. Ou Y, Wang SJ, Li D, Chu B, Gu W Activation of SAT1 engages polyamine metabolism with p53-mediated ferroptotic responses. Proc Natl Acad Sci U.S.A. 2016;113(44):E6806-E6812.[DOI]
-
200. Su Z, Kon N, Yi J, Zhao H, Zhang W, Tang Q, et al. Specific regulation of BACH1 by the hotspot mutant p53R175H reveals a distinct gain-of-function mechanism. Nat Cancer. 2023;4(4):564-581.[DOI]
-
201. Gan Y, Deng J, Hao Q, Huang Y, Han T, Xu JG, et al. UTP11 deficiency suppresses cancer development via nucleolar stress and ferroptosis. Redox Biol. 2023;62:102705.[DOI]
-
202. Seo WJ, Choi S, Roh CK, Cho M, Kim YM, Kim HI, et al. Omentum preservation as an oncologically comparable and surgically superior alternative to total omentectomy during radical gastrectomy for T3-T4 gastric cancer. Surgery. 2021;170(2):610-616.[DOI]
-
203. Elías-Maxil JA, Rigas F, de Velásquez MT, Ramírez-Zamora RM. Optimization of Fenton’s reagent coupled to Dissolved Air Flotation to remove cyanobacterial odorous metabolites and suspended solids from raw surface water. Water Sci Technol. 2011;64(8):1668-1674.[DOI]
-
204. Chen Y, Liu S, Leng SX. Chronic low-grade inflammatory phenotype (CLIP) and senescent immune dysregulation. Clin Ther. 2019;41(3):400-409.[DOI]
-
205. Shin KO, Uchida Y, Park K. Diesel particulate extract accelerates premature skin aging in human fibroblasts via ceramide-1-phosphate-mediated signaling pathway. Int J Mol Sci. 2022;23(5):2691.[DOI]
-
206. Sianturi EI, Latifah E, Probandari A, Effendy C, Taxis K. Daily struggle to take antiretrovirals: a qualitative study in Papuans living with HIV and their healthcare providers. BMJ Open. 2020;10(9):e036832.[DOI]
-
207. Iketani S, Forouhar F, Liu H, Hong SJ, Lin FY, Nair MS, et al. Lead compounds for the development of SARS-CoV-2 3CL protease inhibitors. Nat Commun. 2021;12(1):2708.[DOI]
-
208. Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R, et al. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology. 2016;63(1):173-184.[DOI]
-
209. Lei G, Zhang Y, Hong T, Zhang X, Liu X, Mao C, et al. Ferroptosis as a mechanism to mediate p53 function in tumor radiosensitivity. Oncogene. 2021;40(20):3533-3547.[DOI]
-
210. Chang K, Chen Y, Zhang X, Zhang W, Xu N, Zeng B, et al. DPP9 Stabilizes NRF2 to Suppress Ferroptosis and Induce Sorafenib Resistance in Clear Cell Renal Cell Carcinoma. Cancer Res. 2023;83(23):3940-3955.[DOI]
-
211. Alborzinia H, Chen Z, Yildiz U, Freitas FP, Vogel FC, Varga JP, et al. LRP8-mediated selenocysteine uptake is a targetable vulnerability in MYCN-amplified neuroblastoma. EMBO Mol Med. 2023;15(8):e18014.[DOI]
-
212. Alborzinia H, Flórez AF, Kreth S, Brückner LM, Yildiz U, Gartlgruber M, et al. MYCN mediates cysteine addiction and sensitizes neuroblastoma to ferroptosis. Nat Cancer. 2022;3(4):471-485.[DOI]
-
213. Floros KV, Cai J, Jacob S, Kurupi R, Fairchild CK, Shende M, et al. MYCN-amplified neuroblastoma is addicted to iron and vulnerable to inhibition of the system Xc-/glutathione axis. Cancer Res. 2021;81(7):1896-1908.[DOI]
-
214. Poursaitidis I, Wang X, Crighton T, Labuschagne C, Mason D, Cramer SL, et al. Oncogene-selective sensitivity to synchronous cell death following modulation of the amino acid nutrient cystine. Cell Rep. 2017;18(11):2547-2556.[DOI]
-
215. Prior IA, Hood FE, Hartley JL. The frequency of Ras mutations in cancer. Cancer Res. 2020;80(14):2969-2974.[DOI]
-
216. Kamphorst JJ, Cross JR, Fan J, De Stanchina E, Mathew R, White EP, et al. Hypoxic and Ras-transformed cells support growth by scavenging unsaturated fatty acids from lysophospholipids. Proc Natl Acad Sci U.S.A. 2013;110(22):8882-8887.[DOI]
-
217. Hu K, Li K, Lv J, Feng J, Chen J, Wu H, et al. Suppression of the SLC7A11/glutathione axis causes synthetic lethality in KRAS-mutant lung adenocarcinoma. J Clin Invest. 2020;130(4):1752-1766.[DOI]
-
218. Bartolacci C, Andreani C, Vale G, Berto S, Melegari M, Crouch AC, et al. Targeting de novo lipogenesis and the Lands cycle induces ferroptosis in KRAS-mutant lung cancer. Nat Commun. 2022;13(1):4327.[DOI]
-
219. Padanad MS, Konstantinidou G, Venkateswaran N, Melegari M, Rindhe S, Mitsche M, et al. Fatty acid oxidation mediated by Acyl-CoA synthetase long chain 3 is required for mutant KRAS lung tumorigenesis. Cell Rep. 2016;16(6):1614-1628.[DOI]
-
220. Müller F, Lim JK, Bebber CM, Seidel E, Tishina S, Dahlhaus A, et al. Elevated FSP1 protects KRAS-mutated cells from ferroptosis during tumor initiation. Cell Death Differ. 2023;30(2):442-456.[DOI]
-
221. Chen Y, Yan Q, Ruan S, Cui J, Li Z, Zhang Z, et al. GCLM lactylation mediated by ACAT2 promotes ferroptosis resistance in KRAS(G12D)-mutant cancer. Cell Rep. 2025;44(6):115774.[DOI]
-
222. Li M, Yu X, Liu Y, Ouyang S, Wu L, Chen X, et al. KRAS/ABHD17C/ALOX15B axis promotes pancreatic cancer progression via ferroptosis evasion. Adv Sci. 2025;12(35):e04470.[DOI]
-
223. Glaviano A, Foo AS, Lam HY, Yap KC, Jacot W, Jones RH, et al. PI3K/AKT/mTOR signaling transduction pathway and targeted therapies in cancer. Mol Cancer. 2023;22(1):138.[DOI]
-
224. Zhao L, Zhou X, Xie F, Zhang L, Yan H, Huang J, et al. Ferroptosis in cancer and cancer immunotherapy. Cancer Commun. 2022;42(2):88-116.[DOI]
-
225. Wang Y, Tian Q, Hao Y, Yao W, Lu J, Chen C, et al. The kinase complex mTORC2 promotes the longevity of virus-specific memory CD4+ T cells by preventing ferroptosis. Nat Immunol. 2022;23(2):303-317.[DOI]
-
226. Deepak K, Roy PK, Das A, Mukherjee B, Mandal M. Glucose-6-phosphate dehydrogenase (G6PD) shields pancreatic cancer from autophagy-dependent ferroptosis by suppressing redox imbalance induced AMPK/mTOR signaling. Free Radic Biol Med. 2025;237:195-209.[DOI]
-
227. Gan W, Dai X, Dai X, Xie J, Yin S, Zhu J, et al. LATS suppresses mTORC1 activity to directly coordinate Hippo and mTORC1 pathways in growth control. Nat Cell Biol. 2020;22(2):246-256.[DOI]
-
228. Zhang Y, Swanda RV, Nie L, Liu X, Wang C, Lee H, et al. mTORC1 couples cyst(e)ine availability with GPX4 protein synthesis and ferroptosis regulation. Nat Commun. 2021;12:1589.[DOI]
-
229. Dai E, Zhu Z, Wahed S, Qu Z, Storkus WJ, Guo ZS. Epigenetic modulation of antitumor immunity for improved cancer immunotherapy. Mol Cancer. 2021;20:171.[DOI]
-
230. Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018;25(3):486-541.[DOI]
-
231. Efimova I, Catanzaro E, Van der Meeren L, Turubanova VD, Hammad H, Mishchenko TA, et al. Vaccination with early ferroptotic cancer cells induces efficient antitumor immunity. J Immunother Cancer. 2020;8(2):e001369.[DOI]
-
232. Liu J, Zhu S, Zeng L, Li J, Klionsky DJ, Kroemer G, et al. DCN released from ferroptotic cells ignites AGER-dependent immune responses. Autophagy. 2022;18(9):2036-2049.[DOI]
-
233. Luo X, Gong HB, Gao HY, Wu YP, Sun WY, Li ZQ, et al. Oxygenated phosphatidylethanolamine navigates phagocytosis of ferroptotic cells by interacting with TLR2. Cell Death Differ. 2021;28(6):1971-1989.[DOI]
-
234. Zhivaki D, Borriello F, Chow OA, Doran B, Fleming I, Theisen DJ, et al. Inflammasomes within hyperactive murine dendritic cells stimulate long-lived T cell-mediated anti-tumor immunity. Cell Rep. 2020;33(7):108381.[DOI]
-
235. Conche C, Finkelmeier F, Pešić M, Nicolas AM, Böttger TW, Kennel KB, et al. Combining ferroptosis induction with MDSC blockade renders primary tumours and metastases in liver sensitive to immune checkpoint blockade. Gut. 2023;72(9):1774-1782.[DOI]
-
236. Cheu JW, Lee D, Li Q, Goh CC, Bao MH, Yuen VW, et al. Ferroptosis suppressor protein 1 inhibition promotes tumor ferroptosis and anti-tumor immune responses in liver cancer. Cell Mol Gastroenterol Hepatol. 2023;16:133-159.[DOI]
-
237. Ma S, Liang X, Yang N, Yang J, Zhang J, Pan X, et al. Boosting cancer immunotherapy by biomineralized nanovaccine with ferroptosis-inducing and photothermal properties. Biomater Sci. 2023;11(2):518-532.[DOI]
-
238. Chen Y, Zhang C, Li Y, Tan X, Li W, Tan S, Liu G. Discovery of lung adenocarcinoma tumor antigens and ferroptosis subtypes for developing mRNA vaccines. Sci Rep. 2024;14:3219.[DOI]
-
239. Zhai Q, Wang Z, Tang H, Hu S, Chen M, Ji P. Identification of ferroptosis-associated tumor antigens as the potential targets to prevent head and neck squamous cell carcinoma. Genes Dis. 2024;11(6):101212.[DOI]
-
240. Adebanjo EA, Bakare KM, Matthew UO, Fatai LO, Oyekunle D. Novel therapeutic approaches to cancer immunotherapy and mRNA vaccines technology: A review. Holist Integr Oncol. 2025;4(1):75.[DOI]
-
241. Wiernicki B, Maschalidi S, Pinney J, Adjemian S, Vanden Berghe T, Ravichandran KS, et al. Cancer cells dying from ferroptosis impede dendritic cell-mediated anti-tumor immunity. Nat Commun. 2022;13:3676.[DOI]
-
242. Kim R, Taylor D, Vonderheide RH, Gabrilovich DI. Ferroptosis of immune cells in the tumor microenvironment. Trends Pharmacol Sci. 2023;44(8):542-552.[DOI]
-
243. Wang W, Green M, Choi JE, Gijón M, Kennedy PD, Johnson JK, et al. CD8+ T cells regulate tumour ferroptosis during cancer immunotherapy. Nature. 2019;569(7755):270-274.[DOI]
-
244. Drijvers JM, Gillis JE, Muijlwijk T, Nguyen TH, Gaudiano EF, Harris IS, et al. Pharmacologic screening identifies metabolic vulnerabilities of CD8+ T cells. Cancer Immunol Res. 2021;9(2):184-199.[DOI]
-
245. Matsushita M, Freigang S, Schneider C, Conrad M, Bornkamm GW, Kopf M. T cell lipid peroxidation induces ferroptosis and prevents immunity to infection. J Exp Med. 2015;212(4):555-568.[DOI]
-
246. Ma X, Xiao L, Liu L, Ye L, Su P, Bi E, et al. CD36-mediated ferroptosis dampens intratumoral CD8+ T cell effector function and impairs their antitumor ability. Cell Metab. 2021;33(5):1001-1012.e1005.[DOI]
-
247. Xu S, Chaudhary O, Rodríguez-Morales P, Sun X, Chen D, Zappasodi R, et al. Uptake of oxidized lipids by the scavenger receptor CD36 promotes lipid peroxidation and dysfunction in CD8+ T cells in tumors. Immunity. 2021;54(7):1561-1577.e1567.[DOI]
-
248. Ping Y, Shan J, Qin H, Li F, Qu J, Guo R, et al. PD-1 signaling limits expression of phospholipid phosphatase 1 and promotes intratumoral CD8(+) T cell ferroptosis. Immunity. 2024;57(9):2122-2139.[DOI]
-
249. Yao Y, Chen Z, Zhang H, Chen C, Zeng M, Yunis J, et al. Selenium-GPX4 axis protects follicular helper T cells from ferroptosis. Nat Immunol. 2021;22(9):1127-1139.[DOI]
-
250. Xu C, Sun S, Johnson T, Qi R, Zhang S, Zhang J, et al. The glutathione peroxidase Gpx4 prevents lipid peroxidation and ferroptosis to sustain Treg cell activation and suppression of antitumor immunity. Cell Rep. 2021;35(11):109235.[DOI]
-
251. Arensman MD, Yang XS, Leahy DM, Toral-Barza L, Mileski M, Rosfjord EC, et al. Cystine-glutamate antiporter xCT deficiency suppresses tumor growth while preserving antitumor immunity. Proc Natl Acad Sci U.S.A. 2019;116(19):9533-9542.[DOI]
-
252. Pacheco R, Oliva H, Martinez-Navío JM, Climent N, Ciruela F, Gatell JM, et al. Glutamate released by dendritic cells as a novel modulator of T cell activation. J Immunol. 2006;177(10):6695-6704.[DOI]
-
253. Morgan PK, Pernes G, Huynh K, Giles C, Paul S, Smith AAT, et al. A lipid atlas of human and mouse immune cells provides insights into ferroptosis susceptibility. Nat Cell Biol. 2024;26(4):645-659.[DOI]
-
254. Muri J, Thut H, Bornkamm GW, Kopf M. B1 and marginal zone b cells but not follicular B2 cells require Gpx4 to prevent lipid peroxidation and ferroptosis. Cell Rep. 2019;29(9):2731-2744.e2734.[DOI]
-
255. Kapralov AA, Yang Q, Dar HH, Tyurina YY, Anthonymuthu TS, Kim R, et al. Redox lipid reprogramming commands susceptibility of macrophages and microglia to ferroptotic death. Nat Chem Biol. 2020;16(3):278-290.[DOI]
-
256. Recalcati S, Locati M, Marini A, Santambrogio P, Zaninotto F, De Pizzol M, et al. Differential regulation of iron homeostasis during human macrophage polarized activation. Eur J Immunol. 2010;40(3):824-835.[DOI]
-
257. Tang B, Zhu J, Wang Y, Chen W, Fang S, Mao W, et al. Targeted xCT‐mediated ferroptosis and protumoral polarization of macrophages is effective against HCC and enhances the efficacy of the anti‐PD‐1/L1 response. Adv Sci. 2023;10(2):e2203973.[DOI]
-
258. Hao X, Zheng Z, Liu H, Zhang Y, Kang J, Kong X, et al. Inhibition of APOC1 promotes the transformation of M2 into M1 macrophages via the ferroptosis pathway and enhances anti-PD1 immunotherapy in hepatocellular carcinoma based on single-cell RNA sequencing. Redox Biol. 2022;56:102463.[DOI]
-
259. Wu C, Shen Z, Lu Y, Sun F, Shi H. p53 Promotes ferroptosis in macrophages treated with Fe3O4 nanoparticles. ACS Appl Mater Interfaces. 2022;14(38):42791-42803.[DOI]
-
260. Kim R, Hashimoto A, Markosyan N, Tyurin VA, Tyurina YY, Kar G, et al. Ferroptosis of tumour neutrophils causes immune suppression in cancer. Nature. 2022;612(7939):338-346.[DOI]
-
261. Zhao Y, Liu Z, Liu G, Zhang Y, Liu S, Gan D, et al. Neutrophils resist ferroptosis and promote breast cancer metastasis through aconitate decarboxylase 1. Cell Metab. 2023;35(10):1688-1703.[DOI]
-
262. Chen L, Huang M. Oncometabolites in cancer: From cancer cells to the tumor microenvironment. Holist Integr Oncol. 2024;3(1):26.[DOI]
-
263. Han C, Ge M, Xing P, Xia T, Zhang C, Ma K, et al. Cystine deprivation triggers CD36-mediated ferroptosis and dysfunction of tumor infiltrating CD8+ T cells. Cell Death Dis. 2024;15(2):145.[DOI]
-
264. Wang B, Pei J, Xu S, Liu J, Yu J. A glutamine tug-of-war between cancer and immune cells: recent advances in unraveling the ongoing battle. J Exp Clin Cancer Res. 2024;43(1):74.[DOI]
-
265. Guo C, You Z, Shi H, Sun Y, Du X, Palacios G, et al. SLC38A2 and glutamine signalling in cDC1s dictate anti-tumour immunity. Nature. 2023;620(7972):200-208.[DOI]
-
266. Chang CH, Qiu J, O’Sullivan D, Buck Michael D, Noguchi T, Curtis Jonathan D, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell. 2015;162(6):1229-1241.[DOI]
-
267. Andryszkiewicz W, Gąsiorowska J, Kübler M, Kublińska K, Pałkiewicz A, Wiatkowski A, et al. Glucose metabolism and tumor microenvironment: Mechanistic insights and therapeutic implications. Int J Mol Sci. 2025;26(5):1879.[DOI]
-
268. Yang J, Zhou Y, Xie S, Wang J, Li Z, Chen L, et al. Metformin induces Ferroptosis by inhibiting UFMylation of SLC7A11 in breast cancer. J Exp Clin Cancer Res. 2021;40(1):206.[DOI]
-
269. Shimada K, Skouta R, Kaplan A, Yang WS, Hayano M, Dixon SJ, et al. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat Chem Biol. 2016;12(7):497-503.[DOI]
-
270. Zhou Y, Zeng L, Cai L, Zheng W, Liu X, Xiao Y, et al. Cellular senescence-associated gene IFI16 promotes HMOX1-dependent evasion of ferroptosis and radioresistance in glioblastoma. Nat Commun. 2025;16(1):1212.[DOI]
-
271. Liang C, Zhang X, Yang M, Dong . Recent Progress in Ferroptosis Inducers for Cancer Therapy. Adv Mater. 2019;31(51):1904197.[DOI]
-
272. Liu J, Tang D, Kang R. Targeting GPX4 in ferroptosis and cancer: chemical strategies and challenges. Trends Pharmacol Sci. 2024;45(8):666-670.[DOI]
-
273. Luo T, Zheng Q, Shao L, Ma T, Mao L, Wang M. Intracellular delivery of glutathione peroxidase degrader induces ferroptosis in vivo. Angew Chem Int Ed Engl. 2022;61(39):e202206277.[DOI]
-
274. Duan Y, Hu Z, Han P, Lei B, Wang S, Wang Z, et al. ADSL-generated fumarate binds and inhibits STING to promote tumour immune evasion. Nat Cell Biol. 2025;27(4):668-682.[DOI]
-
275. Li J, Liu J, Zhou Z, Wu R, Chen X, Yu C, et al Tumor-specific GPX4 degradation enhances ferroptosis-initiated antitumor immune response in mouse models of pancreatic cancer. Sci Transl Med. 2023;15(720):eadg3049.[DOI]
-
276. Hassannia B, Wiernicki B, Ingold I, Qu F, Van Herck S, Tyurina YY, et al. Nano-targeted induction of dual ferroptotic mechanisms eradicates high-risk neuroblastoma. J Clin Invest. 2018;128(8):3341-3355.[DOI]
-
277. Yang X, Wang L, Lin P, Ning Y, Lin Y, Xie Y, et al. Discovery of Artesunate (ARS) PROTACs as GPX4 protein degraders for the treatment of bladder cancer. Eur J Med Chem. 2025;293:117710.[DOI]
-
278. Yu D, Hu H, Zhang Q, Wang C, Xu M, Xu H, et al. Acevaltrate as a novel ferroptosis inducer with dual targets of PCBP1/2 and GPX4 in colorectal cancer. Signal Transduct Target Ther. 2025;10(1):211.[DOI]
-
279. Zhao LP, Wang HJ, Hu D, Hu JH, Guan ZR, Yu LH, et al. Β-elemene induced ferroptosis via TFEB-mediated GPX4 degradation in EGFR wide-type non-small cell lung cancer. J Adv Res. 2024;62:257-272.[DOI]
-
280. Liu S, Zhang HL, Li J, Ye ZP, Du T, Li LC, et al. Tubastatin A potently inhibits GPX4 activity to potentiate cancer radiotherapy through boosting ferroptosis. Redox Biol. 2023;62:102677.[DOI]
-
281. Randolph JT, O’Connor MJ, Han F, Hutchins CW, Siu YA, Cho M, et al. Discovery of a potent chloroacetamide GPX4 inhibitor with bioavailability to enable target engagement in mice, a potential tool compound for inducing ferroptosis in vivo. J Med Chem. 2023;66(6):3852-3865.[DOI]
-
282. Rodencal J, Kim N, He A, Li VL, Lange M, He J, et al. Sensitization of cancer cells to ferroptosis coincident with cell cycle arrest. Cell Chem Biol. 2024;31(2):234-248.e213.[DOI]
-
283. Chen T, Leng J, Tan J, Zhao Y, Xie S, Zhao S, et al. Discovery of novel potent covalent glutathione peroxidase 4 inhibitors as highly selective ferroptosis inducers for the treatment of triple-negative breast cancer. J Med Chem. 2023;66(14):10036-10059.[DOI]
-
284. Zhang Y, Tan H, Daniels JD, Zandkarimi F, Liu H, Brown LM, et al. Imidazole ketone erastin induces ferroptosis and slows tumor growth in a mouse lymphoma model. Cell Chem Biol. 2019;26(5):623-633.e629.[DOI]
-
285. Sato M, Onuma K, Domon M, Hasegawa S, Suzuki A, Kusumi R, et al. Loss of the cystine/glutamate antiporter in melanoma abrogates tumor metastasis and markedly increases survival rates of mice. Int J Cancer. 2020;147(11):3224-3235.[DOI]
-
286. Badgley MA, Kremer DM, Maurer HC, DelGiorno KE, Lee HJ, Purohit V, et al. Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science. 2020;368(6486):85-89.[DOI]
-
287. Gout PW, Buckley AR, Simms CR, Bruchovsky N. Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the xc- cystine transporter: A new action for an old drug. Leukemia. 2001;15(10):1633-1640.[DOI]
-
288. NNakamura T, Hipp C, Santos Dias Mourão A, Borggräfe J, Aldrovandi M, Henkelmann B, et al. Phase separation of FSP1 promotes ferroptosis. Nature. 2023;619(7969):371-377.[DOI]
-
289. Hendricks JM, Doubravsky CE, Wehri E, Li Z, Roberts MA, Deol KK, et al. Identification of structurally diverse FSP1 inhibitors that sensitize cancer cells to ferroptosis. Cell Chem Biol. 2023;30(9):1090-1103.e1097.[DOI]
-
290. Liu W, Xie X, Zong H, Li Y, Ding Y, Liu Z, et al. Design, synthesis and biological evaluation of triazolothiadiazole derivatives as FSP1 inhibitors for sensitizing cancer cells to ferroptosis. Eur J Med Chem. 2025;293:117737.[DOI]
-
291. Bruedigam C, Porter AH, Song A, Vroeg in de Wei G, Stoll T, Straube J, et al. Imetelstat-mediated alterations in fatty acid metabolism to induce ferroptosis as a therapeutic strategy for acute myeloid leukemia. Nat Cancer. 2024;5(1):47-65.[DOI]
-
292. Yang Z, Yin S, Yi Z, Li Y, Wu S, Xu P, et al. Erigoster B targeting DECR1 induces ferroptosis of breast cancer cells via promoting phosphatidylcholine/arachidonic acid metabolism. NPJ Precis Oncol. 2025;9(1):162.[DOI]
-
293. Liu Y, Sun Q, Guo J, Yan L, Yan Y, Gong Y, et al. Dual ferroptosis induction in N2-TANs and TNBC cells via FTH1 targeting: A therapeutic strategy for triple-negative breast cancer. Cell Rep Med. 2025;6(1):101915.[DOI]
-
294. Jiang J, Yang L, Xie Q, Liu X, Jiang J, Zhang J, et al. Synthetic vectors for activating the driving axis of ferroptosis. Nat Commun. 2024;15(1):7923.[DOI]
-
295. Lei G, Sun M, Cheng J, Ye R, Lu Z, Horbath A, et al. Radiotherapy promotes cuproptosis and synergizes with cuproptosis inducers to overcome tumor radioresistance. Cancer Cell. 2025;43(6):1076-1092.e1075.[DOI]
-
296. Wu Y, Song Y, Wang R, Wang T. Molecular mechanisms of tumor resistance to radiotherapy. Mol Cancer. 2023;22(1):96.[DOI]
-
297. .Lang X, Green MD, Wang W, Yu J, Choi JE, Jiang L, et al. Radiotherapy and immunotherapy promote tumoral lipid oxidation and ferroptosis via synergistic repression of SLC7A11. Cancer Discov. 2019;9(12):1673-1685.[DOI]
-
298. Afzal Z, Gull AR. The uncertainty of Cholera epidemic. J Pak Med Assoc. 2022;73(1):210.[DOI]
-
299. van de Mheen L, Schuit E, Liem SMS, Lim AC, Bekedam DJ, Goossens SMTA, et al. Second-trimester cervical length as risk indicator for Cesarean delivery in women with twin pregnancy. Ultrasound Obstet Gynecol. 2015;46(5):579-584.[DOI]
-
300. Wan C, Sun Y, Tian Y, Lu L, Dai X, Meng J, et al. Irradiated tumor cell-derived microparticles mediate tumor eradication via cell killing and immune reprogramming. Sci Adv. 2020;6(13):eaay9789.[DOI]
-
301. De Luca A, Flammini G, Vittorini P, Muselli M, Mastrantonio R, Cipollone C, et al. Impact of the healthcare reorganization of the Local Health Authority services in Rieti (Italy) during the SARS-CoV-2 pandemic. Ann Ig. 2023;35(4):441-453.[DOI]
-
302. Teixeira N, Nabais P, de Freitas V, Lopes JA, Melo MJ. In-depth phenolic characterization of iron gall inks by deconstructing representative Iberian recipes. Sci Rep. 2021;11(1):8811.[DOI]
-
303. Ye LF, Chaudhary KR, Zandkarimi F, Harken AD, Kinslow CJ, Upadhyayula PS, et al. Radiation-induced lipid peroxidation triggers ferroptosis and synergizes with ferroptosis inducer. ACS Chem Biol. 2020;15(2):469-484.[DOI]
-
304. Lei G, Zhang Y, Koppula P, Liu X, Zhang J, Lin SH, et al. The role of ferroptosis in ionizing radiation-induced cell death and tumor suppression. Cell Res. 2020;30(2):146-162.[DOI]
-
305. Cao Y, Li J, Chen Y, Wang Y, Liu Z, Huang L, et al. Correction: Monounsaturated fatty acids promote cancer radioresistance by inhibiting ferroptosis through ACSL3. Cell Death Dis. 2025;16(1):430.[DOI]
-
306. Zhang H, Ma J, Hou C, Luo X, Zhu S, Peng Y, et al. A ROS-mediated oxidation-O-GlcNAcylation cascade governs ferroptosis. Nat Cell Biol. 2025;27(8):1288-1300.[DOI]
-
307. Ham S, Kim HJ, Shin N, Hwang JH, Oh SJ, Park JY, et al. Continuous production of gamma aminobutyric acid by engineered and immobilized Escherichia coli whole-cells in a small-scale reactor system. Enzyme Microb Technol. 2023;168:110258.[DOI]
-
308. Prajapati R, Ostwal V, Srinivas S, Engineer R, Bhargava P, Saklani A, et al. Modified FOLFIRINOX (mFOLFIRINOX) as neoadjuvant therapy and ‘salvage’ in patients with high risk locally advanced rectal cancers - tolerance and early outcomes. J Cancer Res Ther. 2024;20(1):199-203.[DOI]
-
309. Sundaresan A, Nuchikkat S, Alee KS. On chip random lasing performance of the acceptor dye in a specially designed linear and zig zag array of microdroplets with intrinsic disorder. Sci Rep. 2022;12(1):3939.[DOI]
-
310. Zhang C, Liu X, Jin S, Chen Y, Guo R. Ferroptosis in cancer therapy: A novel approach to reversing drug resistance. Mol Cancer. 2022;21(1):47.[DOI]
-
311. Deng J, Lin X, Qin J, Li Q, Zhang Y, Zhang Q, et al. SPTBN2 suppresses ferroptosis in NSCLC cells by facilitating SLC7A11 membrane trafficking and localization. Redox Biol. 2024;70:103039.[DOI]
-
312. Han L, Li L, Wu G. Induction of ferroptosis by carnosic acid-mediated inactivation of Nrf2/HO-1 potentiates cisplatin responsiveness in OSCC cells. Mol Cell Probes. 2022;64:101821.[DOI]
-
313. Roh JL, Kim EH, Jang HJ, Park JY, Shin D. Induction of ferroptotic cell death for overcoming cisplatin resistance of head and neck cancer. Cancer Lett. 2016;381(1):96-103.[DOI]
-
314. Fu D, Wang C, Yu L, Yu R. Induction of ferroptosis by ATF3 elevation alleviates cisplatin resistance in gastric cancer by restraining Nrf2/Keap1/xCT signaling. Cell Mol Biol Lett. 2021;26(1):26.[DOI]
-
315. Yu J, Zhong B, Zhao L, Hou Y, Ai N, Lu JJ, et al. Fighting drug-resistant lung cancer by induction of NAD(P)H:quinone oxidoreductase 1 (NQO1)-mediated ferroptosis. Drug Resist Updat. 2023;70:100977.[DOI]
-
316. Mei J, Tian HX, Zhang XY, Chen YS, Wang LY, Zhang Z, et al. Heme oxygenase 1 (HO-1) is a drug target for reversing cisplatin resistance in non-small cell lung cancer. J Adv Res. 2025.[DOI]
-
317. Zeng K, Li W, Wang Y, Zhang Z, Zhang L, Zhang W, et al. Inhibition of CDK1 overcomes oxaliplatin resistance by regulating ACSL4-mediated ferroptosis in colorectal cancer. Adv Sci. 2023;10(25):2301088. [DOI:https://doi.org/10.1002/advs.202301088][DOI]
-
318. Yang C, Zhang Y, Lin S, Liu Y, Li W. Suppressing the KIF20A/NUAK1/Nrf2/GPX4 signaling pathway induces ferroptosis and enhances the sensitivity of colorectal cancer to oxaliplatin. Aging. 2021;13(10):13515.[DOI]
-
319. Zhan M, Ding Y, Huang S, Liu Y, Xiao J, Yu H, et al. Lysyl oxidase-like 3 restrains mitochondrial ferroptosis to promote liver cancer chemoresistance by stabilizing dihydroorotate dehydrogenase. Nat Commun. 2023;14(1):3123.[DOI]
-
320. He H, Liang L, Huang J, Jiang S, Liu Y, Sun X, et al. KIF20A is associated with clinical prognosis and synergistic effect of gemcitabine combined with ferroptosis inducer in lung adenocarcinoma. Front Pharmacol. 2022;13:1007429.[DOI]
-
321. Zhu S, Zhang Q, Sun X, Zeh HJ, III , Lotze MT, Kang R, et al. HSPA5 regulates ferroptotic cell death in cancer cells. Cancer Res. 2017;77(8):2064-2077.[DOI]
-
322. Chen ZW, Shan JJ, Chen M, Wu Z, Zhao YM, Zhu HX, et al. Targeting GPX4 to induce ferroptosis overcomes chemoresistance mediated by the PAX8-AS1/GPX4 axis in intrahepatic cholangiocarcinoma. Adv Sci. 2025;12(30):e01042.[DOI]
-
323. Hu Q, Jiang C, Qin Y, Li B, Wang J, Wang T, et al. Pentose phosphate recycling driven by Gli1 contributes to chemotherapy resistance in cancer cells. Cancer Lett. 2025;618:217633.[DOI]
-
324. Qi R, Bai Y, Li K, Liu N, Xu Y, Dal E, et al. Cancer-associated fibroblasts suppress ferroptosis and induce gemcitabine resistance in pancreatic cancer cells by secreting exosome-derived ACSL4-targeting miRNAs. Drug Resist Updat. 2023;68:100960.[DOI]
-
325. Qu X, Liu B, Wang L, Liu L, Zhao W, Liu C, et al. Loss of cancer-associated fibroblast-derived exosomal DACT3-AS1 promotes malignant transformation and ferroptosis-mediated oxaliplatin resistance in gastric cancer. Drug Resist Updat. 2023;68:100936.[DOI]
-
326. Chen TC, Chuang JY, Ko CY, Kao TJ, Yang PY, Yu CH, et al. AR ubiquitination induced by the curcumin analog suppresses growth of temozolomide-resistant glioblastoma through disrupting GPX4-Mediated redox homeostasis. Redox Biol. 2020;30:101413.[DOI]
-
327. Mansuer M, Zhou L, Wang C, Gao L, Jiang Y. Erianin induces ferroptosis in GSCs via REST/LRSAM1 mediated SLC40A1 ubiquitination to overcome TMZ resistance. Cell Death Dis. 2024;15(7):522.[DOI]
-
328. Ouyang S, Li H, Lou L, Huang Q, Zhang Z, Mo J, et al. Inhibition of STAT3-ferroptosis negative regulatory axis suppresses tumor growth and alleviates chemoresistance in gastric cancer. Redox Biol. 2022;52:102317.[DOI]
-
329. Zhang X, Fang Y, Rong D, Li J, Li Z, Qiu H, et al. A novel taxane SB-T-101141 triggers a noncanonical ferroptosis to overcome Paclitaxel resistance of breast cancer via iron homeostasis-related KHSRP. Cell Death Dis. 2025;16(1):403.[DOI]
-
330. Zhou HH, Chen X, Cai LY, Nan XW, Chen JH, Chen XX, et al. Erastin reverses ABCB1-mediated docetaxel resistance in ovarian cancer. Front Oncol. 2019;9:1398.[DOI]
-
331. Sun X, Niu X, Chen R, He W, Chen D, Kang R, et al. Metallothionein-1G facilitates sorafenib resistance through inhibition of ferroptosis. Hepatology. 2016;64(2):488-500.[DOI]
-
332. Chen Y, Li L, Lan J, Cui Y, Rao X, Zhao J, et al. CRISPR screens uncover protective effect of PSTK as a regulator of chemotherapy-induced ferroptosis in hepatocellular carcinoma. Mol Cancer. 2022;21(1):11.[DOI]
-
333. Gao R, Kalathur RKR, Coto‐Llerena M, Ercan C, Buechel D, Shuang S, et al. YAP/TAZ and ATF4 drive resistance to Sorafenib in hepatocellular carcinoma by preventing ferroptosis. EMBO Mol Med. 2021;13(12):EMMM202114351.[DOI]
-
334. Ren R, Chen Y, Zhou Y, Shen L, Chen Y, Lei J, et al. STIM1 promotes acquired resistance to sorafenib by attenuating ferroptosis in hepatocellular carcinoma. Genes Dis. 2024;11(6):101281.[DOI]
-
335. Ye S, Chen J, Zheng Y, He M, Zhang Y, Cheng Y, et al. Targeting USP18 overcomes acquired resistance in hepatocellular carcinoma by regulating NCOA4 deISGylation and ferroptosis. Cell Death Dis. 2025;16(1):448.[DOI]
-
336. Luo AL, Zheng WY, Zhang Q, Yuan Y, Li MQ, Du K, et al. COPS5 triggers ferroptosis defense by stabilizing MK2 in hepatocellular carcinoma. Adv Sci. 2025;12(22):2416360.[DOI]
-
337. Mu M, Huang CX, Qu C, Li PL, Wu XN, Yao W, et al. Targeting ferroptosis-elicited inflammation suppresses hepatocellular carcinoma metastasis and enhances sorafenib efficacy Cancer Res. 2024;84(6):841-854.[DOI]
-
338. Zheng J, Sato M, Mishima E, Sato H, Proneth B, Conrad M Sorafenib fails to trigger ferroptosis across a wide range of cancer cell lines. Cell Death Dis. 2021;12(7):698.[DOI]
-
339. Hallin J, Engstrom LD, Hargis L, Calinisan A, Aranda R, Briere DM, et al. The KRASG12C inhibitor MRTX849 provides insight toward therapeutic susceptibility of KRAS-mutant cancers in mouse models and patients. Cancer Discov. 2020;10:54-71.[DOI]
-
340. Wang K, Zhang X, Fan Y, Zhou L, Duan Y, Li S, et al. Reactivation of MAPK-SOX2 pathway confers ferroptosis sensitivity in KRASG12C inhibitor resistant tumors. Redox Biol. 2024;78:103419.[DOI]
-
341. Chen P, Li X, Zhang R, Liu S, Xiang Y, Zhang M, et al. Combinative treatment of beta-elemene and cetuximab is sensitive to KRAS mutant colorectal cancer cells by inducing ferroptosis and inhibiting epithelial-mesenchymal transformation. Theranostics. 2020;10(11):5107-5119.[DOI]
-
342. Bian Y, Shan G, Bi G, Xu Z, Liang J, Yan Y, et al. Targeting polyamine metabolism and ferroptosis enhances the efficacy of KRAS-targeted therapy depending on KEAP1 status. Nat Commun. 2025;16(1):9923.[DOI]
-
343. Konishi H, Haga Y, Lin Y, Tsujino H, Higashisaka K, Tsutsumi Y. Osimertinib-tolerant lung cancer cells are susceptible to ferroptosis. Biochem Biophys Res Commun. 2023;641:116-122.[DOI]
-
344. Zhang T, Sun B, Zhong C, Xu K, Wang Z, Hofman P, et al. Targeting histone deacetylase enhances the therapeutic effect of Erastin-induced ferroptosis in EGFR-activating mutant lung adenocarcinoma. Transl Lung Cancer Res. 2021;10(4):1857-1872.[DOI]
-
345. Ni Y, Liu J, Zeng L, Yang Y, Liu L, Yao M, et al. Natural product manoalide promotes EGFR-TKI sensitivity of lung cancer cells by KRAS-ERK pathway and mitochondrial Ca2+ overload-induced ferroptosis. Front Pharmacol. 2022;13:1109822.[DOI]
-
346. Liu S, Yan S, Zhu J, Lu R, Kang C, Tang K, et al. Combination RSL3 treatment sensitizes ferroptosis- and EGFR-inhibition-resistant HNSCCs to cetuximab. Int J Mol Sci. 2022;23(16):9014.[DOI]
-
347. Hong T, Lei G, Chen X, Li H, Zhang X, Wu N, et al. PARP inhibition promotes ferroptosis via repressing SLC7A11 and synergizes with ferroptosis inducers in BRCA-proficient ovarian cancer. Redox Biol. 2021;42:101928.[DOI]
-
348. Zou Y, Zheng S, Xie X, Ye F, Hu X, Tian Z, et al. N6-methyladenosine regulated FGFR4 attenuates ferroptotic cell death in recalcitrant HER2-positive breast cancer. Nat Commun. 2022;13(1):2672.[DOI]
-
349. Kalbasi A, Ribas A. Tumour-intrinsic resistance to immune checkpoint blockade. Nat Rev Immunol. 2020;20(1):25-39.[DOI]
-
350. Xue Y, Lu F, Chang Z, Li J, Gao Y, Zhou J, et al. Intermittent dietary methionine deprivation facilitates tumoral ferroptosis and synergizes with checkpoint blockade. Nat Commun. 2023;14(1):4758.[DOI]
-
351. Zhou L, Lian G, Zhou T, Cai Z, Yang S, Li W, et al. Palmitoylation of GPX4 via the targetable ZDHHC8 determines ferroptosis sensitivity and antitumor immunity. Nat Cancer. 2025;6(5):768-785.[DOI]
-
352. Han Y, Zhang YY, Pan YQ, Zheng XJ, Liao K, Mo HY, et al. IL-1beta-associated NNT acetylation orchestrates iron-sulfur cluster maintenance and cancer immunotherapy resistance. Mol Cell. 2023;83(11):1887-1902.[DOI]
-
353. Meng J, Yang X, Huang J, Tuo Z, Hu Y, Liao Z, et al. Ferroptosis-enhanced immunotherapy with an injectable dextran-chitosan hydrogel for the treatment of malignant ascites in hepatocellular carcinoma. Adv Sci. 2023;10(20):2300517.[DOI]
-
354. Jiang Z, Lim SO, Yan M, Hsu JL, Yao J, Wei Y, et al. TYRO3 induces anti-PD-1/PD-L1 therapy resistance by limiting innate immunity and tumoral ferroptosis. J Clin Invest. 2021;131(8):e139434.[DOI]
-
355. Lin H, Tison K, Du Y, Kirchhoff P, Kim C, Wang W, et al. Itaconate transporter SLC13A3 impairs tumor immunity via endowing ferroptosis resistance. Cancer Cell. 2024;42(12):2032-2044.[DOI]
-
356. Zheng P, Hu Z, Shen Y, Gu L, Ouyang Y, Duan Y, et al. PSAT1 impairs ferroptosis and reduces immunotherapy efficacy via GPX4 hydroxylation. Nat Chem Biol. 2025;21(9):1420-1432.[DOI]
-
357. Zhu Y, Xiao F, Wang Y, Wang Y, Li J, Zhong D, et al. NINJ1 regulates plasma membrane fragility under mechanical strain. Nature. 2025;644(8078):1088-1096.[DOI]
-
358. Green DR. The coming decade of cell death research: Five riddles. Cell. 2019;177(5):1094-1107.[DOI]
-
359. Dondelinger Y, Priem D, Huyghe J, Delanghe T, Vandenabeele P, Bertrand MJM. NINJ1 is activated by cell swelling to regulate plasma membrane permeabilization during regulated necrosis. Cell Death Dis. 2023;14(11):755.[DOI]
-
360. Van Kessel ATM, Cosa G. Lipid-derived electrophiles inhibit the function of membrane channels during ferroptosis. Proc Natl Acad Sci U.S.A. 2024;121(21):e2317616121.[DOI]
-
361. Yang Z, Su W, Wei X, Qu S, Zhao D, Zhou J, et al. HIF-1alpha drives resistance to ferroptosis in solid tumors by promoting lactate production and activating SLC1A1. Cell Rep. 2023;42(8):112945.[DOI]
Copyright
© The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Publisher’s Note
Science Exploration remains a neutral stance on jurisdictional claims in published
maps
and institutional affiliations. The views expressed in this article are solely those
of
the author(s) and do not reflect the opinions of the Editors or the publisher.
Share And Cite



Science Exploration Style
Zhan B, Lin X, Chen P, Zhang J, Guo Y, Deng R, et al. Targeting ferroptosis pathways in cancer: Emerging molecular targets and therapeutic strategies. Ferroptosis Oxid Stress. 2026;2:202510. https://doi.org/10.70401/fos.2025.0011
Tips
Copy completed.
Submit a Manuscript
Author Instructions
Cite this Article
Article Metrics
0
View
0
Download
Cited
Article Updates

- Abstract
- Keywords
- 1. Introduction
- 2. Molecular Mechanisms of Ferroptosis
- 2. 3.6. Radical-trapping antioxidants (RTAs)
- 3. Ferroptosis-Associated Targets in Cancer
- 4. Challenge and Future Direction
- 5. Mechanisms of Cell Death Execution
- 6. Clinical Applications
- Acknowledgements
- Authors contribution
- Conflicts of interest
- Ethical approval
- Consent to participate
- Consent for publication
- Funding
- Copyright
Science Exploration Style
Zhan B, Lin X, Chen P, Zhang J, Guo Y, Deng R, et al. Targeting ferroptosis pathways in cancer: Emerging molecular targets and therapeutic strategies. Ferroptosis Oxid Stress. 2026;2:202510. https://doi.org/10.70401/fos.2025.0011
copy
Share Link
copy