Cracking the neuronal ferroptosis code: In vitro insights into mechanisms and treatment of stroke
Anna C.J. Kalisvaart
1,2,*

,
Rajiv R. Ratan
1,2,*
*Correspondence to:
Anna C.J. Kalisvaart, Burke Neurological Institute, White Plains, NY 10605, USA.
E-mail: akalisva@ualberta.ca; ank4043@med.cornell.edu
Rajiv R. Ratan, Burke Neurological Institute, White Plains, NY 10605, USA. E-mail: rrr2001@med.cornell.edu
Rajiv R. Ratan, Burke Neurological Institute, White Plains, NY 10605, USA. E-mail: rrr2001@med.cornell.edu
Ferroptosis Oxid Stress. 2026;2:202518. 10.70401/fos.2026.0017
Received: November 16, 2025Accepted: January 28, 2026Published: February 03, 2026
Abstract
Ferroptosis, a non-apoptotic regulated cell death culminating in iron-dependent lipid peroxidation, has rapidly emerged as an additional mechanism of neuronal death after stroke. Yet, despite converging molecular features, such as the collapse of antioxidant systems and oxidation of lipids, the upstream biochemical landscapes that trigger ferroptosis differ across ischemic and hemorrhagic injury. In this review, we examine ferroptosis as an emergent property of redox network failure, where the transcriptional, metabolic, and oxidative circuits that normally sustain neuronal resilience are interrupted. Cells continuously operate within a spectrum of oxidative eustress and distress, balancing reactive species that serve as physiological signals and limiting their ability to inflict irreversible damage. Within this continuum, eustress supports adaptive redox signaling, metabolic coupling, and repair, whereas distress reflects exhaustion of reducing power and a shift toward self-propagative oxidation. Drawing on evidence from our lab and others, we explore how distinct stress inputs drive neurons and glia along this continuum toward ferroptotic collapse following each stroke subtype; much of this work originated within reductionist in vitro ferroptotic models and has since been extended to in vivo models. Understanding ferroptosis after stroke not only as a graded failure of redox homeostasis but also an evolutionarily adapted mechanism to couple neuronal injury to the reparative immune response is essential. Effective neuroprotective strategies will therefore depend on identifying and targeting the context-specific triggers and temporal windows that define each stroke subtype.
Keywords
Ischemic stroke, intracerebral hemorrhage, ferroptosis, primary cortical culture, neuroprotection, oxidative stress
1. Introduction
Stroke remains a leading cause of death and long-term disability worldwide[1], yet effective neuroprotective therapies remain elusive. Stroke can be broadly categorized into ischemic stroke and hemorrhagic stroke. Focal ischemia is caused by a vascular occlusion that restricts blood flow and oxygen delivery to downstream brain tissue[2], while intracerebral hemorrhage (ICH) occurs due to extravasation of blood into the brain parenchyma from a ruptured cerebral arteriole[3]. Of the 12 million new strokes that occur globally each year, ~65% are ischemic, ~29% are ICH, and the remainder are subarachnoid hemorrhages, which are not addressed here[1]. While reperfusion strategies have transformed outcomes in a subset of ischemic stroke patients, they remain time-limited[2]. Additionally, the abrupt restoration of blood flow can itself precipitate reperfusion injury, amplifying oxidative and inflammatory damage[2]. Most ICH patients can receive only supportive medical management[3]. While recent trials of minimally invasive hematoma evacuation have shown benefit, this has largely been confined to lobar ICH, representing a minority of cases[4]. In both stroke subtypes, the molecular processes that dictate whether neurons survive or degenerate remain uncontrolled, leading to progressive secondary injury within penumbral or perihematomal tissue.
Among these molecular processes, ferroptosis has emerged as a compelling mechanism linking dyshomeostasis of reactive lipids to cellular degeneration after stroke. First characterized by Dixon et al. in 2012[5], ferroptosis is a regulated form of cell death driven by a failure of cellular antioxidant reserves, resulting in iron-dependent lipid peroxidation and terminal membrane failure[6]. While ischemic and hemorrhagic stroke engage ferroptotic pathways in both animal models and patients[7-14], each does so via distinct upstream mechanisms. During an ischemic stroke, deprivation of oxygen and glucose halts mitochondrial respiration and antioxidant regeneration, predisposing penumbral neurons to oxidative injury upon reperfusion[15]. By contrast, ICH results in oxidation of extravasated hemoglobin from lysed red blood cells, liberating iron-containing heme species that ultimately catalyze neuronal lipid peroxidation within hematomal and perihematomal tissue[16,17].
Despite these diverging biochemical environments, the occurrence of ferroptotic death in both ischemia and ICH reflects a shared failure of dynamic redox homeostasis, in which neurons cannot adequately adapt to insult-specific stressors. Neurons continuously balance oxidation and reduction reactions to sustain their function and plasticity, a dynamic homeostasis maintained through coordinated metabolic and transcriptional programs[18,19]. Stroke pathology overwhelms these homeostatic responses, rendering them insufficient, mistimed, or misdirected. This allows oxidative species to accumulate, shifting their normally modulatory role into a lethal force[20] capable of driving lipid peroxidation and membrane failure. Effective therapeutic strategies will require us to understand how neurons attempt to adapt to insult-specific conditions, why these adaptations fail, and how this failure funnels diverse upstream stressors into ferroptotic death.
Our investigations into the mechanisms underlying neuronal ferroptosis in vitro have demonstrated consistent translational relevance to in vivo stroke models. We employ two in vitro models of ferroptotic neuronal death (Figure 1), one of which originated at Johns Hopkins in the late 1980s through work done by Murphy, Coyle, and colleagues[21]. In this review, we discuss how these in vitro models of ferroptosis have advanced our understanding of therapeutic strategies to mitigate neuronal death after ischemic and hemorrhagic stroke.
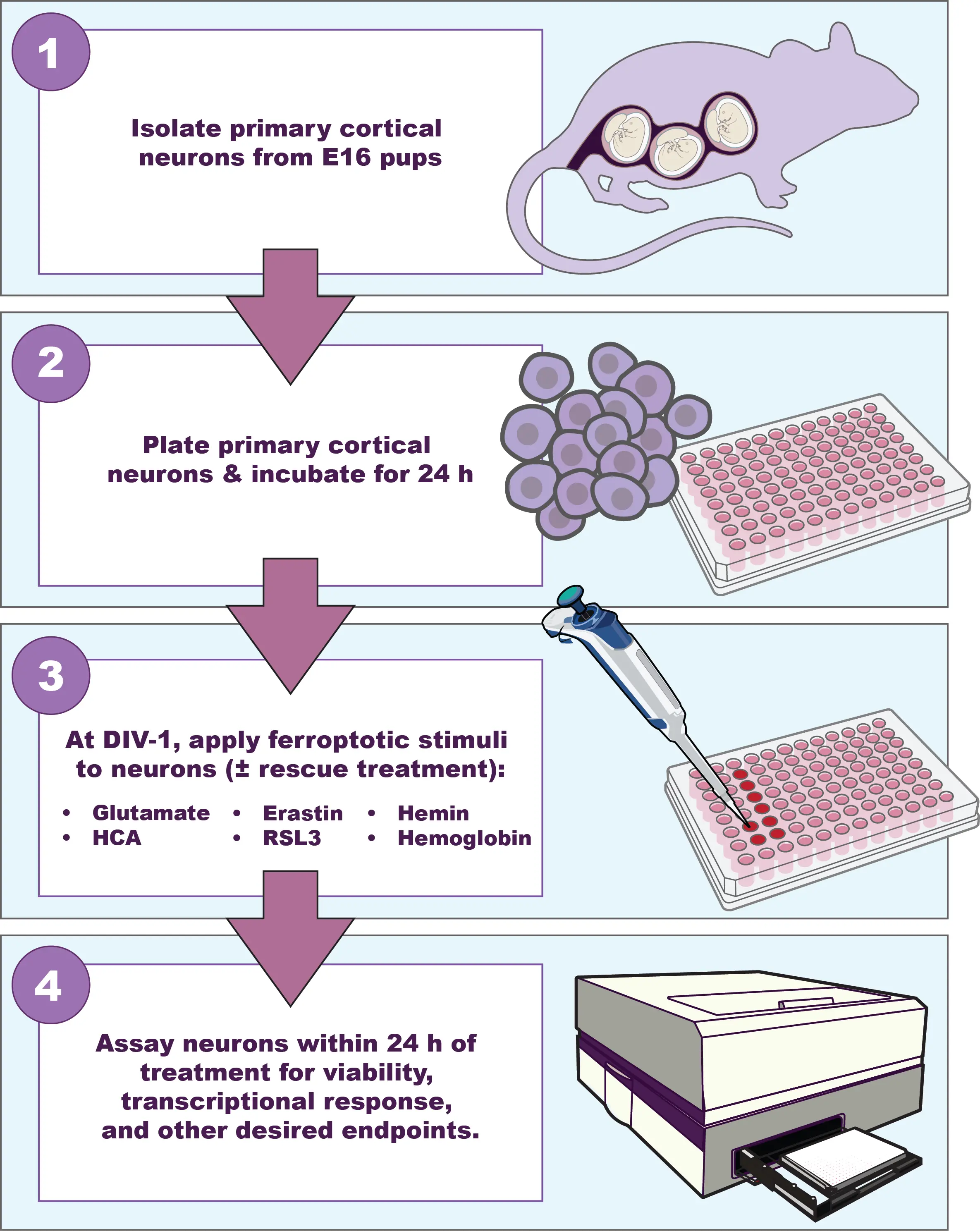
Figure 1. To model ferroptosis in neurons following ischemic or hemorrhagic stroke, our laboratory isolates primary cortical neurons from E16 rat pups, which are incubated for at least 24 hours. After DIV-1, immature neurons are treated with glutamate, HCA, erastin, or RSL3 to model ischemic stroke. Alternatively, cultures are treated with hemin or hemoglobin to model hemorrhagic stroke. Neurons are assayed within 24 hours of treatment for viability, transcriptional responses, and other endpoints. DIV: days in vitro; HCA: homocysteic acid; RSL3: RAS-selective lethal 3.
2. The Emergence of Ferroptosis: Lessons from In Vitro Models
Glutamate is well established as a neurotoxin capable of inducing neuronal death through receptor-mediated excitotoxicity, a framework that has guided decades of neuroprotection research in stroke[22]. However, many candidate therapies have failed in clinical translation[22].
The reasons for these shortcomings are multifactorial, but a key limitation of excitotoxicity-based culture models is their rapid progression: injury occurs so swiftly[22] that causative events cannot be easily distinguished from secondary consequences. Other oxidative stress-based models relevant to stroke often rely on the exogenous application of non-physiological concentrations of oxidants (e.g., H2O2)[23,24], further constraining their translational relevance.
In the late 1980s, Murphy and Coyle observed that glutamate-induced cytotoxicity in the neuroblastoma-retinal hybrid cell line N18-RE-105 could be modulated by cystine availability in the medium[21]. Like immature cortical neurons, N18-RE-105 cells lack N-methyl-D-aspartate (NMDA) receptors, revealing a receptor-independent mechanism of glutamate cytotoxicity[21,25]. This and subsequent work demonstrated that excess extracellular glutamate can kill neurons by blocking the cystine/glutamate antiporter system Xc-, depriving cells of cystine required for glutathione (GSH) synthesis[21,25,26]. As a key co-factor for antioxidant enzymes such as glutathione peroxidase-4 (GPX4; the only known selenoenzyme that can detoxify lipid peroxides), GSH depletion results in progressive lipid peroxidation and neuronal death[26]. In 2001, Tan et al. defined this as a distinct form of cell death termed “oxytosis”[27]. Unlike apoptosis, this NMDA receptor-independent oxidative death proceeds slowly (over the course of ~24 hours), features necrotic membrane rupture rather than caspase activation, and is exacerbated, not rescued, by growth factor withdrawal[28]. Instead of an abrupt execution phase, neuronal demise is driven by a progressive erosion of redox balance, with GSH depletion reaching a steady state by ~6 hours. This delay between the onset of death and terminal execution is a defining ferroptotic feature that reflects the gradual collapse of antioxidant capacity over time.
Together, these ideas foreshadowed what would later be recognized as ferroptosis, as defined by Dixon and Stockwell in 2012[5]. While screening for compounds selectively lethal to oncogenic RAS-mutant tumor cells, Stockwell’s group identified erastin, a small molecule that reproduced the same cystine-starvation and glutathione depletion observed in glutamate-induced death by directly inhibiting system Xc-. The same screen revealed RAS-selective lethal 3 (RSL3), which acts downstream of erastin by directly inhibiting GPX4[5]. By introducing pharmacologically tractable tools and a unified biochemical framework, Dixon and Stockwell’s definition of ferroptosis quickly gained widespread acceptance. Together, the discovery of erastin and RSL3 established ferroptosis as a hierarchical process that is initiated by depletion of reducing power and amplified by loss of enzymatic peroxide control.
This conceptual foundation proved essential for translating ferroptosis into models of ischemic and hemorrhagic stroke. Following ischemia, ferroptosis is likely driven by a combination of glutamate-mediated inhibition of system Xc- and bioenergetic stress, as oxygen and glucose deprivation within oligemic or hypo-perfused tissue work in parallel to diminish nicotinamide adenine dinucleotide phosphate (NADPH) and glutathione regeneration, a situation exacerbated by reperfusion[29]. Therefore, application of glutamate, erastin, or RSL3 can be used to model ferroptosis following ischemia in vitro by exhausting reducing capacity through cystine starvation or GPX4 inhibition (Figure 1). In contrast, ICH introduces an exogenous source of catalytic stress. The rupture of small arterioles floods the parenchyma with erythrocytes, whose lysis releases hemoglobin and hemin (the oxidized form of heme)[30]. Both promote ferroptosis through iron-catalyzed lipid peroxidation and consumption of GSH and NADPH[30,31]. Hemin intercalates into lipid membranes and undergoes redox cycling, while hemoglobin degradation liberates ferrous iron, expanding the labile iron pool and overwhelming antioxidant defenses[30,31]. Accordingly, hemin and hemoglobin can be applied to primary neurons in vitro to model ferroptosis after ICH (Figure 1).
Together, the conceptual evolution from glutamate-induced cytotoxicity to erastin, RSL3, and finally to hemin and hemoglobin mirrors growing recognition of ferroptosis as a spectrum of redox network collapse induced by heterogeneous upstream stimuli.
3. Redox Networks as Determinants of Ferroptotic Susceptibility
By coupling energy production to redox signaling, cells are able to sense and adapt to fluctuations in oxygen, nutrients, and metabolic status. In neurons and other aerobic cells, these redox signals arise primarily from the mitochondrial electron transport chain, where a small fraction of electrons reduce oxygen to superoxide during oxidative phosphorylation. Additional oxidants come from regulated enzyme systems (e.g., NADPH oxidases, nitric oxide synthases, and peroxisomal metabolism), creating a baseline “redox tone” that is buffered via thiol metabolism (e.g., glutathione and thioredoxin systems)[18,19]. The reversible oxidation of cysteine and thiol-based switches within cytosolic and nuclear proteins transduce these redox signals, coordinating gene expression, protein synthesis, antioxidant defence, and energy metabolism via a series of feedback loops- a concept known as dynamic homeostasis[18,32]. Yet these same reactions can become destructive when regulation fails[32].
The dual capacity of oxidants to serve as both signaling molecules/modifiers and as agents of injury is referred to as the “redox paradox”[33]. In biological systems, low-level oxidative activity (e.g., “oxidative eustress”) sustains signaling and cellular plasticity, whereas excessive or unbuffered oxidative activity (e.g., “oxidative distress”) drives molecular damage (Figure 2). As an illustrative example, we have found that low-level H2O2 produced in astrocytes over a defined period (~7 hours) induces protective antioxidant signaling in neighbouring neurons, whereas high-level H2O2 production results in neuronal death[23]. This protective effect is dependent on intracellular generation of H2O2, and correlates with selective, reversible oxidation of protein tyrosine phosphatases[23].
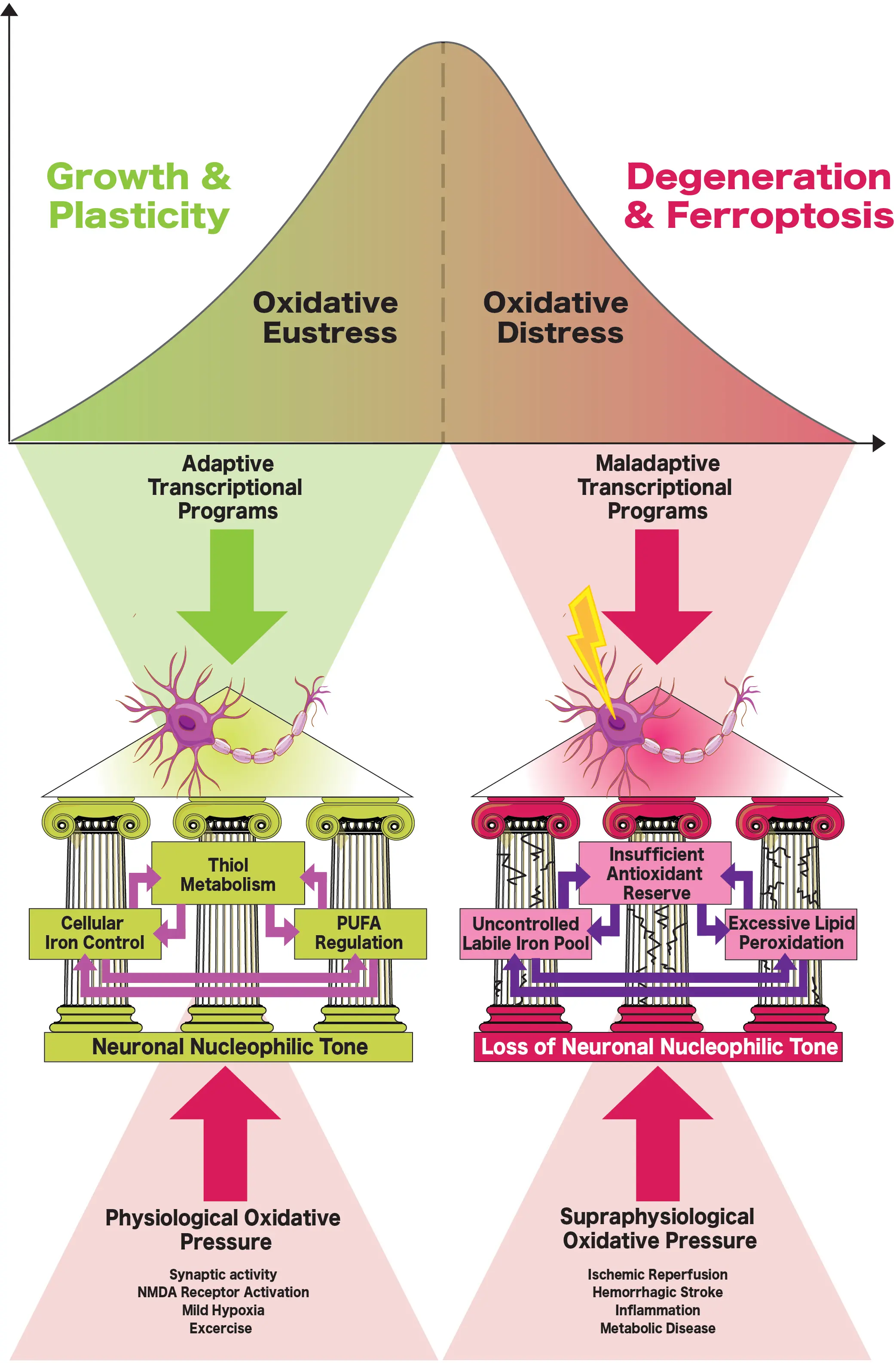
Figure 2. In order to balance physiological oxidative pressure and maintain growth and plasticity (e.g., oxidative eustress), the “three pillars” of oxidative metabolism must remain intact: cellular iron control, thiol metabolism (e.g., cysteine and GSH), and regulation of PUFAs. Loss of any one pillar under supraphysiological oxidative pressure (such as following stroke) disrupts regulation of the others, resulting in maladaptive transcriptional programs that sway neurons towards uncontrolled oxidative distress, and eventually, cellular degeneration and ferroptosis. Following ischemic stroke, thiol metabolism is the first “lost pillar”, as energetic deficits impair GSH synthesis and recycling, which in turn disrupts labile iron control and allows lipid peroxidation to proceed unchecked. In hemorrhagic stroke, the first “lost pillar” is cellular iron control, as excessive iron from blood breakdown products overwhelms antioxidant defenses, permitting runaway lipid peroxidation. GSH: glutathione; PUFAs: polyunsaturated fatty acids.
Whether a cell experiences oxidative eustress or distress depends not only on absolute oxidant levels, but also on the source, duration, and pre-existing cellular state[34]. Resistance to oxidative pressure relies on how electrons are distributed and recycled among competing reactions. This redox economy is sustained by a network of reducing systems that together define the cell’s nucleophilic tone (e.g. the collective ability to donate electrons and maintain macromolecules in a reduced functional state). In this manner, ferroptosis can be understood as a systemic failure of three different “pillars” of metabolism (Figure 2), as described by Bayír and colleagues: iron control, the cysteine-glutathione axis (e.g., thiol metabolism), and regulation of polyunsaturated phospholipids (PUFAs)[35]. Normally, these systems operate together to sustain redox homeostasis: iron shuttles electrons, the cysteine-glutathione axis buffers them, and phospholipids act as both substrates and sensors of oxidative activity[35]. The integrity of these pillars feeds forward to transcriptional programs, as redox-sensitive signaling translates metabolic cues into gene expression and post-translational modifications that reinforce antioxidant defences. When this coordination is lost, adaptive signaling (e.g., oxidative eustress) collapses into self-propagating oxidative distress, maladaptive transcriptional responses emerge, and neurons are primed for ferroptotic death (Figure 2).
To dissect how this integrated neuronal redox network fails following stroke, our laboratory has examined the actions of canonical ferroptosis inhibitors that stabilize discrete control points[14,17]. Deferoxamine (DFO) limits iron-catalyzed radical formation and preserves NADPH-dependent buffering, while N-acetylcysteine (NAC) replenishes cystine-derived thiols and supports GSH synthesis. Radical trapping antioxidants (RTAs) such as vitamin E and ferrostatin-1 intercept lipid radicals to preserve membrane integrity. Finally, the MEK-ERK inhibitor U0126 reduces the activity of cMyc and transglutaminases, regulating iron import genes and epigenetically modulating homeostatic gene programs. Building on the mechanisms of these canonical inhibitors, we have explored additional strategies to stabilize these key nodes within the redox network and refine potential therapeutic targets. Together, this work has made it clear that neuronal ferroptotic death arises not from a single cellular event, but from the coordinated collapse of numerous different metabolic and transcriptional defences. Understanding these vulnerabilities will provide a roadmap for interventions aimed at restoring redox balance and protecting neurons after stroke.
3.1 Iron chelation and catalytic control
By definition, ferroptosis is iron-dependent, suppressed by structurally diverse iron chelators such as DFO[5]. To understand why iron plays such a decisive role, it must be viewed within its broader biological context. Across nearly all known life forms, the ability of iron to shuttle electrons has made it a cornerstone of metabolism, driving mitochondrial respiration, DNA synthesis, and lipid metabolism. Yet the same redox flexibility that makes iron indispensable also renders it hazardous when unregulated.
Within cells, controlled iron redox cycling supports oxidative eustress: localized, transient oxidation that tunes enzymatic activity, transcription, and metabolic flux. Indeed, these transient fluctuations in labile iron are not inherently harmful to neurons. Iron-mediated signaling facilitates neuronal growth, myelination, structural plasticity, synaptic plasticity, and a myriad of other physiological processes through transient modification of redox-sensitive enzymes and transcription factors[36-40]. When iron regulation escapes control, however, the same chemistry drives a shift toward oxidative distress, in which uncontrolled electron transfer drives lipid peroxidation and metabolic failure. Therefore, iron homeostasis is tightly maintained through a coordinated cycle of uptake, utilization, and storage[41]. Within the interstitium of the central nervous system (CNS), ferric iron (Fe3+) is bound by transferrin, which is synthesized by the choroid plexus and circulates within blood[42,43]. The transferrin-iron complex enters cells via transferrin receptor 1 (TfR1)-mediated endocytosis (Figure 3a), where it is reduced to ferrous iron (Fe2+) by STEAP (six-transmembrane epithelial antigen of the prostate) oxidoreductases and released into the cytosol through divalent metal transporter 1 (DMT1)[41]. This Fe2+ replenishes the labile iron pool (LIP), a metabolically active but potentially cytotoxic reservoir supplying iron for heme synthesis, mitochondrial iron-sulfur (Fe-S) cluster assembly, and other essential reactions[41,44,45]. To prevent uncontrolled redox activity, Fe2+ within the cytosol is transiently buffered by poly(rC)-binding protein 1 (PCBP1), a GSH-dependent iron chaperone that delivers Fe2+ safely to ferritin or metalloenzymes (Figure 3a)[46-50].
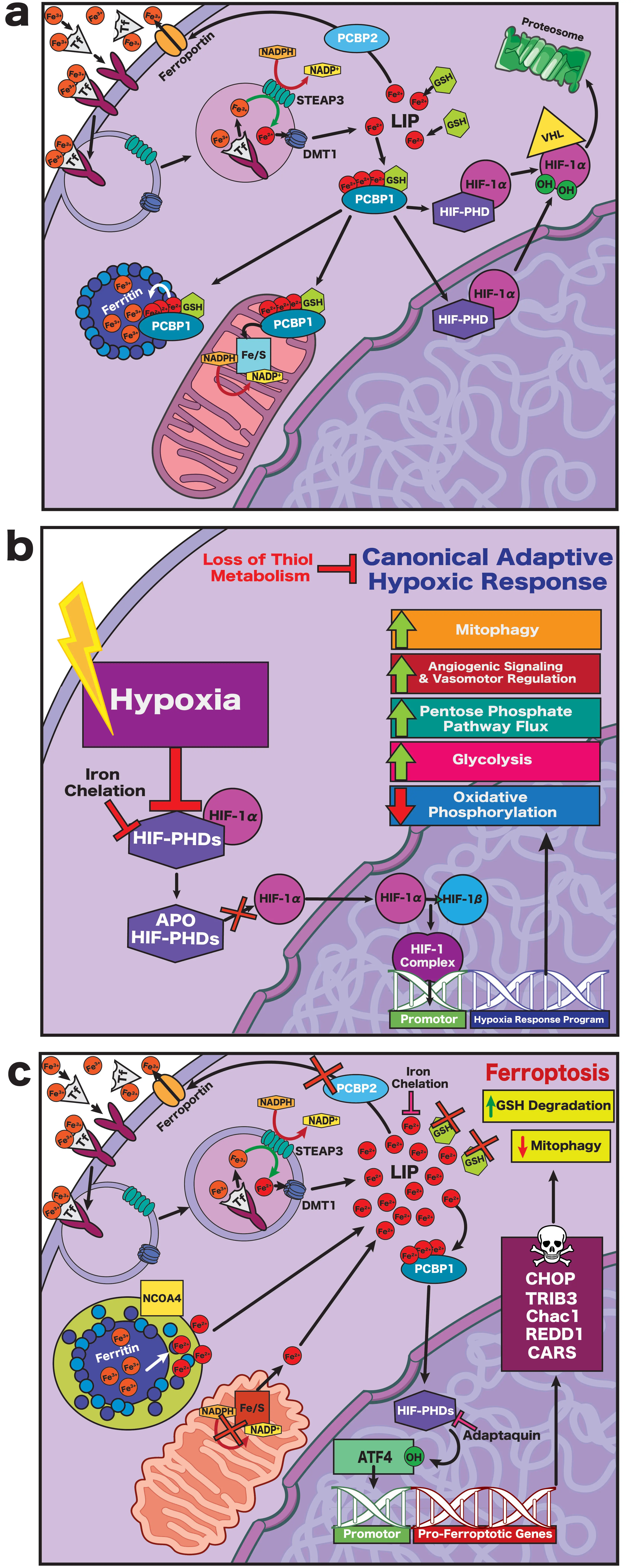
Figure 3. (a) Under normal physiological circumstances, ferrous iron (Fe3+) is imported into neurons via transferrin-mediated endocytosis. Inside the endosome, ferric iron is reduced to ferrous iron (Fe2+) by STEAP3 and exported into the cytosol via DMT1, contributing to the LIP. The LIP is tightly regulated; ferrous iron within the LIP is stabilized by forming a complex with GSH, with excess ferrous iron exported via PCBP2 and ferroportin. Iron chaperone PCBP1 delivers ferrous iron from the LIP either to ferritin for storage, to the mitochondria for iron-sulfur cluster formation, or to metalloenzymes such as the hypoxia-inducible factor. Under normal circumstances, HIF PHDs hydroxylate HIF-1α, tagging it for proteasomal degradation upon ubiquitination by VHL; (b) When neurons undergo hypoxia, the lack of oxygen inactivates HIF PHDs, leaving them unable to hydroxylate HIF-1α. This prevents the degradation of HIF-1α, allowing it to translocate to the nucleus where it binds to HIF-1β to form the HIF-1 complex. The HIF-1 complex binds to hypoxia response elements, promoting expression of a coordinated gene program that allows the cell to adapt to hypoxic conditions- as long as thiol metabolism remains intact. Because the HIF PHDs require iron as a co-factor, iron chelation also permits HIF-1α stabilization; (c) When thiol metabolism is compromised and GSH is depleted, control of the LIP is lost, tipping neurons into oxidative distress. Ferrous iron export is downregulated, and import is upregulated. Ferritinophagy and dissociation of the Fe-S clusters further expands the LIP, loading PCBP1 with ferrous iron and redirecting it to the HIF PHDs. Once loaded with iron within an oxidative environment, the HIF PHDs (likely HIF PHD1) preferentially hydroxylate ATF4, resulting in the transcription of a pro-ferroptotic gene cassette. STEAP3: six-membrane epithelial antigen of prostate 3; DMT1: divalent metal ion transporter 1; LIP: labile iron pool; GSH: glutathione; PCBP2: poly(rc)-binding protein 2; PCBP1: poly(rc)-binding protein 1; HIF PHDs: hypoxia-inducible prolyl hydroxylases; VHL: von Hippel-Lindau tumor suppressor protein; HIF-1α: hypoxia-inducible factor 1α; HIF-1β: hypoxia-inducible factor 1β.
Under physiological conditions, most intracellular iron is tightly sequestered within ferritin, cytochromes, or Fe-S enzymes, maintaining the LIP within a narrow range[51]. When mitochondrial redox balance is preserved, Fe-S clusters remain stable (Figure 3a)[40,52]. However, following ischemia, the impaired electron transport chain limits NADPH regeneration and GSH synthesis[29], creating an oxidizing mitochondrial environment that directly destabilizes Fe-S clusters (Figure 3c)[40]. The loss of Fe-S clusters reduces the activity of associated metalloenzymes, weakening cellular redox buffering[52-54]. In hemorrhagic stroke, heme degradation and ferrireductase activity introduce additional Fe2+ to the LIP[30], fueling mitochondrial oxidative dysfunction and compounding Fe-S cluster instability. As the Fe-S clusters disassemble, Fe2+ is released back into the LIP, expanding the pool of redox-active iron (Figure 3c). Since IRP1 and IRP2 (iron response protein 1/2) sense iron status through Fe-S cluster integrity rather than total iron[55], Fe-S loss is misinterpreted as iron deficiency. Iron uptake is increased via upregulation of TfR1 and DMT1, iron export via ferroportin is blocked, and ferritin storage is suppressed despite an increasing LIP (Figure 3c)[56]. Concurrently, ferritin is targeted for nuclear receptor coactivator 4 (NCOA4)-mediated ferritinophagy, further reinforcing LIP expansion (Figure 3c)[57,58]. This feed-forward sequence aligns with elevated total iron observed within peri-infarct regions of focal ischemia patients on neuroimaging[59,60]. Consistent with this model, proteins associated with Fe-S cluster stability and coordination, such as CDGSH iron sulfur domain 2 (CISD2) decline after ischemia[61,62], and CISD2 overexpression preserves Fe-S integrity and limits ferroptosis in both ischemic and hemorrhagic stroke models[62,63].
Once the LIP exceeds the cell’s buffering capacity, Fe2+ catalyzes Fenton reactions with peroxides, producing hydroxyl radicals and ferryl intermediates that attack membrane lipids (Figure 4a)[35,64]. These reactions are not random; local intracellular iron coordination and redox conditions determine whether oxidation occurs diffusely or at specific sites[34]. Thus, ferroptosis arises not from chance oxidation but through a collapse in iron handling and peroxide metabolism. Only later did such mechanistic understanding clarify what early interventions like DFO had already exploited: the therapeutic potential of stabilizing redox-active iron in the CNS. Originally derived from the microbial siderophore ferrioxamine B, DFO was first used clinically in 1962 to treat thalassemia and later shown to reverse aluminum-induced dialysis encephalopathy[65-67], demonstrating the importance of iron homeostasis within the CNS.
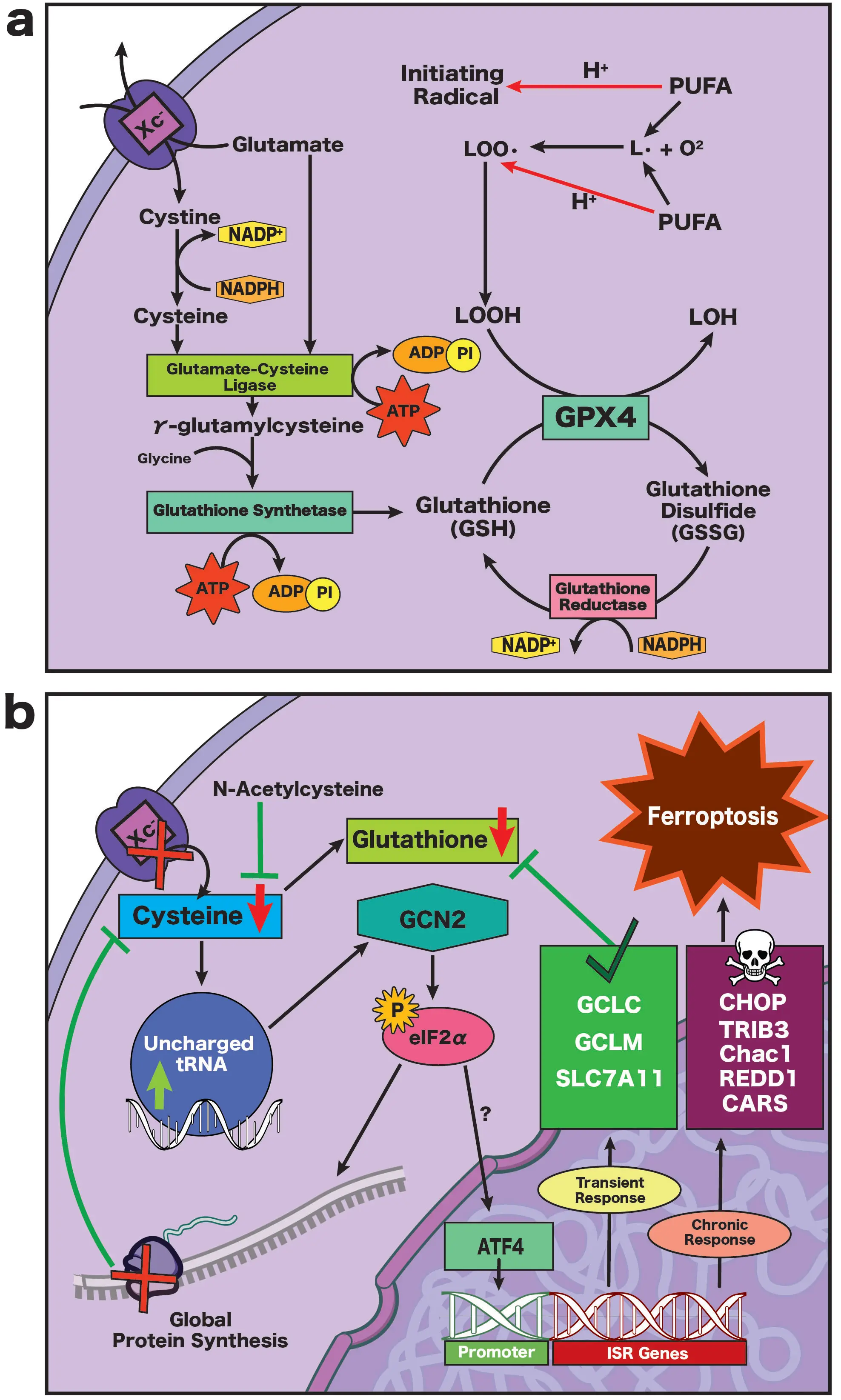
Figure 4. (a) In astrocytes and immature neurons, cystine is imported into the cell in exchange for glutamate via the system Xc- transporter. Cystine is converted to cysteine, which is then converted to γ-glutamylcysteine by glutamate-cysteine ligase. Glutathione synthetase then acts on γ-glutamylcysteine and glycine to produce GSH. GPX4 uses GSH as a reducing agent to neutralize toxic lipid hydroperoxides, forming GSSG. Glutathione reductase then recycles GSSG back into GSH in an NADPH-dependent reaction; (b) Upon depletion of cellular cysteine supply, uncharged tRNA builds up within the neuron, activating GCN2, which in turn activates eIF2. Activation of eIF2α triggers the integrated stress response, globally reducing protein synthesis, and redirects cysteine away from translation towards glutathione production. While the upstream ferroptotic triggers for ATF4 activation in neurons are incompletely understood, they may include eIF2α phosphorylation or association with an unidentified co-factor. Data from other models (e.g., immortalized neuronal cell lines, non-neuronal cells) suggest that transient ATF4 activation is beneficial, resulting in expression of antioxidant gene programs which act to increase cellular cysteine and GSH levels. Chronic ATF4 activation, however, results in pro-ferroptotic transcription and eventual death. Notably, delivering cysteine to the cell in the form of N-acetylcysteine prevents neuronal ferroptosis. GSH: glutathione; GPX4: glutathione peroxidase 4; GSSG: glutathione disulfide; NADPH: nicotinamide adenine dinucleotide phosphate; GCN2: general control nonderepressible 2; eIF2α: eukaryotic initiation factor 2 subunit 1; ATF4: activating transcription factor 4.
Our laboratory first demonstrated that iron chelators such as DFO prevent ferroptotic death in immature cortical neurons not only by quenching hydroxyl radicals or interrupting Fenton chemistry, but also by activating the transcriptional response normally triggered by hypoxia (Figure 3b)[68]. Structurally diverse iron chelators enhanced activation of hypoxia-inducible factor 1α (HIF-1α), a master transcription factor that orchestrates cellular adaptation to low oxygen by inducing genes involved in glycolysis, erythropoiesis, angiogenesis, and antioxidant defense (Figure 3b)[68]. Under normoxic conditions, HIF-1α is continuously synthesized but rapidly degraded: HIF prolyl hydroxylase domain (PHD) enzymes hydroxylate proline residues on HIF-1α in an iron- and 2-oxoglutarate-dependent reaction, marking it for recognition by the von Hippel-Lindau (VHL) E3 ubiquitin ligase (Figure 3a)[69-71]. In turn, VHL ubiquitinates HIF-1α and targets it for proteasomal degradation[69]. This work by Kaelin, Ratcliffe, and Semenza provided the mechanistic foundation for this phenomenon, revealing how these oxygen- and iron-sensing PHDs couple metabolic state to HIF-1α stability (winning them the Nobel Prize in 2019)[69-71]. By chelating ferrous iron and limiting expansion of the LIP, DFO inhibits PHD activity (Figure 3c). This effectively prevents HIF-1α hydroxylation and degradation, thereby mimicking hypoxia and activating a coordinated transcriptional program that can enhance neuronal redox resilience and survival (Figure 3b)[68,70]. Subsequent work from our laboratory revealed that the neuroprotective actions of iron chelators arise from selective inhibition of HIF PHD1. Genetic or pharmacologic suppression of PHD1, but not PHD2 or PHD3, protected neurons from death by glutathione depletion, identifying PHD1 as a key redox-sensitive regulator of metabolic resilience even under normoxic conditions[72].
Yet, as we later found, HIF-1α activation is not uniformly protective in a ferroptotic context. Forced expression of an oxygen-stabilized HIF-1α in a hippocampal neuroblast (HT-22) cell line enhanced resistance to DNA damage and endoplasmic reticulum (ER) stress, but unexpectedly potentiated ferroptotic death, indicating that the outcome of HIF stabilization depends on the nature of the stressor and the ongoing cellular context[73]. This paradox arises when oxidative stress and iron loading convert HIF-1α from an adaptive transcription factor into a pro-death effector by inducing the apoptotic proteins BNIP3 (BCL2/adenovirus E1B 19 kDA protein-interacting protein 3) and p53 upregulated modulator of apoptosis (PUMA)[74]. These genes are canonical HIF targets, as they contain hypoxia-response elements, yet their induction alone is not lethal; a secondary oxidative signal (e.g., GSH depletion) activates their mitochondrial death functions[74]. Importantly, antioxidants or pharmacologic inhibition of HIF PHDs prevents this switch, suggesting that active, iron-loaded PHDs are permissive for HIF to engage its pro-death transcriptional program, whereas apo (iron-free) PHDs favor canonical HIF signaling[74].
Together, these findings posit that the iron-loading state of the HIF PHDs dictates their regulatory control over transcriptional pathways. Under conditions of iron chelation or hypoxia, HIF PHDs remain in their apo (iron-free) form, favouring HIF stabilization (Figure 3b)[24]. In contrast, during normoxic oxidative stress, the expansion of the LIP appears to preferentially load the HIF PHDs (via iron chaperone PCBP1; Figure 3c)[49], redirecting their activity to non-HIF substrates such as activating transcription factor 4 (ATF4)[24]. Normally, ATF4 functions as part of the integrated stress response (ISR), which serves to balance protein synthesis, metabolism, and redox homeostasis when the cell is under stress[75]. Transient activation of the ISR pathway supports adaptive cellular responses to redox stress. However, our model suggests that under sustained oxidative or metabolic stress, iron-loaded HIF PHDs hydroxylate ATF4, enhancing its stability and transcriptional output toward pro-ferroptotic death genes such as C/EBP homologous protein (CHOP), tribbles homolog 3 (TRIB3), cysteinyl-tRNA synthetase 1 (CARS), regulated in development and DNA damage 1 (REDD1), and cation transport regulator-like 1 (CHAC1; Figure 3c)[17,76].
To block this maladaptive loop, our laboratory developed adaptaquin, a brain-penetrant 8-hydroxyquinoline that binds the catalytic iron center of HIF PHDs and inhibits their hydroxylase activity[77,78]. Unlike broad chelators, adaptaquin spares bulk iron metabolism while suppressing ATF4-dependent transcription (Figure 3c)[77]. In primary cortical neurons, adaptaquin potently suppressed ferroptotic death triggered by glutathione depletion, hemin toxicity, or excitotoxic oxidative stress. Protective concentrations (~1 μM) were several-fold lower than those needed to stabilize HIF-1α, and transcriptomic analyses revealed suppression of an ATF4-dependent gene cassette (e.g., CHOP, TRIB3, and CHAC1) rather than activation of canonical HIF targets[77]. These genes likely amplify oxidative injury by promoting degradation of Parkin and degrading intracellular glutathione, respectively, thereby reinforcing redox and mitochondrial collapse[17,76,79]. In vivo, adaptaquin recapitulated the effects of genetic HIF PHD knockdown, improving neuronal survival and behavioral recovery in collagenase- and autologous blood-induced models of intracerebral hemorrhage[77], as well as ischemic stroke (unpublished data, Ratan lab). Post-injury treatment reduced perihematomal degeneration without altering total brain iron or zinc, confirming selective PHD inhibition rather than wholesale metal chelation as the mechanism of protection. Notably, adaptaquin remained effective when delivered up to six hours after hemorrhage onset[77].
By contrast, systemic chelators like DFO, though protective in vitro, exhibit poor brain penetration and narrow safety margins, a consequence of wholesale iron chelation. While numerous preclinical studies have found neuroprotective efficacy with DFO treatment following both focal ischemia and hemorrhage, others found no benefit or evidence of harm[80-88]. Randomized controlled trials in ICH have been similarly mixed; the Phase II i-DEF trial did not meet their efficacy endpoint[89], while the Phase II Hi-DEF trial (e.g., high dose DFO) was terminated early due to evidence of harm[90]. Post-hoc analyses of the i-DEF trial have since indicated that DFO may accelerate recovery trajectories[91,92], but there remains room for therapeutic refinement. Deferiprone has been posited as a more bioavailable alternative to DFO in the treatment of CNS disorders; however, recent clinical findings indicating potential harm in both patients with Alzheimer’s disease and Parkinson’s disease warrant extreme caution[93-95]. Our findings regarding adaptaquin have established targeted inhibition of iron-dependent dioxygenases as a potential alternative to global iron chelation, intercepting maladaptive ATF4 signaling while preserving physiological iron metabolism.
Over the past three decades, this work has collectively illustrated that disturbances in iron handling set the stage for neuronal ferroptosis by transcriptionally altering the cell’s redox balance. When iron-driven reactions diminish glutathione recycling, redox homeostasis falters. In this manner, the boundary between oxidative eustress and distress is defined by the efficiency of cellular thiol metabolism, which plays a major role in buffering the reactivity of iron.
3.2 Thiol metabolism and the cystine-glutathione axis
At the center of cellular redox control is GSH, the most abundant endogenous antioxidant that establishes the baseline redox potential of the cell[96]. GSH is synthesized from glutamate, cysteine, and glycine via glutathione-cysteine ligase catalytic subunit/glutamate-cysteine ligase modifier subunit (GCLC/GCLM) and glutathione synthetase (Figure 4a)[97]. Its core function is to provide reducing equivalents for GPX4, enabling detoxification of lipid hydroperoxides and thereby limiting ferroptotic lipid damage (Figure 4a)[98]. Beyond supporting GPX4, GSH maintains protein cysteine redox states through reversible S-glutathionylation and fuels glutaredoxin-dependent Fe-S cluster assembly and repair[54,99]. Through glutathione S-transferases, GSH also conjugates electrophilic lipid aldehydes formed during membrane peroxidation, preventing secondary oxidative injury[16]. In addition, GSH can directly bind Fe2+ within the labile iron pool, forming a complex that PCBP1 chaperones toward Fe-S cluster synthesis (Figure 4a)[48]. Sustaining a robust GSH pool is metabolically demanding, and highly dependent on NADPH availability. In most cells, NADPH is primarily generated via the pentose phosphate pathway, with cytosolic malic enzyme 1 (ME1) and NADP⁺-dependent isocitrate dehydrogenases (IDH1/2) providing auxiliary sources that couple mitochondrial and cytosolic metabolism to the tricarboxylic acid (TCA) cycle[100].
Neurons and astrocytes, however, differ markedly in how they balance these reactions. Neurons prioritize oxidative phosphorylation and ATP generation to meet the energetic demands of firing action potentials, whereas astrocytes maintain high glycolytic flux and robust pentose phosphate pathway (PPP) activity, producing abundant NADPH[101]. Accordingly, the burden of GSH production is shared asymmetrically. Astrocytes, with their strong capacity for NADPH production and abundant expression of system Xc-, are able to sustain cystine import and cysteine reduction under oxidative load (e.g., eustress), releasing GSH and GSH-derived dipeptides that neurons can import via γ-glutamyl or amino-acid transporters[102,103]. This division of labor preserves neuronal redox balance under normal conditions but becomes precarious when energy metabolism falters (e.g., due to stroke). In this scenario, astrocytes must use their own GSH supply to combat oxidative distress; increased astrocytic GSH consumption reduces their rate of GSH export to neurons and other cell types[103]. The resulting depletion of neuronal GSH supply weakens iron coordination (as discussed above), accelerating Fe-S cluster instability and the expansion of the labile iron pool (Figure 3c).
Beyond its direct metabolic consequences, cystine depletion also initiates a distinct cellular stress program (Figure 4b). When cystine import fails, and/or GSH stores decline, cells interpret this loss as a signal of amino acid starvation[104]. As cysteine becomes scarce, uncharged tRNAs bind to the general control nonderepressible 2 (GCN2) kinase, activating this sensor of amino acid deprivation[105]. Once engaged, GCN2 phosphorylates eukaryotic initiation factor 2 subunit (eIF2α), triggering the integrated stress response and globally reducing protein synthesis (Figure 3)[106]. Transient activation of this circuit is likely adaptive, as it conserves amino acid supply while permitting selective translation of ATF4, which in turn upregulates amino-acid transporters, GSH-synthesis enzymes, and other stress response genes (Figure 4b)[75,107]. However, if cysteine scarcity persists, ATF4 signaling appears to transition from restorative to destructive, driving a pro-death transcriptional cassette that accelerates ferroptosis (Figure 4b; as discussed above in Section 3.1)[76]. Indeed, early studies from our lab showed that inhibition of macromolecular synthesis rescues neurons from glutamate-induced oxidative death by redirecting cysteine flux away from protein synthesis and toward glutathione production, reinforcing antioxidant defense[108-110], as similarly corroborated by others[111].
Together, these mechanisms define the cystine-glutathione axis as a key inflection point that determines whether redox stress remains adaptive or becomes fatal. When cystine uptake and NADPH supply are adequate, neurons can sustain oxidative eustress that supports signaling and plasticity. Once this coordination fails, however, cysteine depletion, iron dysregulation, and the resulting lipid peroxidation form a self-reinforcing cycle of oxidative distress. Encouragingly, this trajectory can be intercepted after hemorrhagic stroke by restoring cysteine availability. We have shown that treatment with NAC, a clinically approved cysteine prodrug, bypasses system Xc- to replenish intracellular cysteine (Figure 4b)[16]. This restores GSH synthesis and enables GPX4-dependent detoxification of oxidized lipids, independent of iron modulation[16]. In primary cortical cultures exposed to hemin, NAC prevents ferroptotic cell death by neutralizing arachidonate-derived lipid species produced by 5-lipoxygenase (LOX-5), an enzyme also upregulated in human ICH brain tissue samples[16]. Systemic NAC administration initiated two hours post-stroke improved behavioral outcomes after collagenase ICH, a phenotype mirrored by LOX-5 deletion[16].
These findings emphasize that sustaining thiol metabolism (and by extension, the antioxidant economy) restrains ferroptosis. When this control fails, LOX and other lipid-oxidizing enzymes begin to execute the terminal phases of ferroptosis, opposed only by the membrane-associated and radical-trapping antioxidant systems described below in Section 3.3.
3.3 Membrane integrity and radical trapping antioxidants
Once the buffering capacity of thiol metabolism is exhausted, cellular membranes become the final “battleground” of ferroptosis. The same polyunsaturated phospholipids (PUFAs) that endow neuronal membranes with fluidity and signaling versatility also render them intrinsically vulnerable to oxidation[112,113]. Each PUFA chain contains bis-allylic hydrogens (e.g., chemically fragile sites with low bond-dissociation energy)[35]. When a reactive species abstracts one of these hydrogens, the resulting lipid radical reacts with oxygen to form lipid peroxyl radicals that propagate across the membrane in a chain reaction (Figure 4a)[35]. Under physiological conditions, these transient oxidation events are tightly regulated and contribute to redox signaling, membrane remodeling, and prostanoid synthesis[35,112]. During oxidative distress, however, this chemistry becomes self-propagating, driving structural disintegration, ionic collapse, and cell death[35].
As described above, ferroptotic lipid peroxidation is not random but enzyme-directed, shaped by the same iron-dependent dioxygenases that regulate normal lipid signaling. Among these, the lipoxygenases (LOXs) play a central role[35]. Under basal conditions, LOXs act on free arachidonic acid (AA) released by cytosolic phospholipase A2 (cPLA2) to generate hydroperoxyeicosatetraenoic acids (HPETEs), intermediates in prostaglandin and leukotriene biosynthesis[35,112]. During ferroptotic stress, substrate preference shifts from free AA to membrane-bound phosphatidylethanolamine (PE) species, a transition thought to be orchestrated by phosphatidylethanolamine-binding protein 1 (PEBP1; also known as Raf-kinase inhibitory protein, or RKIP; Figure 5a)[35,114]. When oxidative distress and mitochondrial dysfunction trigger Ca2+ transients, ERK1/2-phosphorylated cPLA2 hydrolyzes membrane phospholipids, releasing AA that is rapidly re-esterified by ACSL4 and LPCAT3 into arachidonoyl-PE (AA-PE), enriching membranes with oxidizable phospholipids[115-117]. As oxidative load escalates, PEBP1 is thought to disengage from Raf-1 and bind 12/15-lipoxygenase (12/15-LOX), forming a PEBP1-15-LOX complex that converts AA-PE to 15-HpETE-PE (15-hydroperoxy-eicosatetraenoyl-PE; Figure 5a)[114,116,118].
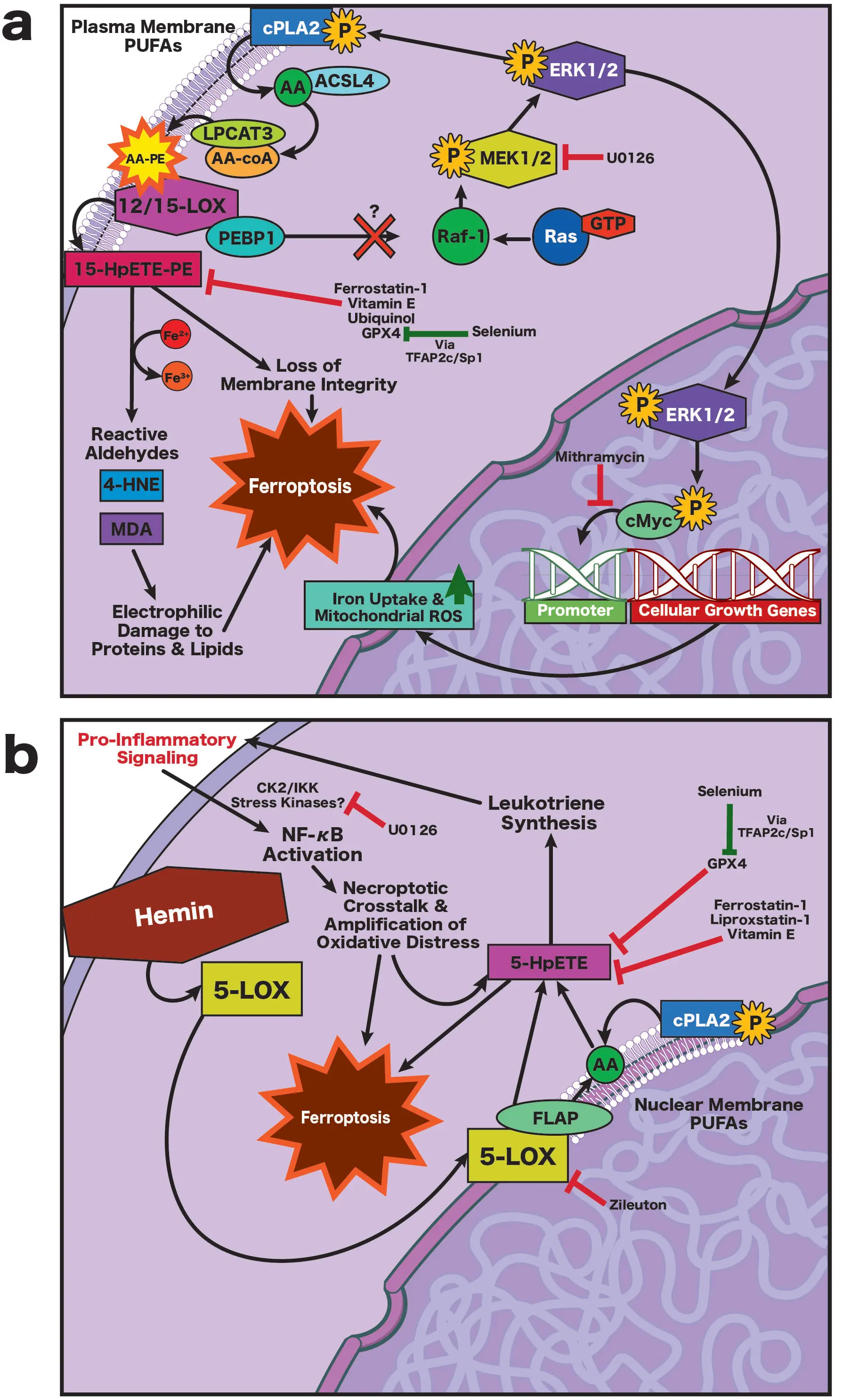
Figure 5. (a) Ischemia-induced ferroptosis. Oxidative stress dissociates PEBP1 from Raf-1, enabling Ras-Raf-MEK1/2-ERK1/2 signaling that activates cPLA2, releasing AA from membrane PUFAs. AA is reincorporated into PE by ACSL4 and LPCAT3 and oxidized by 12/15-LOX to form 15-HpETE-PE, compromising membrane integrity and generating toxic aldehydes (4-HNE, MDA). ERK1/2 also activates cMyc, promoting iron overload and mitochondrial ROS, thereby accelerating ferroptosis. This pathway is inhibited by U0126 and mithramycin; (b) Hemorrhage-induced ferroptosis. Hemin inserts into membranes, promoting formation of a 5-LOX/FLAP complex at the nuclear envelope, where free AA is converted to 5-HpETEs and leukotrienes, amplifying inflammation via CK2/IKK-NF-κB signaling, lipid peroxidation, and generation of toxic aldehydes. This process is independent of ERK1/2 activity; the protective effect of U0126 in this context reflects off-target inhibition of CK2/IKK signaling. In both models, ferroptosis is suppressed by radical-trapping antioxidants or upregulation of GPX4, which is enhanced by selenium via TFAP2c/Sp1-dependent GPX4 induction. AA: arachidonic acid; ACSL4: long-chain fatty acid-CoA ligase 4; cMyc: cellular Myc proto-oncogene; cPLA2: cytosolic phospholipase A2; CK2: casein kinase 2; ERK1/2: extracellular signal-regulated kinases 1 and 2; FLAP: 5-lipoxygenase–activating protein; GPX4: glutathione peroxidase 4; 4-HNE: 4-hydroxynonenal; IKK: IκB kinase; LPCAT3: lysophosphatidylcholine acyltransferase 3; LOX: lipoxygenase; 12/15-LOX: 12/15-lipoxygenase; 5-LOX: 5-lipoxygenase; MDA: malondialdehyde; MEK1/2: mitogen-activated protein kinase kinase 1 and 2; NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells; PE: phosphatidylethanolamine; PEBP1: phosphatidylethanolamine-binding protein 1; PUFAs: polyunsaturated fatty acids; Raf-1: proto-oncogene serine/threonine-protein kinase Raf-1; Ras: rat sarcoma; ROS, reactive oxygen species; TFAP2c: transcription factor AP-2 gamma; Sp1: specificity protein 1; 15-HpETE-PE: 15-hydroperoxy-eicosatetraenoyl-phosphatidylethanolamine; 5-HpETE: 5-hydroperoxy-eicosatetraenoic acid.
The dominant LOX isoform that mediates neuronal ferroptotic damage depends on the injury context: during ischemia, an increase in oxidative tone activates 12/15-LOX-PEBP1 complexes at the plasma membrane, oxidizing PE to 15-HpETE-PE and effectively coupling membrane rupture to antioxidant failure (Figure 5a)[119]. Inhibition of 12/15-LOX with LOXBlock-1 or newer compounds like ML351 is protective across both permanent and transient middle cerebral artery occlusion (MCAO) models[120-122]. In contrast, after ICH, hemin and other iron-rich hemoglobin products preferentially activate 5-lipoxygenase (5-LOX) at the nuclear envelope (Figure 5b)[16]. Once activated, 5-LOX acts on free AA to generate 5-HpETE, a highly unstable lipid hydroperoxide. The 5-HpETEs are the precursors for leukotrienes that amplify oxidative distress indirectly via NADPH oxidase activation, cytokine release, and inflammatory recruitment (Figure 5b)[123,124]; together, this cascade sustains lipid peroxidation and promotes ferroptosis in surrounding cells[16]. Therefore, ischemia and hemorrhage drive ferroptotic membrane injury through distinct spatial and biochemical routes, though both converge on oxidative collapse. In support of this, we find that structurally diverse 5-LOX inhibitors (e.g., zileuton) are protective against hemin-induced neurotoxicity in vitro, while selective inhibitors of 12/15-LOX are not[16]. While zileuton is found to be protective following collagenase ICH in mice[125], it also confers benefit after transient and permanent MCAO in mice[126-129], an effect likely reflective of attenuated leukotriene-driven neuroinflammation rather than direct suppression of ferroptotic lipid peroxidation.
The core enzymatic defense against lipid peroxidation is GPX4, the only enzyme capable of directly reducing membrane-embedded phospholipid hydroperoxides (PUFA-OOH) into lipid alcohols (Figure 4a)[98,130]. GPX4 defines the decisive checkpoint of ferroptosis, analogous to Bcl-2 in apoptosis[17]. Genetic deletion or inhibition of GPX4 (e.g., with RSL3) commits cells to oxidative death, whereas forced GPX4 expression fortifies neurons against ferroptosis in vitro and in vivo following stroke[131]. Under physiological conditions, GPX4 allows controlled lipid oxidation to support signaling and membrane dynamics; when its activity declines, lipid peroxides accumulate, increasing membrane permeability and generating reactive aldehydes such as 4-hydroxynonenal (4-HNE) and malondialdehyde (MDA)[132,133]. At low concentrations, these products induce plasticity and adaptive transcriptional responses (e.g., endogenous antioxidant programs); at high concentrations, however, they form adducts with histones, induce inflammatory and pro-death signaling, and cross-link proteins, resulting in stiff membranes, osmotic failure, and ferroptotic collapse[133,134]. The threshold at which peroxidation becomes irreversible likely depends on the spatial extent and kinetics of oxidation within different intracellular compartments, as well as events occurring in adjacent cells[34]. It is intriguing to note that ferroptosis in one cell can propagate to adjacent bystanders, potentially via cell-cell contacts[135]; future work may help us understand how this phenomenon may be leveraged therapeutically.
Although GPX4 is the principal defense against lipid peroxidation, cells deploy additional antioxidant circuits that act directly at the membrane interface (Figure 5a,b). One such pathway involves ferroptosis suppressor protein 1 (FSP1), which regenerates ubiquinol (CoQ10H2) at the plasma membrane to trap lipid peroxyl radicals and preserve membrane integrity in parallel to GPX4 (Figure 4)[136]. Structural studies indicate that FSP1 can use NADPH as an electron donor, aligning it with the GSH-GPX4 axis[137]. However, recent work also suggests that a membrane-associated NADH pool, regulated in part by ALDH7A1, can support FSP1 activity under ferroptotic stress, implying that cofactor usage may be compartmentalized or contextually-dependent[138,139]. This FSP1-CoQ10 circuit could be particularly vital in neurons, whose PUFA-rich membranes and limited GSH turnover heighten vulnerability[113]. Another recently discovered endogenous RTA system is the GCH1 (GTP-cyclohydrolase 1)-BH4 axis, which supplies the antioxidant BH4 in an NADPH-dependent process[140]. While these mechanisms are yet to be exploited to treat stroke in vivo, they represent important areas for future investigation.
Other RTAs, including vitamin E (α-tocopherol), liproxstatin-1, and ferrostatin-1 (Figure 4) act through similar radical-quenching chemistry[141]. While vitamin E and its derivatives potently inhibit ferroptosis in vitro[141], there is mixed preclinical evidence regarding their benefit when administered prior to stroke[142-146], and few have investigated efficacy with treatment delay following stroke. Additionally, the increased risk of hemorrhagic stroke with vitamin E supplementation substantially limits translational potential for stroke neuroprotection[147,148]. Interestingly, the canonical ferroptosis inhibitor ferrostatin-1 acts not only as an RTA but also inhibits formation of the 12/15-LOX/PEBP1 complex (Figure 5a), limiting formation of 15-HpETE-PE[149]. Administration of ferrostatin-1 following rodent ICH and focal ischemia has shown neuroprotective efficacy[7,9,150-152], but the translational feasibility of ferrostatin-1 is currently limited by its short half-life and limited CNS penetrance[153]. Work on similar alternatives is ongoing[153,154], as the efficacy of lipophilic RTAs both in vitro and in preclinical models thus far has validated lipid peroxidation and ferroptosis as tractable therapeutic targets following stroke.
While RTAs buy time, they act transiently at the end stage of oxidative injury. A potentially more durable approach is to enhance endogenous enzymatic repair, especially those governed by selenoproteins that restore antioxidant capacity from within. As an essential micronutrient, selenium is incorporated as selenocysteine into a family of selenoproteins, such as GPX4, thioredoxin reductase 1 (TXNRD1), and Selenoprotein P (SelP)[131,155]. Collectively, these selenoproteins sustain redox balance under ferroptotic stress. Our group demonstrated that ferroptotic stimuli such as hemin or glutamate analogues elicit a frustrated adaptive transcriptional response in neurons, characterized by modest upregulation of these same selenoproteins[131]. We found that selenium supplementation amplifies this response, driving a broad selenome transcriptional network via coordinated activation of the transcription factors transcription factor AP-2 Gamma (TFAP2c) and Specificity Protein 1 (Sp1)[131]. This adaptive pathway increases expression of both nuclear and mitochondrial GPX4 isoforms (Figure 5a,b), expanding the cellular capacity to detoxify lipid hydroperoxides and suppressing ferroptosis even when initiated hours after insult. Following collagenase ICH in mice, a single intracerebroventricular dose of selenium or systemic delivery of a brain-penetrant selenocysteine peptide (Tat-SelPep) upregulated GPX4, reduced neuronal death, and restored behavioral function without affecting hematoma size[131]. Following transient MCAO, Tat-SelPep also reduced infarct volume independently of excitotoxicity when delivered two hours post-stroke[131]. Mechanistically, this selenium-driven transcriptional reinforcement counterbalances the pro-ferroptotic ATF4 axis activated during sustained glutathione depletion (Figure 3c, Figure 4b), converging with nuclear factor erythroid 2- related factor 2 (Nrf2)-dependent antioxidant gene expression to restore redox equilibrium.
Together, these antioxidant systems illustrate that the outcome of ferroptosis depends not only on the chemistry of lipid peroxidation, but also on the capacity of the cell to coordinate both enzymatic and transcriptional responses to oxidative stress.
3.4 Transcriptional control of redox tone
When antioxidant defenses begin to falter, the cell must decide whether to restore redox balance or commit to degeneration. This decision is governed by transcriptional hierarchies that sense oxidative pressure and redistribute metabolic resources toward either recovery or collapse. At the apex of this system is Nrf2, the principal transcriptional regulator of dynamic redox homeostasis[17]. Under basal conditions, Nrf2 maintains constitutive expression of genes that preserve redox tone. Normally sequestered by Kelch-like ECH-associated protein 1 (Keap1) and targeted for proteasomal degradation, Nrf2 is released when electrophiles oxidize Keap1’s cysteine residues. Freed Nrf2 then translocates to the nucleus, where it activates antioxidant-response elements (AREs), orchestrating a broad cytoprotective program. This includes upregulation of GCLC and GCLM (for glutathione synthesis), SLC7A11 (the cystine importer of system Xc⁻), NQO1 (which prevents quinone redox cycling), HO-1 (which catabolizes heme), and ferritin subunits FTH1 and FTL1, which sequester iron[17]. Collectively, this program stabilizes iron homeostasis and reinforces NADPH production through induction of the pentose-phosphate pathway and ME1, coupling antioxidant defenses to metabolic supply[156].
When oxidative pressure rises, Nrf2 activity is amplified through stabilization and nuclear accumulation, expanding metabolic and antioxidant capacity. Therefore, while Nrf2 maintains dynamic basal antioxidant tone, it also acts as a rapid-response sensor when redox balance tips toward oxidation. Importantly, within the CNS, Nrf2 expression is cell-type specific. Mature neurons exhibit low basal Nrf2 levels, but astrocytes and microglia have robust expression, allowing them to dynamically adjust to local oxidative load[102,157]. Astrocytic Nrf2 activation enhances glutathione synthesis and export (along with glutathione precursors), which are taken up by neurons to support antioxidant defence[102,158]. Experimental data support this compartmentalization: in primary cultures, the forced expression of Nrf2 in astrocytes protects against neuronal ferroptosis, even when constituting less than 2% of the total culture population[102].
This cell-type specific expression likely has developmental origins: low levels of Nrf2 activation enhance the capacity for oxidative eustress, which is required for neuronal differentiation, neurite outgrowth, and synapse formation[159-161]. Similarly, in post-mitotic neurons, oxidative eustress plays an important role in shaping structural plasticity via JNK/c-Jun signaling[159]. Therefore, compared to astrocytes, neurons must maintain a relatively oxidized intracellular state (via higher expression of Keap1)[157], while astrocytes retain their stress-inducible Nrf2 signaling into adulthood. In this manner, Nrf2 links transcriptional control to metabolic supply, ensuring that antioxidant defenses and NADPH regeneration scale with redox demand. Under normal conditions, this coordination maintains oxidative eustress within a physiological range. During acute injuries such as stroke, however, oxidative load overwhelms metabolic capacity.
As Nrf2-dependent programs fail to meet rising oxidative demand, secondary signaling cascades such as stress-activated MAPKs are recruited in an attempt to restore homeostasis. In particular, the Ras/Raf/MEK/ERK pathway plays a pivotal role in this transition (Figure 5a). Initially, transient ERK activation supports metabolic compensation and cytoprotective gene expression, but sustained activation becomes maladaptive when reducing power is exhausted[162,163]. This concept became apparent in early ferroptosis studies, which demonstrated that unlike apoptosis[164], growth factor stimulation enhanced neuronal susceptibility to ferroptotic death[28]. As discussed above, under normal physiological conditions, PEBP1 (RKIP) binds to Raf-1 and prevents its association with Ras, restraining ERK activation. During canonical growth factor signaling, protein kinase C (PKC) is activated by tyrosine receptor kinases, phosphorylating PEBP1 and dissociating the PEBP1-Raf-1 complex; together, these events activate the downstream MEK-ERK1/2 cascade[165]. In the setting of ferroptosis, an analogous shift appears to occur, though the precise trigger remains uncertain (Figure 5a). Oxidative modifications of PEBP1, phospholipid interactions, or PKC-dependent phosphorylation could each contribute to Raf-1 release, freeing PEBP1 to associate with 12/15-LOX. This effectively links stress-activated kinase signaling to the onset of lipid peroxidation.
Once activated, ERK1/2 translocates to the nucleus to phosphorylate its transcriptional targets, including cMyc, Elk1, and cfos (Figure 5a)[166]. Under normal conditions, this activity is transient, terminated by the phosphatase MKP3, which dephosphorylates ERK1/2 and returns it to the cytoplasm. During oxidative distress, however, MPK3 itself is vulnerable, as its redox-sensitive residues can be oxidized or covalently modified by lipid hydroperoxides such as 15-HpETE-PE, rendering the enzyme inactive[167]. The resulting persistence of nuclear ERK1/2 sustains phosphorylation of c-Myc, stabilizing it as a pro-ferroptotic transcriptional amplifier (Figure 5a)[168,169]. Normally, c-Myc coordinates energy metabolism and ribosome biogenesis, matching neuronal activity with metabolic demand. Many of its canonical targets enhance iron uptake and turnover to sustain oxidative metabolism, such as the ferritin subunits, Trf1, natural resistance-associated macrophage protein-1 (Nramp1) and iron-regulatory protein-2 (IRP2), among others[170]. When reducing power is sufficient, this response is adaptive and self-limiting. Yet under sustained ERK1/2 activation and redox imbalance, the same program drives excessive iron import and mitochondrial ROS production, outpacing antioxidant capacity[168]. This creates a feed-forward loop of lipid peroxidation and iron-driven oxidative stress (Figure 5a), further committing neurons to ferroptotic death.
Sustained oxidative stress also triggers chromatin remodeling (e.g., histone glutamylation, acetylation, and methylation), altering the accessibility of antioxidant and metabolic gene promoters[171]. These epigenetic shifts likely determine whether redox signals are resolved through adaptive transcription or a transition to ferroptotic commitment, coupling metabolic state to chromatin configuration. Indeed, pharmacologic dissection of the ferroptotic transcriptional programs discussed in this section has revealed several points of intervention. Mithramycin, an antibiotic that binds to GC-rich motifs of the c-Myc promoter, prevents cofactor Sp1 from binding and silences the ERK/c-Myc amplification loop that drives iron-handling and metabolic genes during oxidative stress (Figure 5a), which we have found to protect neurons from glutamate-induced ferroptosis in vitro[172]. While we have not tested mithramycin following stroke in vivo, others have found that it confers cytoprotective benefit following transient global ischemia[173], though its translational potential is low due to a high risk of toxicity, thrombocytopenia, and platelet dysfunction[174].
Evidence from our lab and others indicates that Sp1 acts in a contextually and temporally dependent manner via post-translational modifications[175]; pro-survival when acetylated early after the onset of oxidative stress, to sustain antioxidant tone, but pro-death when engaged downstream of ERK during sustained oxidative distress. For instance, pan-histone deacetylase (HDAC) inhibitors such as trichostatin A or suberoylanilide hydroxamic acid (SAHA; also known as vorinostat) enhance p300-mediated Sp1 acetylation, promoting expression of adaptive antioxidant genes like MnSOD and HO-1 in vitro during glutamate-induced ferroptosis[176,177]. Additionally, we have found that inhibition of specific HDAC isoforms, such as HDAC6, may confer not only neuroprotection, but also encourage neurite extension in vitro[178]. Beyond transcriptional modulation, sustained ERK activation engages an additional layer of regulation through transglutaminases. Dramatically upregulated both following ischemic stroke in vivo and glutamate exposure in vitro, transglutaminase inhibitors block ferroptosis up to 14 hours after exposure onset in vitro, while U0126 does not prevent death mediated by forced transglutaminase expression, placing transglutaminase activity functionally downstream of ERK signaling[179].
In vivo, both pan-HDAC inhibitors and class 1 HDAC inhibitors reduce cell death and provide functional benefit when administered both immediately and up to several days following MCAO or focal cortical stroke[180,181]. Selective class 1 HDAC inhibitors also confer benefit when administered immediately following autologous whole blood ICH[182]. However, selective class IIa HDAC inhibition has been shown to impair remodeling, exacerbate lesion volume, and increase mortality following transient MCAO in rats[183]. Therefore, despite strong mechanistic rationale, the delivery of HDAC inhibitors as stroke therapeutics remains translationally challenging due to their complex pharmacodynamics, contextual dependence, narrow temporal window of efficacy, and risk of adverse and off-target effects (including metal chelation)[184,185].
While erastin- and glutamate-induced ferroptosis activate the ERK/c-Myc axis, we find that hemin-induced ferroptosis proceeds independently of ERK (Figure 5b)[14]. Yet ferroptosis triggered by erastin, glutamate, and hemin are blocked by U0126, a nominally MEK-selective inhibitor (Figure 5b), even though other ERK inhibitors fail to protect against hemin toxicity. This apparent contradiction prompted us to conduct a comparative phosphoproteomic analysis of neurons exposed to erastin or hemin (with or without U0126), revealing distinct kinase signatures: erastin promotes the canonical proline-directed MAPK motifs characteristic of ERK activation, whereas hemin exposure is associated with Ca2+- and CK2/IKK-type signatures, implicating a broader stress-kinase network[14]. These data suggest that U0126 protects against hemin toxicity not by directly inhibiting ERK, but by dampening alternative phosphorylation cascades that link lipid peroxidation to inflammatory signaling. Although the precise downstream mediators remain uncertain, enrichment of CK2/IKK motifs strongly suggest NF-κB (nuclear factor kappa-light-chain enhancer of activated B cells) involvement[186], a pathway known to intersect with both redox and lipid metabolism across numerous cell types[187]. Consistent with this interpretation, we find that hemin-induced neuronal death in vitro and after hemorrhage in vivo uniquely displays features of both ferroptosis and necroptosis, with protection achieved by inhibitors of either pathway, including U0126[14,188].
As discussed above, hemin exposure and 5-LOX-mediated lipid peroxidation result in neuronal synthesis and release of leukotrienes, which can act in an autocrine or paracrine manner, activating their cognate G-protein coupled receptors on neighbouring neurons and glia. Leukotriene signaling is known to engage NF-κB either directly through their cognate receptors or indirectly via amplification of cytokines associated with NF-κB activation and necroptosis, such as TNF-α[124,189,190]. Although the precise upstream signaling cascade responsible for CK2/IKK stress kinase activation following neuronal hemin exposure must be further explored, it is possible that leukotriene signaling promotes IKK-dependent phosphorylation of IκBα, permitting nuclear translocation of NF-κB[191]. Once in the nucleus, NF-κB engages a transcriptional program that primes key components of the necroptotic machinery (e.g., “the necrosome”), including RIPK1, RIPK3, and MLKL[191], licensing neurons for execution via crosstalk between necroptotic and ferroptotic processes- a phenomenon that must be explored further.
Within this framework, ferroptotic lipid peroxidation constitutes the initiating insult, while leukotriene-driven inflammatory signaling provides the molecular bridge to necroptosis, functionally coupling these two pathways. Depending on the cellular context, the convergence of ferroptotic and necroptotic signals may push neurons beyond a shared threshold for necrotic death. Consistent with this model, inhibition of either pathway alone during hemin exposure is sufficient to restore survival, though hemin-induced death can still be operationally defined as ferroptosis given its sensitivity to ferroptosis-selective inhibitors[14]. While further experimental exploration is required, analogous crosstalk between ferroptosis and necroptosis has been reported in other pathological contexts, including acute kidney injury[192]. More broadly, the integration of lipid peroxidation with NF-κB dependent inflammatory signaling raises the possibility that ferroptosis evolved not only as a mode of cell death, but also as a mechanism to convert oxidative collapse into signals that promote tissue repair and immune recruitment[15,158], a concept we explore further below.
Taken together, these findings point to transcriptional reprogramming as a potential inflection point between adaptive redox responses and ferroptotic collapse. As antioxidant reserves wane, signaling networks that normally sustain recovery instead appear to reinforce oxidative and inflammatory stress. Although the precise hierarchy of these pathways remains uncertain, their interplay suggests a gradual shift from metabolic coordination to loss of homeostatic control. Clarifying when and where this transition occurs across neurons, astrocytes, and immune cells will be key to identifying therapeutic windows in which ferroptosis remains reversible after each stroke subtype.
4. Translational Frontiers: Timing, Spatial Dynamics, and Cellular Crosstalk
Importantly, ferroptosis does not occur in isolation; it evolves within a shifting tissue landscape where neurons and glia are weathering rapid changes on numerous fronts, from intracellular metabolic instability to an ongoing and dynamic inflammatory response. Despite major advances in defining the molecular framework of ferroptosis, its translation into effective stroke therapies remains constrained by a limited understanding of how, when, and where it unfolds in animal models and patients.
4.1 Divergent redox trajectories in ischemia and hemorrhage
Although the temptation to categorize ischemia and hemorrhage together as insults that cause “oxidative stress” is understandable, this oversimplification obscures their distinct biochemical trajectories. Both focal ischemia and ICH share a common initiation phase in which damage-associated molecular patterns (DAMPs) released by acute necrotic cell death immediately begin to shape redox and inflammatory signaling[193-195]. This early DAMP signaling triggers NADPH-oxidase dependent superoxide generation, primes microglia for cytokine release, and establishes an oxidizing extracellular milieu that spreads into surrounding viable tissue[196]. However, against this shared backdrop, ischemic and hemorrhagic strokes diverge in how they sustain, amplify, and resolve oxidative injury.
Within penumbral tissue ranging from oligemic (cerebral blood flow in the range of 22-60 mL/100 g/min) to ischemic (below 22 mL/100 g/min)[197], partial oxygen and glucose deprivation slows mitochondrial respiration, diminishes ATP and NADPH generation, increases lactate production, and produces a hyper-reduced redox state (“reductive stress”) due to mitochondrial accumulation of reduced electron carriers[96,198-200]. Upon reperfusion, accumulated reducing equivalents generate reactive oxygen species (ROS)[15], and intracellular calcium levels rise[201], rapidly transforming reductive stress into oxidative distress. Transient activation of transcriptional programs by HIF-1α and ATF4 initially supports adaptation by promoting glycolytic metabolism, but the concurrent suppression of the PPP reduces neuronal NADPH regeneration, impairs glutathione cycling, and expands the labile iron pool (LIP)[75,202-204]. Together, this creates a delayed ferroptotic vulnerability at the ischemic penumbra that is likely exacerbated by development of edema, given the sensitivity of ferroptotic processes to osmoprotectants in non-CNS cell types[205]. Therefore, following focal ischemia, ferroptosis appears to emerge around the time of reperfusion[13,206], likely within penumbral tissue where oxidative flux suddenly returns and outpaces the restoration of cellular NADPH and GSH pools.
In contrast, following ICH, neurons in the perihematomal zone that escape necrosis from direct mechanical injury experience early and intense oxidative distress driven by iron toxicity, shifting hydrostatic pressure, osmotic gradients, thrombin release, and complement activation[3]. Preclinical models indicate that early limited erythrocyte lysis within the hematoma begins immediately following ICH, reaching a peak by ~24-48 hours post-stroke[207-209]. Given average hemoglobin levels within whole blood[31], a conservative estimate of the hemoglobin within the hematoma is ~2.3 mM. Erythrocytes must lyse for hemoglobin to be released. Of note, Hgb induces death of cortical and striatal neurons in vitro at concentrations of 1.5 mM[14,188], suggesting that only 1 out of 2,000 erythrocytes need to lyse in order to trigger local cell death. These results may explain why anti-ferroptotic agents are efficacious early after ICH onset.
It is clear that ischemic and hemorrhagic strokes impose distinct redox trajectories that converge on the same endpoints: exhaustion of reducing power, collapse of glutathione and NADPH systems, and lipid peroxidation-driven membrane failure. The challenge is to determine which components of this cascade are causative, reversible, or adaptive, their timing, and whether they represent feasible therapeutic targets. To tackle this challenge, we must better understand the interplay between acute metabolic status and the spatiotemporal progression of ferroptosis within the penumbral and perihematomal zones, along with the relationships between constituent cell types. Recent evidence suggests that the vascular compartment is an early and critical site of injury following focal ischemia: brain endothelial cells highly express 12/15-LOX, which directly contributes to blood-brain barrier breakdown, vasogenic edema, and subsequent neuroinflammation, along with hemorrhagic transformation[120]. Astrocytes can buffer oxidative stress and deliver glutathione precursors, along with healthy mitochondria, to neurons, yet this protective capacity may convert to pro-oxidant signaling under sustained activation[102,158,210]. Compensatory ischemic glycolysis, iron overload, and lipid peroxidation in microglia can drive them toward a pro-inflammatory state, amplifying cell death and limiting plasticity and repair following stroke through the release of cytokines and reactive oxygen species[210,211]. Oligodendrocytes, rich in iron-handling and lipid-synthesis machinery, are particularly vulnerable to secondary ferroptotic degeneration once local redox buffering collapses[10,212-214]. It is likely that cell-type specific antioxidant capacities also vary across regions, shaping susceptibility to ferroptosis as a function of local redox tone and activity level[215]. Future work using spatial transcriptomics, in vivo redox imaging, and single-cell metabolomics could define how ferroptotic vulnerability propagates across tissue compartments.
Many other unresolved questions remain. What is the precise timing of ferroptotic death relative to reperfusion or red blood cell lysis? How long does the reversible window of ferroptosis remain open in vivo? What molecular cues determine where/when ferroptosis begins within peri-infarct or peri-hematoma tissue, and what are the irreversible terminal events? Clarifying such kinetic parameters is needed to align mechanistic insights with realistic treatment windows.
4.2 Crosstalk and immunological context
Ferroptosis exists within a broader network of oxidative and inflammatory death programs that co-occur after stroke. It overlaps mechanistically and functionally with autophagy, necroptosis, parthanatos, pyroptosis, and apoptosis, dependent on the cellular context in which the inducing stimuli occur. In ischemic tissue, PARP-1 activation and NAD⁺ depletion bridge ferroptosis to parthanatos, while MLKL-dependent necroptosis often co-occurs downstream of TNF-α and RIPK1/3 signaling[216,217]. As discussed above, in hemorrhagic stroke models, we have found ferroptosis and necroptosis to coincide[14], suggesting shared metabolic checkpoints involving mitochondrial dysfunction, iron dysregulation, and amplification of reactive oxygen species. Others have demonstrated possible overlap between necroptosis and ferroptosis upon ischemic reperfusion following transient middle cerebral artery occlusion in mice[13].
Intriguingly, in innate immune cells, induction of NF-κB signaling via toll-like receptors (TLRs) increases intracellular iron sequestration by ferritin, resulting in HIF PHD inhibition and HIF1α stabilization[218]. This helps dictate their pro-inflammatory function under both hypoxic and normoxic circumstances[218,219]. While it is unclear whether such a relationship exists in neurons, sustained NF-κB signaling is shown to suppress Nrf2 in neurons and glial cells (and vice versa); conversely, acute/activity-dependent NF-κB induction signals oxidative eustress and induces plasticity in healthy cells[20,32,220-223]. It is possible that the frustrated Nrf2 response after stroke and other acute CNS injuries reflects astrocytic sensitivity to sustained NF-κB signals, impairing their antioxidant support to other cell types and amplifying neuroinflammation[220-222,224]. Such crosstalk between these and other pathways is simultaneously influenced by ongoing changes in the mechanical and osmotic environment (e.g., edema, ionic imbalance, and elevated intracranial pressure) that occur following stroke in both preclinical models and patients[225-229]. These biomechanical forces distort oxygen delivery and redox flux, both triggering and amplifying inflammatory signaling, ferroptosis and other forms of cell death[205,230,231].
Indeed, a theorized evolutionary function of ferroptosis is to act as a source of bioactive inflammatory signals, providing a warning to the immune system that redox homeostasis has been lost within a given region[232]. Following stroke, the DAMPs released from acutely necrotic cells in the infarct core or hematoma initiate early oxidative distress through TLRs, RAGE, and inflammasome activation[193-195]. As ferroptosis progresses in penumbral or perihematomal cells, secondary release of oxidized phospholipids and aldehydes such as 4-HNE and MDA further propagate this inflammatory circuit. These lipid peroxidation products function as immunotransmitters that signal the failure of local redox homeostasis, recruiting and polarizing microglia and infiltrating macrophages to contain the damage and protect wider organ integrity[233].
Some lipid peroxidation products can also help resolve the inflammatory response[123]. The membrane 12/15-LOX activity characteristic of focal ischemia yields 15-HpETE-PE, but when redox balance recovers, this pathway can shift toward production of pro-resolving lipoxins and prostaglandins that guide tissue repair[233]. Conversely, in ICH pathology, nuclear 5-LOX activity dominates, producing leukotrienes that massively amplify neutrophil extravasation and sustain chronic inflammation and oxidative distress[233]. These distinct repertoires of lipid mediators may explain why ischemic injury more readily transitions toward resolution, whereas hemorrhagic injury often results in chronic, ongoing cell death. Yet how and when these oxidized lipids serve primarily as pro-death signals or as adaptive cues for debris clearance and tissue remodeling remains unclear. Disentangling how ferroptotic signaling interfaces with innate immunity, mechanical/osmotic stress, and parallel death programs will be necessary to determine how and when ferroptosis should be suppressed, modulated, or repurposed as part of a controlled injury-resolution process following stroke.
5. Conclusion: The Path to Translation
It is striking that many anti-ferroptotic agents that inhibit cell death induced by ferroptotic stimuli in vitro also reduce cell death and improve behavioral outcomes following ischemic or hemorrhagic stroke in vivo (Table 1). This suggests that anti-ferroptotic agents protect the brain in a manner that preserves integrated functional benefit. Therefore, there is hope that blocking ferroptosis in the first days following stroke could reduce neuronal and glial cell loss, preserve functional circuits, and promote improved motor, cognitive, and sensory recovery in patients- though for many therapeutic agents, translational challenges remain (Table 1). Additionally, while our understanding of ferroptosis is advancing rapidly, there are numerous therapeutic “nodes” that are yet to be exploited in the context of stroke.
Table 1. Selected pharmacological modulators of ferroptotic pathways validated in stroke models.
| Therapeutic Agent | Therapeutic Target | Mechanism of Action | Representative Preclinical/Clinical Evidence | Translational Considerations/Challenges |
| Deferoxamine | LIP | Chelates intracellular iron, reducing iron-driven lipid peroxidation by limiting expansion of the LIP; reduced intracellular iron can relieve HIF-1α inhibition (contextually dependent effects). | Preclinical: Reduced peri-hematomal edema, iron deposition, cell death, and provided functional benefit in multiple porcine/rodent ICH models with 0-6 h treatment delay[81,86-88], though evidence of benefit is mixed in the collagenase model[80]. Functional benefit and reduced infarct volume, neuronal death, and BBB disruption when administered 1-3 h[85] or 24 h[83] following reperfusion upon transient MCAO in rodents. Others find evidence of harm, impeding repair and prolonging neurological deficits following permanent rodent MCAO with subacute administration (72 h treatment delay)[84]. Clinical: Evaluated in Phase II i-DEF trial for ICH; failed to reach primary endpoint of functional improvement[89], though post-hoc analyses suggest earlier and more rapid recovery trajectories in treated patients[91,92]. A Phase I trial for high dose treatment (HI-DEF) in ICH patients was terminated early due to evidence of harm[90]. Evaluated in Phase I trials for focal ischemic stroke patients alongside tPA infusion; found to be safe[242] | FDA-approved for iron overload; poor CNS penetrance (requires high doses), short half-life, and risk of systemic hypotension/toxicity. Trial failures suggest a need for better CNS-targeting, or alternatives to wholesale iron chelation. |
| Adaptaquin | HIF PHDs | Selective inhibition of HIF PHDs suppresses ATF4-driven pro-ferroptotic transcription while sparing bulk iron metabolism. | Functional benefit and reduced cell death in multiple mouse ICH models (2 h treatment delay)[77]. | Unknown pharmacokinetic and safety profile in humans. |
| Zileuton | 5-LOX | Suppresses hemin-mediated production of 5-HpETE lipid hydroperoxides by 5-LOX and leukotriene-driven inflammatory amplification. | Functional benefit, reduced levels of leukotriene B4, and attenuated neutrophil infiltration after collagenase ICH (3 h treatment delay)[125]. | FDA-approved for asthma; possible risk of hepatotoxicity at doses required for CNS penetrance in humans; short half-life; poor solubility. Likely restricted to acute treatment window due to risk of interfering with the lipid mediators that help resolve inflammation during the sub-acute recovery phase (e.g., lipoxins). |
| ML351 | 12/15-LOX | Suppresses production of 15-HpETE-PE lipid hydroperoxides by 12/15-LOX. | Functional benefit, reduced infarct volume, reduced lipid peroxidation, and attenuated pro-inflammatory signalling when administered immediately following reperfusion or onset in transient and permanent MCAO models[120,121]. | Unknown pharmacokinetic and safety profile in humans; short half-life; poor solubility. Work on alternative formulations ongoing. |
| Selenium; Tat-SelPep | GPX4/Selenoproteome | Transcriptional upregulation of GPX4 and antioxidant selenoproteins to maintain reduction and detoxification of lipid hydroperoxides | Functional benefit and reduced cell death in multiple mouse ICH models with 2 h (Se, Tat-SelPep) or 6 h (Tat-SelPep) treatment delay; reduced infarct volume following transient MCAO with 2 h treatment delay (Tat-Se Pep)[131]. | Selenium itself has limited CNS penetrance and a U-shaped dose response curve, with risk for toxicity. Delivery via engineered peptides or nanoparticles[131,243,244] address these issues but have unknown pharmacokinetic and safety profile in humans. |
| NAC | System Xc-/Cystine-Glutathione (GSH) Axis | Bypasses cystine transport via system Xc- to replenish intracellular cysteine and GSH. | Functional benefit and reduced cell death following collagenase ICH with 2 h treatment delay[16]. | FDA-approved for acetaminophen overdose and as a mucolytic therapy; limited CNS penetrance with systemic administration. |
| Ferrostatin-1 | 12/15-LOX/PEBP1 Complex; Peroxyl Radicals | Suppresses production of 15-HpETE-PE lipid hydroperoxides by blocking the formation of the 12/15-LOX/PEBP1 complex; also acts as a RTA, quenching peroxyl radicals. | Functional benefit, reduced cell death, reduced edema, faster hematoma resolution, phagocytic microglial polarization, and reduced brain atrophy across multiple ICH models with 2-3 h treatment delay[7,9,152]. Benefit observed both in young and aged mice following ICH[9]. Functional benefit, reduced infarct volume, and reduced cell death when administered immediately upon reperfusion following MCAO[150,151]. | Poor CNS penetrance (may be improved by nanoparticle delivery)[150]; very short half-life; poor solubility. Work on alternative formulations ongoing. |
| Liproxstatin-1 | Peroxyl Radicals | Acts as an RTA to quench peroxyl radicals. | Functional benefit and reduced cell death when administered 4 h following collagenase ICH[9]. | Limited CNS penetrance, low solubility. |
This table summarizes representative agents targeting distinct nodes of the ferroptotic cascade. Inclusion is restricted to compounds demonstrating ferroptotic mechanistic specificity in vitro and subsequent functional or histological efficacy in preclinical models of focal ischemia or intracerebral hemorrhage, with current potential for clinical translation. Studies using pre-treatments delivered prior to stroke were not included. LIP: labile iron pool; HIF-1α: hypoxia-inducible factor 1α; ICH: intracerebral hemorrhage; MCAO: middle cerebral artery occlusion; CNS: central nervous system; PHDs: prolyl hydroxylases; 5-LOX: 5-lipoxygenase; HpETE: 5-HpETE: 5-hydroperoxy-eicosatetraenoic acid.; 15-HpETE-PE: 15-hydroperoxy-eicosatetraenoyl-phosphatidylethanolamine; 12/15-LOX: 12/15-lipoxygenase; MCAO: middle cerebral artery occlusion; GPX4: glutathione peroxidase 4; NAC: N-acetylcysteine; RTA: radical trapping antioxidant.
Moving forward, to effectively translate these findings into clinical practice, it will be essential first to identify reliable biomarkers in preclinical studies that connect cellular protection and behavioral improvement in animal models to measurable targets in humans, providing a rationale for patient selection and trial endpoints. Progress will depend on demonstrating that agents shown to provide protection across multiple laboratories and stroke models can effectively engage their targets in vivo in humans. Successful clinical translation will also require careful consideration of how these agents are administered to stroke patients, including optimal timing, dosing, and route of delivery. Achieving this goal will require creative strategies to identify and measure electrophilic lipids and drug concentrations in the cerebrospinal fluid (CSF) in both preclinical and clinical studies, laying the groundwork for biomarker-driven clinical trials. Early-phase human studies could then leverage these biomarkers, using lumbar puncture, microdialysis, or CSF drains to confirm target engagement, guide dosing strategies, and inform patient stratification. Together, these steps are necessary to move anti-ferroptotic therapies from experimental models to safe and effective clinical use.
Equally important is understanding how patient context (e.g., age, sex, and comorbidities such as hypertension, diabetes, or hyperlipidemia) reshapes ferroptotic vulnerability after stroke. Sex hormones differentially regulate iron handling and lipid metabolism[234,235], while aging and chronic vascular or metabolic disease alter NADPH availability, microglial priming, and inflammatory tone[236-238]. Each of these factors likely shifts the redox “set point” at which ferroptotic signaling becomes pathogenic. Understanding these variables could allow stratification of patients in clinical trials to maximize efficacy and minimize adverse effects, moving toward a personalized medicine approach. To enhance the chances of successful translation, future preclinical studies must model these systemic modifiers and evaluate how existing treatments (e.g., reperfusion therapy, minimally invasive hematoma evacuation, and rehabilitation-induced plasticity) interact with ferroptotic signaling. In addition to use of robust methods[239] and rigorous experimental and statistical practices (e.g., a priori experimental planning, power analyses)[240,241], integrating these layers will be essential to align ferroptosis research with stroke biology.
Ultimately, embedding ferroptosis within the broader network of secondary injury, repair, and recovery is crucial for realizing its therapeutic potential. Strategies that engage redox network stress sensors to modulate homeostatic programs involving hundreds of protective genes at the cellular, local, and systemic levels may help preserve brain tissue and function, offering a path toward clinically actionable neuroprotection in human stroke patients.
Supplementary materials
The supplementary material for this article is available at: Supplementary materials.
Acknowledgements
We thank past and present members of the Ratan Lab for their contributions to this work. We also thank Dr. Caroline Philpott for her input. We thank Logan Schalk for editorial support.
Public domain image sources for artwork included the National Institutes of Health BioArt Source and Wikimedia Commons. Links to all artwork sources used can be found in Supplementary materials. All Figures were created using Adobe Illustrator 2026.
Authors contributions
Kalisvaart ACJ: Conceptualization, visualization, writing-original draft, writing-review & editing.
Ratan RR: Conceptualization, writing-review & editing.
All authors reviewed and approved the final manuscript prior to submission.
Conflicts of interest
Rajiv R. Ratan serves on Scientific Boards for Alevian and Neuronasal, with equity interest in Neuronasal. Neuronasal has intranasal N-acetylcysteine in development as an anti-ferroptotic agent to treat central nervous system injury and Parkinson’s disease.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
Not available.
Funding
Our work is supported by grants from the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation, the Burke Foundation, the Cure Alzheimer’s Fund, and the NIH (Grant No. 1R01NS140765).
Copyright
© The Author(s) 2026.
References
-
1. Feigin VL, Brainin M, Norrving B, Martins SO, Pandian J, Lindsay P, et al. World stroke organization: Global stroke fact sheet 2025. Int J Stroke. 2025;20(2):132-144.[DOI]
-
2. Catanese L, Tarsia J, Fisher M. Acute ischemic stroke therapy overview. Circ Res. 2017;120(3):541-558.[DOI]
-
3. Magid-Bernstein J, Girard R, Polster S, Srinath A, Romanos S, Awad IA, et al. Cerebral hemorrhage: Pathophysiology, treatment, and future directions. Circ Res. 2022;130(8):1204-1229.[DOI]
-
4. Pradilla G, Ratcliff JJ, Hall AJ, Saville BR, Allen JW, Paulon G, et al. Trial of early minimally invasive removal of intracerebral hemorrhage. N Engl J Med. 2024;390(14):1277-1289.[DOI]
-
5. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060-1072.[DOI]
-
6. Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171(2):273-285.[DOI]
-
7. Chen B, Chen Z, Liu M, Gao X, Cheng Y, Wei Y, et al. Inhibition of neuronal ferroptosis in the acute phase of intracerebral hemorrhage shows long-term cerebroprotective effects. Brain Res Bull. 2019;153:122-132.[DOI]
-
8. Fan W, Zheng J, Jiang F. Analysis of ferroptosis-related genes in cerebral ischemic stroke via immune infiltration and single-cell RNA-sequencing. BMC Med Genomics. 2025;18(1):31.[DOI]
-
9. Li Q, Han X, Lan X, Gao Y, Wan J, Durham F, et al. Inhibition of neuronal ferroptosis protects hemorrhagic brain. JCI Insight. 2017;2(7):e90777.[DOI]
-
10. Shen D, Wu W, Liu J, Lan T, Xiao Z, Gai K, et al. Ferroptosis in oligodendrocyte progenitor cells mediates white matter injury after hemorrhagic stroke. Cell Death Dis. 2022;13(3):259.[DOI]
-
11. Shi CL, Han XL, Chen JC, Pan QF, Gao YC, Guo PY, et al. Single-nucleus transcriptome unveils the role of ferroptosis in ischemic stroke. Heliyon. 2024;10(12):e32727.[DOI]
-
12. Liu T, Li X, Cui Y, Meng P, Zeng G, Wang Y, et al. Bioinformatics analysis identifies potential ferroptosis key genes in the pathogenesis of intracerebral hemorrhage. Front Neurosci. 2021;15:661663.[DOI]
-
13. Du B, Deng Z, Chen K, Yang Z, Wei J, Zhou L, et al. Iron promotes both ferroptosis and necroptosis in the early stage of reperfusion in ischemic stroke. Genes Dis. 2024;11(6):101262.[DOI]
-
14. Zille M, Karuppagounder SS, Chen Y, Gough PJ, Bertin J, Finger J, et al. Neuronal death after hemorrhagic stroke in vitro and in vivo shares features of ferroptosis and necroptosis. Stroke. 2017;48(4):1033-1043.[DOI]
-
15. Sorby-Adams A, Prime TA, Miljkovic JL, Prag HA, Krieg T, Murphy MP. A model of mitochondrial superoxide production during ischaemia-reperfusion injury for therapeutic development and mechanistic understanding. Redox Biol. 2024;72:103161.[DOI]
-
16. Karuppagounder SS, Alin L, Chen Y, Brand D, Bourassa MW, Dietrich K, et al. N-acetylcysteine targets 5 lipoxygenase-derived, toxic lipids and can synergize with prostaglandin E2 to inhibit ferroptosis and improve outcomes following hemorrhagic stroke in mice. Ann Ann Neurol. 2018;84(6):854-872.[DOI]
-
17. Ratan RR. The chemical biology of ferroptosis in the central nervous system. Cell Chem Biol. 2020;27(5):479-498.[DOI]
-
18. Jones DP, Sies H. The redox code. Antioxid Redox Signal. 2015;23(9):734-746.[DOI]
-
19. Li B, Ming H, Qin S, Nice EC, Dong J, Du Z, et al. Redox regulation: Mechanisms, biology and therapeutic targets in diseases. Sig Transduct Target Ther. 2025;10(1):72.[DOI]
-
20. Sies H. Oxidative eustress: On constant alert for redox homeostasis. Redox Biol. 2021;41:101867.[DOI]
-
21. Murphy TH, Miyamoto M, Sastre A, Schnaar RL, Coyle JT. Glutamate toxicity in a neuronal cell line involves inhibition of cystine transport leading to oxidative stress. Neuron. 1989;2(6):1547-1558.[DOI]
-
22. Choi DW. Excitotoxicity: Still hammering the ischemic brain in 2020. Front Neurosci. 2020;14:579953.[DOI]
-
23. Haskew-Layton RE, Payappilly JB, Smirnova NA, Ma TC, Chan KK, Murphy TH, et al. Controlled enzymatic production of astrocytic hydrogen peroxide protects neurons from oxidative stress via an Nrf2-independent pathway. Proc Natl Acad Sci U S A. 2010;107(40):17385-17390.[DOI]
-
24. Ratan RR. Does iron loading of oxygen-sensing prolyl hydroxylases rather than random Fenton-driven radical formation drive programmed ferroptosis and degeneration in neurological diseases? Curr Opin Physiol. 2019;7:60-65.[DOI]
-
25. Murphy TH, Schnaar RL, Coyle JT. Immature cortical neurons are uniquely sensitive to glutamate toxicity by inhibition of cystine uptake. FASEB J. 1990;4(6):1624-1633.[PubMed]
-
26. Li Y, Maher P, Schubert D. A role for 12-lipoxygenase in nerve cell death caused by glutathione depletion. Neuron. 1997;19(2):453-463.[DOI]
-
27. Tan S, Schubert D, Maher P. Oxytosis: A novel form of programmed cell death. Curr Top Med Chem. 2001;1(6):497-506.[DOI]
-
28. Ratan RR, Lee PJ, Baraban JM. Serum deprivation inhibits glutathione depletion-induced death in embryonic cortical neurons: Evidence against oxidative stress as a final common mediator of neuronal apoptosis. Neurochem Int. 1996;29(2):153-157.[DOI]
-
29. Li M, Zhou ZP, Sun M, Cao L, Chen J, Qin YY, et al. Reduced nicotinamide adenine dinucleotide phosphate, a pentose phosphate pathway product, might be a novel drug candidate for ischemic stroke. Stroke. 2016;47(1):187-195.[DOI]
-
30. Derry PJ, Vo ATT, Gnanansekaran A, Mitra J, Liopo AV, Hegde ML, et al. The chemical basis of intracerebral hemorrhage and cell toxicity with contributions from eryptosis and ferroptosis. Front Cell Neurosci. 2020;14:603043.[DOI]
-
31. Robinson SR, Dang TN, Dringen R, Bishop GM. Hemin toxicity: A preventable source of brain damage following hemorrhagic stroke. Redox Rep. 2009;14(6):228-235.[DOI]
-
32. Sies H, Mailloux RJ, Jakob U. Fundamentals of redox regulation in biology. Nat Rev Mol Cell Biol. 2024;25(9):701-719.[DOI]
-
33. Chiarugi P. Survival or death: The redox paradox. Antioxid Redox Signal. 2009;11(11):2651-2654.[DOI]
-
34. Sies H. Dynamics of intracellular and intercellular redox communication. Free Radic Biol Med. 2024;225:933-939.[DOI]
-
35. Bayır H, Anthonymuthu TS, Tyurina YY, Patel SJ, Amoscato AA, Lamade AM, et al. Achieving life through death: Redox biology of lipid peroxidation in ferroptosis. Cell Chem Biol. 2020;27(4):387-408.[DOI]
-
36. Connor JR, Menzies SL. Relationship of iron to oligodendrocytes and myelination. Glia. 1996;17(2):83-93.[DOI]
-
37. Bastian TW, von Hohenberg WC, Mickelson DJ, Lanier LM, Georgieff MK. Iron deficiency impairs developing hippocampal neuron gene expression, energy metabolism, and dendrite complexity. Dev Neurosci. 2016;38(4):264-276.[DOI]
-
38. Carlson ES, Tkac I, Magid R, O’Connor MB, Andrews NC, Schallert T, et al. Iron is essential for neuron development and memory function in mouse hippocampus. J Nutr. 2009;139(4):672-679.[DOI]
-
39. Muñoz P, Humeres A, Elgueta C, Kirkwood A, Hidalgo C, Núñez MT. Iron mediates N-methyl-D-aspartate receptor-dependent stimulation of calcium-induced pathways and hippocampal synaptic plasticity. J Biol Chem. 2011;286(15):13382-13392.[DOI]
-
40. Outten FW, Theil EC. Iron-based redox switches in biology. Antioxid Redox Signal. 2009;11(5):1029-1046.[DOI]
-
41. David S, Jhelum P, Ryan F, Jeong SY, Kroner A. Dysregulation of iron homeostasis in the central nervous system and the role of ferroptosis in neurodegenerative disorders. Antioxid Redox Signal. 2022;37(1-3):150-170.[DOI]
-
42. Rouault TA, Zhang DL, Jeong SY. Brain iron homeostasis, the choroid plexus, and localization of iron transport proteins. Metab Brain Dis. 2009;24(4):673-684.[DOI]
-
43. Levi S, Ripamonti M, Moro AS, Cozzi A. Iron imbalance in neurodegeneration. Mol Psychiatry. 2024;29(4):1139-1152.[DOI]
-
44. Kakhlon O, Cabantchik ZI. The labile iron pool: Characterization, measurement, and participation in cellular processes. Free Radic Biol Med. 2002;33(8):1037-1046.[DOI]
-
45. Jacobs A. An intracellular transit iron pool. Ciba Found Symp. 1976;51:91-106.[DOI]
-
46. Shi H, Bencze KZ, Stemmler TL, Philpott CC. A cytosolic iron chaperone that delivers iron to ferritin. Science. 2008;320(5880):1207-1210.[DOI]
-
47. Philpott CC, Protchenko O, Wang Y, Novoa-Aponte L, Leon-Torres A, Grounds S, et al. Iron-tracking strategies: Chaperones capture iron in the cytosolic labile iron pool. Front Mol Biosci. 2023;10:1127690.[DOI]
-
48. Patel SJ, Frey AG, Palenchar DJ, Achar S, Bullough KZ, Vashisht A, et al. A PCBP1-BolA2 chaperone complex delivers iron for cytosolic [2Fe-2S] cluster assembly. Nat Chem Biol. 2019;15(9):872-881.[DOI]
-
49. Nandal A, Ruiz JC, Subramanian P, Ghimire-Rijal S, Sinnamon RA, Stemmler TL, et al. Activation of the HIF prolyl hydroxylase by the iron chaperones PCBP1 and PCBP2. Cell Metab. 2011;14(5):647-657.[DOI]
-
50. Patel SJ, Protchenko O, Shakoury-Elizeh M, Baratz E, Jadhav S, Philpott CC. The iron chaperone and nucleic acid-binding activities of poly(rC)-binding protein 1 are separable and independently essential. Proc Natl Acad Sci U S A. 2021;118(25):e2104666118.[DOI]
-
51. Philpott CC, Ryu MS. Special delivery: Distributing iron in the cytosol of mammalian cells. Front Pharmacol. 2014;5:173.[DOI]
-
52. Maio N, Rouault TA. Outlining the complex pathway of mammalian Fe-S cluster biogenesis. Trends Biochem Sci. 2020;45(5):411-426.[DOI]
-
53. Vallières C, Benoit O, Guittet O, Huang ME, Lepoivre M, Golinelli-Cohen MP, et al. Iron-sulfur protein odyssey: Exploring their cluster functional versatility and challenging identification. Metallomics. 2024;16(5):mfae025.[DOI]
-
54. Talib EA, Outten CE. Iron-sulfur cluster biogenesis, trafficking, and signaling: Roles for CGFS glutaredoxins and BolA proteins. Biochim Biophys Acta Mol Cell Res. 2021;1868(1):118847.[DOI]
-
55. Terzi EM, Sviderskiy VO, Alvarez SW, Whiten GC, Possemato R. Iron-sulfur cluster deficiency can be sensed by IRP2 and regulates iron homeostasis and sensitivity to ferroptosis independent of IRP1 and FBXL5. Sci Adv. 2021;7(22):eabg4302.[DOI]
-
56. Guo J, Tuo QZ, Lei P. Iron, ferroptosis, and ischemic stroke. J Neurochem. 2023;165(4):487-520.[DOI]
-
57. Shi Z, Chen K, Wang Y, Du H. The crosstalk between ferritinophagy and ferroptosis in ischemic stroke: Regulatory mechanisms and therapeutic implications. Cell Mol Neurobiol. 2025;45(1):73.[DOI]
-
58. Mancias JD, Wang X, Gygi SP, Harper JW, Kimmelman AC. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature. 2014;509(7498):105-109.[DOI]
-
59. van Etten ES, van der Grond J, Dumas EM, van den Bogaard SJ, van Buchem MA, Wermer MJ. MRI susceptibility changes suggestive of iron deposition in the thalamus after ischemic stroke. Cerebrovasc Dis. 2015;40(1-2):67-72.[DOI]
-
60. Kataike VM, Desmond PM, Steward C, Mitchell PJ, Davey C, Yassi N, et al. Iron changes within infarct tissue in ischemic stroke patients after successful reperfusion quantified using QSM. Neuroradiology. 2024;66(12):2233-2242.[DOI]
-
61. Feng CK, Chen WJ, Kung WM, Sun YY, Lin MS. CDGSH iron-sulfur domain 2 as a therapeutic target for stroke: An opinion article. Front Mol Neurosci. 2024;17:1386118.[DOI]
-
62. Hu M, Huang J, Chen L, Sun XR, Yao ZM, Tong XH, et al. Upregulation of CDGSH iron sulfur doma in 2 attenuates cerebral ischemia/reperfusion injury. Neural Regen Res. 2023;18(7):1512-1520.[DOI]
-
63. Li R, Zhang X, Gu L, Yuan Y, Luo X, Shen W, et al. CDGSH iron sulfur domain 2 over-expression alleviates neuronal ferroptosis and brain injury by inhibiting lipid peroxidation via AKT/mTOR pathway following intracerebral hemorrhage in mice. J Neurochem. 2023;165(3):426-444.[DOI]
-
64. Philpott CC, Patel SJ, Protchenko O. Management versus miscues in the cytosolic labile iron pool: The varied functions of iron chaperones. Biochim Biophys Acta Mol Cell Res. 2020;1867(11):118830.[DOI]
-
65. Ackrill P, Ralston AJ, Day JP, Hodge KC. Successful removal of aluminium from patient with dialysis encephalopathy. Lancet. 1980;2(8196):692-693.[DOI]
-
66. Smith RS. Iron excretion in thalassaemia major after administration of chelating agents. Br Med J. 1962;2(5319):1577-1580.[DOI]
-
67. Swayambhu G, Bruno M, Gulick AM, Pfeifer BA. Siderophore natural products as pharmaceutical agents. Curr Opin Biotechnol. 2021;69:242-251.[DOI]
-
68. Zaman K, Ryu H, Hall D, O’Donovan K, Lin KI, Miller MP, et al. Protection from oxidative stress-induced apoptosis in cortical neuronal cultures by iron chelators is associated with enhanced DNA binding of hypoxia-inducible factor-1 and ATF-1/CREB and increased expression of glycolytic enzymes, p21(Waf1/cip1), and erythropoietin. J Neurosci. 1999;19(22):9821-9830.[DOI]
-
69. Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, et al. HIFα targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science. 2001;292(5516):464-468.[DOI]
-
70. Wang GL, Semenza GL. General involvement of hypoxia-inducible factor 1 in transcriptional response to hypoxia. Proc Natl Acad Sci U S A. 1993;90(9):4304-4308.[DOI]
-
71. Epstein AC, Gleadle JM, McNeill LA, Hewitson KS, O’Rourke J, Mole DR, et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell. 2001;107(1):43-54.[DOI]
-
72. Siddiq A, Aminova LR, Troy CM, Suh K, Messer Z, Semenza GL, et al. Selective inhibition of hypoxia-inducible factor (HIF) prolyl-hydroxylase 1 mediates neuroprotection against normoxic oxidative death via HIF- and CREB-independent pathways. J Neurosci. 2009;29(27):8828-8838.[DOI]
-
73. Aminova LR, Chavez JC, Lee J, Ryu H, Kung A, Lamanna JC, et al. Prosurvival and prodeath effects of hypoxia-inducible factor-1alpha stabilization in a murine hippocampal cell line. J Biol Chem. 2005;280(5):3996-4003.[DOI]
-
74. Aminova LR, Siddiq A, Ratan RR. Antioxidants, HIF prolyl hydroxylase inhibitors or short interfering RNAs to BNIP3 or PUMA, can prevent prodeath effects of the transcriptional activator, HIF-1alpha, in a mouse hippocampal neuronal line. Antioxid Redox Signal. 2008;10(12):1989-1998.[DOI]
-
75. Kreß JKC, Jessen C, Hufnagel A, Schmitz W, Xavier da Silva TN, Ferreira Dos Santos A, et al. The integrated stress response effector ATF4 is an obligatory metabolic activator of NRF2. Cell Rep. 2023;42(7):112724.[DOI]
-
76. Lange PS, Chavez JC, Pinto JT, Coppola G, Sun CW, Townes TM, et al. ATF4 is an oxidative stress-inducible, prodeath transcription factor in neurons in vitro and in vivo. J Exp Med. 2008;205(5):1227-1242.[DOI]
-
77. Karuppagounder SS, Alim I, Khim SJ, Bourassa MW, Sleiman SF, John R, et al. Therapeutic targeting of oxygen-sensing prolyl hydroxylases abrogates ATF4-dependent neuronal death and improves outcomes after brain hemorrhage in several rodent models. Sci Transl Med. 2016;8(328):328ra29.[DOI]
-
78. Smirnova NA, Rakhman I, Moroz N, Basso M, Payappilly J, Kazakov S, et al. Utilization of an in vivo reporter for high throughput identification of branched small molecule regulators of hypoxic adaptation. Chem Biol. 2010;17(4):380-391.[DOI]
-
79. Aimé P, Karuppagounder SS, Rao A, Chen Y, Burke RE, Ratan RR, et al. The drug adaptaquin blocks ATF4/CHOP-dependent pro-death Trib3 induction and protects in cellular and mouse models of Parkinson’s disease. Neurobiol Dis. 2020;136:104725.[DOI]
-
80. Auriat AM, Silasi G, Wei Z, Paquette R, Paterson P, Nichol H, et al. Ferric iron chelation lowers brain iron levels after intracerebral hemorrhage in rats but does not improve outcome. Exp Neurol. 2012;234(1):136-143.[DOI]
-
81. Wu H, Wu T, Xu X, Wang J, Wang J. Iron toxicity in mice with collagenase-induced intracerebral hemorrhage. J Cereb Blood Flow Metab. 2011;31(5):1243-1250.[DOI]
-
82. Hatakeyama T, Okauchi M, Hua Y, Keep RF, Xi G. Deferoxamine reduces neuronal death and hematoma lysis after intracerebral hemorrhage in aged rats. Transl Stroke Res. 2013;4(5):546-553.[DOI]
-
83. Freret T, Valable S, Chazalviel L, Saulnier R, MacKenzie ET, Petit E, et al. Delayed administration of deferoxamine reduces brain damage and promotes functional recovery after transient focal cerebral ischemia in the rat. Eur J Neurosci. 2006;23(7):1757-1765.[DOI]
-
84. Guo X, Jin X, Han K, Kang S, Tian S, Lv X, et al. Iron promotes neurological function recovery in mice with ischemic stroke through endogenous repair mechanisms. Free Radic Biol Med. 2022;182:59-72.[DOI]
-
85. Ugale R, Vatte S, Girdhar P, Anandani D. Deferoxamine prevents BBB disruption, neuroinflammation and apoptotic changes in early hours of ischemic reperfusion injury. Neurochem Int. 2025;188:106009.[DOI]
-
86. Song S, Hua Y, Keep RF, He Y, Wang J, Wu J, et al. Deferoxamine reduces brain swelling in a rat model of hippocampal intracerebral hemorrhage. Acta Neurochir Suppl. 2008;105:13-18.[DOI]
-
87. Wan Y, Xie Q, Hua Y, Xi G, Keep RF, Pandey A. Comparing white and gray matter responses to lobar intracerebral hemorrhage in piglets and the effects of deferoxamine. Exp Neurol. 2025;383:115041.[DOI]
-
88. Nakamura T, Keep RF, Hua Y, Schallert T, Hoff JT, Xi G. Deferoxamine-induced attenuation of brain edema and neurological deficits in a rat model of intracerebral hemorrhage. J Neurosurg. 2004;100(4):672-678.[DOI]
-
89. Selim M, Foster LD, Moy CS, Xi G, Hill MD, Morgenstern LB, et al. Deferoxamine mesylate in patients with intracerebral haemorrhage (i-DEF): A multicentre, randomised, placebo-controlled, double-blind phase 2 trial. Lancet Neurol. 2019;18(5):428-438.[DOI]
-
90. Yeatts SD, Palesch YY, Moy CS, Selim M. High dose deferoxamine in intracerebral hemorrhage (HI-DEF) trial: Rationale, design, and methods. Neurocrit Care. 2013;19(2):257-266.[DOI]
-
91. Foster L, Robinson L, Yeatts SD, Conwit RA, Shehadah A, Lioutas V, et al. Effect of deferoxamine on trajectory of recovery after intracerebral hemorrhage: A post hoc analysis of the i-DEF trial. Stroke. 2022;53(7):2204-2210.[DOI]
-
92. Polymeris AA, Lioutas VA, Foster LD, Incontri D, Heistand EC, Marchal J, et al. Early neurological improvement with deferoxamine after intracerebral hemorrhage: A post hoc analysis of the i-DEF trial. Int J Stroke. 2025;20(10):1235-1245.[DOI]
-
93. Ayton S, Moreau C, Devos D, Bush AI. Iron on trial: Recasting the role of iron in neurodegeneration. Brain. 2025;148(12):4241-4247.[DOI]
-
94. Devos D, Labreuche J, Rascol O, Corvol JC, Duhamel A, Guyon Delannoy P, et al. Trial of deferiprone in Parkinson’s disease. N Engl J Med. 2022;387(22):2045-2055.[DOI]
-
95. Ayton S, Barton D, Brew B, Brodtmann A, Clarnette R, Desmond P, et al. Deferiprone in Alzheimer disease: A randomized clinical trial. JAMA Neurol. 2025;82(1):11-18.[DOI]
-
96. Xiao W, Loscalzo J. Metabolic responses to reductive stress. Antioxid Redox Signal. 2020;32(18):1330-1347.[DOI]
-
97. Lu SC. Glutathione synthesis. Biochim Biophys Acta. 2013;1830(5):3143-3153.[DOI]
-
98. Seiler A, Schneider M, Förster H, Roth S, Wirth EK, Culmsee C, et al. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab. 2008;8(3):237-248.[DOI]
-
99. Dalle-Donne I, Rossi R, Colombo G, Giustarini D, Milzani A. Protein S-glutathionylation: A regulatory device from bacteria to humans. Trends Biochem Sci. 2009;34(2):85-96.[DOI]
-
100. Chandel NS. NADPH—The Forgotten Reducing Equivalent. Cold Spring Harb Perspect Biol. 2021;13(6):a040550.[DOI]
-
101. Bolaños JP, Magistretti PJ. The neuron-astrocyte metabolic unit as a cornerstone of brain energy metabolism in health and disease. Nat Metab. 2025;7(12):2414-2423.[DOI]
-
102. Shih AY, Johnson DA, Wong G, Kraft AD, Jiang L, Erb H, et al. Coordinate regulation of glutathione biosynthesis and release by Nrf2-expressing Glia Potently protects neurons from oxidative stress. J Neurosci. 2003;23(8):3394-3406.[DOI]
-
103. Dringen R, Arend C. Glutathione metabolism of the brain-the role of astrocytes. J Neurochem. 2025;169(5):e70073.[DOI]
-
104. Chen MS, Wang SF, Hsu CY, Yin PH, Yeh TS, Lee HC, et al. CHAC1 degradation of glutathione enhances cystine-starvation-induced necroptosis and ferroptosis in human triple negative breast cancer cells via the GCN2-eIF2α-ATF4 pathway. Oncotarget. 2017;8(70):114588-114602.[DOI]
-
105. Dong J, Qiu H, Garcia-Barrio M, Anderson J, Hinnebusch AG. Uncharged tRNA activates GCN2 by displacing the protein kinase moiety from a bipartite tRNA-binding domain. Mol Cell. 2000;6(2):269-279.[DOI]
-
106. Ryoo HD. The integrated stress response in metabolic adaptation. J Biol Chem. 2024;300(4):107151.[DOI]
-
107. Lewerenz J, Maher P. Basal levels of eIF2alpha phosphorylation determine cellular antioxidant status by regulating ATF4 and xCT expression. J Biol Chem. 2009;284(2):1106-1115.[DOI]
-
108. Ratan RR, Murphy TH, Baraban JM. Oxidative stress induces apoptosis in embryonic cortical neurons. J Neurochem. 1994;62(1):376-379.[DOI]
-
109. Ratan RR, Murphy TH, Baraban JM. Macromolecular synthesis inhibitors prevent oxidative stress-induced apoptosis in embryonic cortical neurons by shunting cysteine from protein synthesis to glutathione. J Neurosci. 1994;14(7):4385-4392.[DOI]
-
110. Esch F, Lin KI, Hills A, Zaman K, Baraban JM, Chatterjee S, et al. Purification of a multipotent antideath activity from bovine liver and its identification as arginase: Nitric oxide-independent inhibition of neuronal apoptosis. J Neurosci. 1998;18(11):4083-4095.[DOI]
-
111. Tan S, Sagara Y, Liu Y, Maher P, Schubert D. The regulation of reactive oxygen species production during programmed cell death. J Cell Biol. 1998;141(6):1423-1432.[DOI]
-
112. Stachowicz K. The role of polyunsaturated fatty acids in neuronal signaling in depression and cognitive processes. Arch Biochem Biophys. 2023;737:109555.[DOI]
-
113. Cobley JN, Fiorello ML, Bailey DM. 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 2018;15:490-503.[DOI]
-
114. Wenzel SE, Tyurina YY, Zhao J, St Croix CM, Dar HH, Mao G, et al. PEBP1 wardens ferroptosis by enabling lipoxygenase generation of lipid death signals. Cell. 2017;171(3):628-641.[DOI]
-
115. Evans JH, Fergus DJ, Leslie CC. Inhibition of the MEK1/ERK pathway reduces arachidonic acid release independently of cPLA2 phosphorylation and translocation. BMC Biochem. 2002;3:30.[DOI]
-
116. Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol. 2017;13(1):81-90.[DOI]
-
117. Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 2017;13(1):91-98.[DOI]
-
118. Anthonymuthu TS, Kenny EM, Shrivastava I, Tyurina YY, Hier ZE, Ting HC, et al. Empowerment of 15-lipoxygenase catalytic competence in selective oxidation of membrane ETE-PE to ferroptotic death signals, HpETE-PE. J Am Chem Soc. 2018;140(51):17835-17839.[DOI]
-
119. Wang X, Shao Q, Gao Y. The emerging role of 12/15-lipoxygenase in ischemic stroke. Brain Res Bull. 2025;221:111194.[DOI]
-
120. Cakir-Aktas C, Bodur E, Yemisci M, van Leyen K, Karatas H. 12/15-lipoxygenase inhibition attenuates neuroinflammation by suppressing inflammasomes. Front Cell Neurosci. 2023;17:1277268.[DOI]
-
121. Rai G, Joshi N, Jung JE, Liu Y, Schultz L, Yasgar A, et al. Potent and selective inhibitors of human reticulocyte 12/15-lipoxygenase as anti-stroke therapies. J Med Chem. 2014;57(10):4035-4048.[DOI]
-
122. Yigitkanli K, Pekcec A, Karatas H, Pallast S, Mandeville E, Joshi N, et al. Inhibition of 12/15-lipoxygenase as therapeutic strategy to treat stroke. Ann Neurol. 2013;73(1):129-135.[DOI]
-
123. Gilbert NC, Newcomer ME, Werz O. Untangling the web of 5-lipoxygenase-derived products from a molecular and structural perspective: The battle between pro- and anti-inflammatory lipid mediators. Biochem Pharmacol. 2021;193:114759.[DOI]
-
124. Firdous SM, Pal S. Modulation of several downstream cascades served by enzymes in the pathogenesis of stroke. In: Mathew B, Parambi DGT, editors. Enzymes in Neurodegenerative Disorders. Singapore: Springer; 2024. p. 153-169. [10.1007/978-981-97-6822-6_9]
-
125. Hijioka M, Anan J, Ishibashi H, Kurauchi Y, Hisatsune A, Seki T, et al. Inhibition of leukotriene B4 action mitigates intracerebral hemorrhage-associated pathological events in mice. J Pharmacol Exp Ther. 2017;360(3):399-408.[DOI]
-
126. Tu XK, Yang WZ, Wang CH, Shi SS, Zhang YL, Chen CM, et al. Zileuton reduces inflammatory reaction and brain damage following permanent cerebral ischemia in rats. Inflammation. 2010;33(5):344-352.[DOI]
-
127. Tu XK, Zhang HB, Shi SS, Liang RS, Wang CH, Chen CM, et al. 5-LOX inhibitor zileuton reduces inflammatory reaction and ischemic brain damage through the activation of PI3K/Akt signaling pathway. Neurochem Res. 2016;41(10):2779-2787.[DOI]
-
128. Tu XK, Yang WZ, Shi SS, Chen CM, Wang CH. 5-lipoxygenase inhibitor zileuton attenuates ischemic brain damage: Involvement of matrix metalloproteinase 9. Neurol Res. 2009;31(8):848-852.[DOI]
-
129. Silva BC, de Miranda AS, Rodrigues FG, Silveira AL, Resende GH, Moraes MF, et al. The 5-lipoxygenase (5-LOX) inhibitor zileuton reduces inflammation and infarct size with improvement in neurological outcome following cerebral ischemia. Curr Neurovasc Res. 2015;12(4):398-403.[DOI]
-
130. Ma T, Du J, Zhang Y, Wang Y, Wang B, Zhang T. GPX4-independent ferroptosis: A new strategy in disease’s therapy. Cell Death Discov. 2022;8:434.[DOI]
-
131. Alim I, Caulfield JT, Chen Y, Swarup V, Geschwind DH, Ivanova E, et al. Selenium drives a transcriptional adaptive program to block ferroptosis and treat stroke. Cell. 2019;177(5):1262-1279.[DOI]
-
132. Forcina GC, Dixon SJ. GPX4 at the crossroads of lipid homeostasis and ferroptosis. Proteomics. 2019;19(18):e1800311.[DOI]
-
133. Gęgotek A, Skrzydlewska E. Biological effect of protein modifications by lipid peroxidation products. Chem Phys Lipids. 2019;221:46-52.[DOI]
-
134. Chen JJ, Yu BP. Alterations in mitochondrial membrane fluidity by lipid peroxidation products. Free Radic Biol Med. 1994;17(5):411-418.[DOI]
-
135. Roeck BF, Lotfipour Nasudivar S, Vorndran MRH, Schueller L, Yapici FI, Rübsam M, et al. Ferroptosis spreads to neighboring cells via plasma membrane contacts. Nat Commun. 2025;16(1):2951.[DOI]
-
136. Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. 2019;575(7784):688-692.[DOI]
-
137. Zhang S, Gou S, Zhang Q, Yong X, Gan B, Jia D. FSP1 oxidizes NADPH to suppress ferroptosis. Cell Res. 2023;33(12):967-970.[DOI]
-
138. Yang JS, Morris AJ, Kamizaki K, Chen J, Stark J, Oldham WM, et al. ALDH7A1 protects against ferroptosis by generating membrane NADH and regulating FSP1. Cell. 2025;188(10):2569-2585.[DOI]
-
139. Megarioti AH, Deol KK, Olzmann JA. Cell death: Dynamics and compartmentalization of NADH and FSP1 in ferroptosis. Curr Biol. 2025;35(11):R406-R409.[DOI]
-
140. Kraft VAN, Bezjian CT, Pfeiffer S, Ringelstetter L, Müller C, Zandkarimi F, et al. GTP cyclohydrolase 1/tetrahydrobiopterin counteract ferroptosis through lipid remodeling. ACS Cent Sci. 2020;6(1):41-53.[DOI]
-
141. Scarpellini C, Klejborowska G, Lanthier C, Hassannia B, Vanden Berghe T, Augustyns K. Beyond ferrostatin-1: A comprehensive review of ferroptosis inhibitors. Trends Pharmacol Sci. 2023;44(12):902-916.[DOI]
-
142. Chaudhary G, Sinha K, Gupta YK. Protective effect of exogenous administration of alpha-tocopherol in middle cerebral artery occlusion model of cerebral ischemia in rats. Fundam Clin Pharmacol. 2003;17(6):703-707.[DOI]
-
143. Murad LB, Guimarães MR, Paganelli A, de Oliveira CA, Vianna LM. Alpha-tocopherol in the brain tissue preservation of stroke-prone spontaneously hypertensive rats. J Physiol Biochem. 2014;70(1):49-60.[DOI]
-
144. Jiao Y, Shang J, Ohta Y, Yan H, Liu X, Li X, et al. Neuroprotective effects of tocovid pretreatment in a mouse stroke model. J Stroke Cerebrovasc Dis. 2018;27(8):2166-2174.[DOI]
-
145. Khanna S, Roy S, Slivka A, Craft TK, Chaki S, Rink C, et al. Neuroprotective properties of the natural vitamin E alpha-tocotrienol. Stroke. 2005;36(10):2258-2264.[DOI]
-
146. Khanna S, Heigel M, Weist J, Gnyawali S, Teplitsky S, Roy S, et al. Excessive α-tocopherol exacerbates microglial activation and brain injury caused by acute ischemic stroke. FASEB J. 2015;29(3):828-836.[DOI]
-
147. Schürks M, Glynn RJ, Rist PM, Tzourio C, Kurth T. Effects of vitamin E on stroke subtypes: Meta-analysis of randomised controlled trials. BMJ. 2010;341:c5702.[DOI]
-
148. Maggio E, Bocchini VP, Carnevale R, Pignatelli P, Violi F, Loffredo L. Vitamin E supplementation (alone or with other antioxidants) and stroke: A meta-analysis. Nutr Rev. 2024;82(8):1069-1078.[DOI]
-
149. Anthonymuthu TS, Tyurina YY, Sun WY, Mikulska-Ruminska K, Shrivastava IH, Tyurin VA, et al. Resolving the paradox of ferroptotic cell death: Ferrostatin-1 binds to 15LOX/PEBP1 complex, suppresses generation of peroxidized ETE-PE, and protects against ferroptosis. Redox Biol. 2021;38:101744.[DOI]
-
150. Shi W, Yuan S, Cheng G, Zhang H, Liu KJ, Ji X, et al. Blood brain barrier-targeted lipid nanoparticles improved the neuroprotection of Ferrostatin-1 against cerebral ischemic damage in an experimental stroke model. Exp Neurol. 2024;379:114849.[DOI]
-
151. Liu X, Du Y, Liu J, Cheng L, He W, Zhang W. Ferrostatin-1 alleviates cerebral ischemia/reperfusion injury through activation of the AKT/GSK3β signaling pathway. Brain Res Bull. 2023;193:146-157.[DOI]
-
152. Huang L, Zhang Y, Zhao L, Chen Q, Li L. Ferrostatin-1 polarizes microglial cells toward M2 phenotype to alleviate inflammation after intracerebral hemorrhage. Neurocrit Care. 2022;36(3):942-954.[DOI]
-
153. Morrow JP, Mazrad ZAI, Bush AI, Kempe K. Poly (2-oxazoline)–Ferrostatin-1 drug conjugates inhibit ferroptotic cell death. J Control Release. 2022;350:193-203.[DOI]
-
154. Dar HH, Mikulska-Ruminska K, Tyurina YY, Luci DK, Yasgar A, Samovich SN, et al. Discovering selective antiferroptotic inhibitors of the 15LOX/PEBP1 complex noninterfering with biosynthesis of lipid mediators. Proc Natl Acad Sci U S A. 2023;120(25):e2218896120.[DOI]
-
155. Conrad M, Proneth B. Selenium: Tracing another essential element of ferroptotic cell death. Cell Chem Biol. 2020;27(4):409-419.[DOI]
-
156. Luchkova A, Mata A, Cadenas S. Nrf2 as a regulator of energy metabolism and mitochondrial function. FEBS Lett. 2024;598(17):2092-2105.[DOI]
-
157. Levings DC, Pathak SS, Yang YM, Slattery M. Limited expression of Nrf2 in neurons across the central nervous system. Redox Biol. 2023;65:102830.[DOI]
-
158. Shih AY, Erb H, Sun X, Toda S, Kalivas PW, Murphy TH. Cystine/glutamate exchange modulates glutathione supply for neuroprotection from oxidative stress and cell proliferation. J Neurosci. 2006;26(41):10514-10523.[DOI]
-
159. Ebding J, Mazzone F, Kins S, Pielage J, Maritzen T. How neurons cope with oxidative stress. Biol Chem. 2025;406(5-7):219-228.[DOI]
-
160. Wilson C, Muñoz-Palma E, González-Billault C. From birth to death: A role for reactive oxygen species in neuronal development. Semin Cell Dev Biol. 2018;80:43-49.[DOI]
-
161. Hu Q, Khanna P, Ee Wong BS, Lin Heng ZS, Subhramanyam CS, Thanga LZ, et al. Oxidative stress promotes exit from the stem cell state and spontaneous neuronal differentiation. Oncotarget. 2018;9(3):4223-4238.[DOI]
-
162. Stanciu M, Wang Y, Kentor R, Burke N, Watkins S, Kress G, et al. Persistent activation of ERK contributes to glutamate-induced oxidative toxicity in a neuronal cell line and primary cortical neuron cultures. J Biol Chem. 2000;275(16):12200-12206.[DOI]
-
163. Luo Y, DeFranco DB. Opposing roles for ERK1/2 in neuronal oxidative toxicity: Distinct mechanisms of ERK1/2 action at early versus late phases of oxidative stress. J Biol Chem. 2006;281(24):16436-16442.[DOI]
-
164. Martin DP, Schmidt RE, DiStefano PS, Lowry OH, Carter JG, Johnson EMJ. Inhibitors of protein synthesis and RNA synthesis prevent neuronal death caused by nerve growth factor deprivation. J Cell Biol. 1988;106(3):829-844.[DOI]
-
165. Rajkumar K, Nichita A, Anoor PK, Raju S, Singh SS, Burgula S. Understanding perspectives of signalling mechanisms regulating PEBP1 function. Cell Biochem Funct. 2016;34(6):394-403.[DOI]
-
166. Giltnane JM, Balko JM. Rationale for targeting the Ras/MAPK pathway in triple-negative breast cancer. Discov Med. 2014;17(95):275-283.[PubMed]
-
167. Levinthal DJ, Defranco DB. Reversible oxidation of ERK-directed protein phosphatases drives oxidative toxicity in neurons. J Biol Chem. 2005;280(7):5875-5883.[DOI]
-
168. Lu Y, Yang Q, Su Y, Ji Y, Li G, Yang X, et al. MYCN mediates TFRC-dependent ferroptosis and reveals vulnerabilities in neuroblastoma. Cell Death Dis. 2021;12(6):511.[DOI]
-
169. Alborzinia H, Flórez AF, Kreth S, Brückner LM, Yildiz U, Gartlgruber M, et al. MYCN mediates cysteine addiction and sensitizes neuroblastoma to ferroptosis. Nat Cancer. 2022;3(4):471-485.[DOI]
-
170. Wu KJ, Polack A, Dalla-Favera R. Coordinated regulation of iron-controlling genes, H-ferritin and IRP2 by c-MYC. Science. 1999;283(5402):676-679.[DOI]
-
171. Kreuz S, Fischle W. Oxidative stress signaling to chromatin in health and disease. Epigenomics. 2016;8(6):843-862.[DOI]
-
172. Chatterjee S, Zaman K, Ryu H, Conforto A, Ratan RR. Sequence-selective DNA binding drugs mithramycin A and chromomycin A3 are potent inhibitors of neuronal apoptosis induced by oxidative stress and DNA damage in cortical neurons. Ann Neurol. 2001;49(3):345-354.[PubMed]
-
173. Osada N, Kosuge Y, Oguchi S, Miyagishi H, Ishige K, Ito Y. Protective action of mithramycin against neurodegeneration and impairment of synaptic plasticity in the hippocampal CA1 area after transient global ischemia. Neurochem Int. 2012;60(1):47-54.[DOI]
-
174. Kennedy BJ. Metabolic and toxic effects of mithramycin during tumor therapy. Am J Med. 1970;49(4):494-503.[DOI]
-
175. Yu Q, Liu W, Chen Z, Zhang M. Specificity protein 1: A protein with a two-sided role in ischemic stroke. Front Cell Neurosci. 2021;15:757670.[DOI]
-
176. Langley B, D’Annibale MA, Suh K, Ayoub I, Tolhurst A, Bastan B, et al. Pulse inhibition of histone deacetylases induces complete resistance to oxidative death in cortical neurons without toxicity and reveals a role for cytoplasmic p21(Waf1/cip1) in cell cycle-independent neuroprotection. J Neurosci. 2008;28(1):163-176.[DOI]
-
177. Ryu H, Lee J, Olofsson BA, Mwidau A, Dedeoglu A, Escudero M, et al. Histone deacetylase inhibitors prevent oxidative neuronal death independent of expanded polyglutamine repeats via an Sp1-dependent pathway. Proc Natl Acad Sci U S A. 2003;100(7):4281-4286.[DOI]
-
178. Rivieccio MA, Brochier C, Willis DE, Walker BA, D’Annibale MA, McLaughlin K, et al. HDAC6 is a target for protection and regeneration following injury in the nervous system. i U S A. 2009;106(46):19599-19604.[DOI]
-
179. Basso M, Berlin J, Xia L, Sleiman SF, Ko B, Haskew-Layton R, et al. Transglutaminase inhibition protects against oxidative stress-induced neuronal death downstream of pathological ERK activation. J Neurosci. 2012;32(19):6561-6569.[DOI]
-
180. Kim HJ, Rowe M, Ren M, Hong JS, Chen PS, Chuang DM. Histone deacetylase inhibitors exhibit anti-inflammatory and neuroprotective effects in a rat permanent ischemic model of stroke: Multiple mechanisms of action. J Pharmacol Exp Ther. 2007;321(3):892-901.[DOI]
-
181. Lin YH, Dong J, Tang Y, Ni HY, Zhang Y, Su P, et al. Opening a new time window for treatment of stroke by targeting HDAC2. J Neurosci. 2017;37(28):6712-6728.[DOI]
-
182. Jiang Z, Yang H, Ni W, Gao X, Pei X, Jiang H, et al. Attenuation of neuronal ferroptosis in intracerebral hemorrhage by inhibiting HDAC1/2: Microglial heterogenization via the Nrf2/HO1 pathway. CNS Neurosci Ther. 2024;30(3):e14646.[DOI]
-
183. Kassis H, Shehadah A, Li C, Zhang Y, Cui Y, Roberts C, et al. Class IIa histone deacetylases affect neuronal remodeling and functional outcome after stroke. Neurochem Int. 2016;96:24-31.[DOI]
-
184. Sleiman SF, Basso M, Mahishi L, Kozikowski AP, Donohoe ME, Langley B, et al. Putting the ‘HAT’ back on survival signalling: The promises and challenges of HDAC inhibition in the treatment of neurological conditions. Expert Opin Investig Drugs. 2009;18(5):573-584.[DOI]
-
185. Sleiman SF, Olson DE, Bourassa MW, Karuppagounder SS, Zhang YL, Gale J, et al. Hydroxamic acid-based histone deacetylase (HDAC) inhibitors can mediate neuroprotection independent of HDAC inhibition. J Neurosci. 2014;34(43):14328-14337.[DOI]
-
186. Borgo C, D’Amore C, Sarno S, Salvi M, Ruzzene M. Protein kinase CK2: A potential therapeutic target for diverse human diseases. Sig Transduct Target Ther. 2021;6(1):183.[DOI]
-
187. Capece D, Verzella D, Flati I, Arboretto P, Cornice J, Franzoso G. NF-κB: Blending metabolism, immunity, and inflammation. Trends Immunol. 2022;43(9):757-775.[DOI]
-
188. Zille M, Oses-Prieto JA, Savage SR, Karuppagounder SS, Chen Y, Kumar A, et al. Hemin-induced death models hemorrhagic stroke and is a variant of classical neuronal ferroptosis. J Neurosci. 2022;42(10):2065-2079.[DOI]
-
189. Thompson C, Cloutier A, Bossé Y, Poisson C, Larivée P, McDonald PP, et al. Signaling by the cysteinyl-leukotriene receptor 2 INVOLVEMENT IN CHEMOKINE GENE TRANSCRIPTION. J Biol Chem. 2008;283(4):1974-1984.[DOI]
-
190. Heckmann BL, Tummers B, Green DR. Crashing the computer: Apoptosis vs. necroptosis in neuroinflammation. Cell Death Differ. 2019;26(1):41-52.[DOI]
-
191. Yu Z, Jiang N, Su W, Zhuo Y. Necroptosis: A novel pathway in neuroinflammation. Front Pharmacol. 2021;12:701564.[DOI]
-
192. Belavgeni A, Meyer C, Stumpf J, Hugo C, Linkermann A. Ferroptosis and necroptosis in the kidney. Cell Chem Biol. 2020;27(4):448-462.[DOI]
-
193. Iadecola C, Anrather J. The immunology of stroke: From mechanisms to translation. Nat Med. 2011;17(7):796-808.[DOI]
-
194. Bozza MT, Jeney V. Pro-inflammatory actions of heme and other hemoglobin-derived DAMPs. Front Immunol. 2020;11:1323.[DOI]
-
195. Schuhmann MK, Kollikowski AM, März AG, Bieber M, Pham M, Stoll G. Danger-associated molecular patterns are locally released during occlusion in hyper-acute stroke. Brain Behav Immun Health. 2021;15:100270.[DOI]
-
196. Sakai S, Shichita T. Role of alarmins in poststroke inflammation and neuronal repair. Semin Immunopathol. 2023;45(3):427-435.[DOI]
-
197. Wu L, Wu W, Tali ET, Yuh WT. Oligemia, penumbra, infarction: Understanding hypoperfusion with neuroimaging. Neuroimaging Clin N Am. 2018;28(4):599-609.[DOI]
-
198. de Kok MJC, Schaapherder AFM, Bloeme-Ter Horst JR, Faro MLL, de Vries DK, Ploeg RJ, et al. Clinical ischemia-reperfusion injury: Driven by reductive rather than oxidative stress? A narrative review. Biochim Biophys Acta Bioenerg. 2025;1866(2):149539.[DOI]
-
199. Ravi , Singh J. Redox imbalance and hypoxia-inducible factors: A multifaceted crosstalk. FEBS J. 2025;292(15):3833-3848.[DOI]
-
200. Wang Y, Ge M, Wang J, Xu Y, Wang N, Xu S. Metabolic reprogramming in ischemic stroke: When glycolytic overdrive meets lipid storm. Cell Death Dis. 2025;16(1):788.[DOI]
-
201. Li YW, Liu Y, Luo SZ, Huang XJ, Shen Y, Wang WS, et al. The significance of calcium ions in cerebral ischemia-reperfusion injury: Mechanisms and intervention strategies. Front Mol Biosci. 2025;12:1585758.[DOI]
-
202. Vetrovoy O, Sarieva K, Lomert E, Nimiritsky P, Eschenko N, Galkina O, et al. Pharmacological HIF1 inhibition eliminates downregulation of the pentose phosphate pathway and prevents neuronal apoptosis in rat hippocampus caused by severe hypoxia. J Mol Neurosci. 2020;70(5):635-646.[DOI]
-
203. Ryan DG, Yang M, Prag HA, Blanco GR, Nikitopoulou E, Segarra-Mondejar M, et al. Disruption of the TCA cycle reveals an ATF4-dependent integration of redox and amino acid metabolism. Elife. 2021;10:e72593.[DOI]
-
204. Kathagen-Buhmann A, Schulte A, Weller J, Holz M, Herold-Mende C, Glass R, et al. Glycolysis and the pentose phosphate pathway are differentially associated with the dichotomous regulation of glioblastoma cell migration versus proliferation. Neuro Oncol. 2016;18(9):1219-1229.[DOI]
-
205. Riegman M, Sagie L, Galed C, Levin T, Steinberg N, Dixon SJ, et al. Ferroptosis occurs through an osmotic mechanism and propagates independently of cell rupture. Nat Cell Biol. 2020;22(9):1042-1048.[DOI]
-
206. Tang LJ, Luo XJ, Tu H, Chen H, Xiong XM, Li NS, et al. Ferroptosis occurs in phase of reperfusion but not ischemia in rat heart following ischemia or ischemia/reperfusion. Naunyn Schmiedebergs Arch Pharmacol. 2021;394(2):401-410.[DOI]
-
207. Xia F, Keep RF, Ye F, Holste KG, Wan S, Xi G, et al. The fate of erythrocytes after cerebral hemorrhage. Transl Stroke Res. 2022;13(5):655-664.[DOI]
-
208. Dang G, Yang Y, Wu G, Hua Y, Keep RF, Xi G. Early erythrolysis in the hematoma after experimental intracerebral hemorrhage. Transl Stroke Res. 2017;8(2):174-182.[DOI]
-
209. Cao S, Zheng M, Hua Y, Chen G, Keep RF, Xi G. Hematoma changes during clot resolution after experimental intracerebral hemorrhage. Stroke. 2016;47(6):1626-1631.[DOI]
-
210. Zhang C, Cheng J, Yan X, Zheng Y, Lan X, Zhao Y, et al. Ferroptosis in ischemic stroke: From a Glia-neuron crosstalk perspective. Prog Neurobiol. 2025;257:102868.[DOI]
-
211. Liu Z, Shen X, Li M, Liu P, Ge Z, Jin J. Exploring the nexus: How ferroptosis, microglia, and neuroinflammation converge in ischemic stroke pathogenesis. Mol Neurobiol. 2025;62(7):8965-8976.[DOI]
-
212. Philips T, Rothstein JD. Oligodendroglia: Metabolic supporters of neurons. J Clin Investig. 2017;127(9):3271-3280.[DOI]
-
213. Giacci MK, Bartlett CA, Smith NM, Iyer KS, Toomey LM, Jiang H, et al. Oligodendroglia are particularly vulnerable to oxidative damage after neurotrauma in vivo. J Neurosci. 2018;38(29):6491-6504.[DOI]
-
214. Cheli VT, Correale J, Paez PM, Pasquini JM. Iron metabolism in oligodendrocytes and astrocytes, implications for myelination and remyelination. ASN Neuro. 2020;12:1759091420962681.[DOI]
-
215. Vinokurov AY, Stelmashuk OA, Ukolova PA, Zherebtsov EA, Abramov AY. Brain region specificity in reactive oxygen species production and maintenance of redox balance. Free Radic Biol Med. 2021;174:195-201.[DOI]
-
216. Dvoriantchikova G, Lypka KR, Adis EV, Ivanov D. Multiple types of programmed necrosis such as necroptosis, pyroptosis, oxytosis/ferroptosis, and parthanatos contribute simultaneously to retinal damage after ischemia-reperfusion. Sci Rep. 2022;12(1):17152.[DOI]
-
217. Zhang X, Li H, Zhao Y, Zhao T, Wang Z, Tang Q. Neuronal injury after ischemic stroke: Mechanisms of crosstalk involving necroptosis. J Mol Neurosci. 2025;75(1):15.[DOI]
-
218. Palazon A, Goldrath AW, Nizet V, Johnson RS. HIF transcription factors, inflammation, and immunity. Immunity. 2014;41(4):518-528.[DOI]
-
219. Siegert I, Schödel J, Nairz M, Schatz V, Dettmer K, Dick C, et al. Ferritin-mediated iron sequestration stabilizes hypoxia-inducible factor-1α upon LPS activation in the presence of ample oxygen. Cell Rep. 2015;13(10):2048-2055.[DOI]
-
220. Sivandzade F, Prasad S, Bhalerao A, Cucullo L. NRF2 and NF-қB interplay in cerebrovascular and neurodegenerative disorders: Molecular mechanisms and possible therapeutic approaches. Redox Biol. 2019;21:101059.[DOI]
-
221. Pan H, Wang H, Wang X, Zhu L, Mao L. The absence of Nrf2 enhances NF-κB-dependent inflammation following scratch injury in mouse primary cultured astrocytes. Mediators Inflamm. 2012;2012:217580.[DOI]
-
222. Correa F, Mallard C, Nilsson M, Sandberg M. Dual TNFα-induced effects on NRF2 mediated antioxidant defence in astrocyte-rich cultures: Role of protein kinase activation. Neurochem Res. 2012;37(12):2842-2855.[DOI]
-
223. Mattson MP, Camandola S. NF-κB in neuronal plasticity and neurodegenerative disorders. J Clin Invest. 2001;107(3):247-254.[DOI]
-
224. Zhao W, Gasterich N, Clarner T, Voelz C, Behrens V, Beyer C, et al. Astrocytic Nrf2 expression protects spinal cord from oxidative stress following spinal cord injury in a male mouse model. J Neuroinflammation. 2022;19(1):134.[DOI]
-
225. Warren KE, Hood RJ, Walker FR, Spratt NJ. Intracranial pressure is elevated at 24 h post-stroke in mice. Neuroprotection. 2024;2(1):60-64.[DOI]
-
226. Hood RJ, Beard DJ, McLeod DD, Murtha LA, Spratt NJ. Intracranial pressure elevation post-stroke: Mechanisms and consequences. Front Stroke. 2023;2:1119120.[DOI]
-
227. Kalisvaart ACJ, Bahr NA, Colbourne F. Relationship between edema and intracranial pressure following intracerebral hemorrhage in rat. Front Stroke. 2023;2:1155937.[DOI]
-
228. Kalisvaart ACJ, Abrahart AH, Coney AT, Gu S, Colbourne F. Intracranial pressure dysfunction following severe intracerebral hemorrhage in middle-aged rats. Transl Stroke Res. 2023;14(6):970-986.[DOI]
-
229. Wilkinson CM, Kalisvaart ACJ, Kung TFC, Abrahart AH, Khiabani E, Colbourne F. Tissue compliance and intracranial pressure responses to large intracerebral hemorrhage in young and aged spontaneously hypertensive rats. Hypertension. 2024;81(1):151-161.[DOI]
-
230. Kim YJ, Hyun J. Mechanosensitive ion channels in apoptosis and ferroptosis: Focusing on the role of Piezo1. BMB Rep. 2023;56(2):145-152.[DOI]
-
231. Ke W, Liao Z, Liang H, Tong B, Song Y, Li G, et al. Stiff substrate induces nucleus pulposus cell ferroptosis via YAP and N-cadherin mediated mechanotransduction. Adv Healthc Mater. 2023; 12(23): e2300458.[DOI]
-
232. Yu Y, Yan Y, Niu F, Wang Y, Chen X, Su G, et al. Ferroptosis: A cell death connecting oxidative stress, inflammation and cardiovascular diseases. Cell Death Discov. 2021;7(1):193.[DOI]
-
233. Dias IHK, Milic I, Heiss C, Ademowo OS, Polidori MC, Devitt A, et al. Inflammation, lipid (per)oxidation, and redox regulation. Antioxid Redox Signal. 2020;33(3):166-190.[DOI]
-
234. Tonnus W, Maremonti F, Gavali S, Schlecht MN, Gembardt F, Belavgeni A, et al. Multiple oestradiol functions inhibit ferroptosis and acute kidney injury. Nature. 2025;645(8082):1011-1019.[DOI]
-
235. Ide S, Ide K, Abe K, Kobayashi Y, Kitai H, McKey J, et al. Sex differences in resilience to ferroptosis underlie sexual dimorphism in kidney injury and repair. Cell Rep. 2022;41(6):111610.[DOI]
-
236. Bonomini F, Rodella LF, Rezzani R. Metabolic syndrome, aging and involvement of oxidative stress. Aging Dis. 2015;6(2):109-120.[DOI]
-
237. Niraula A, Sheridan JF, Godbout JP. Microglia priming with aging and stress. Neuropsychopharmacology. 2017;42(1):318-333.[DOI]
-
238. Ferrucci L, Fabbri E. Inflammageing: Chronic inflammation in ageing, cardiovascular disease, and frailty. Nat Rev Cardiol. 2018;15(9):505-522.[DOI]
-
239. Mishima E, Nakamura T, Doll S, Proneth B, Fedorova M, Pratt DA, et al. Recommendations for robust and reproducible research on ferroptosis. Nat Rev Mol Cell Biol. 2025;26(8):615-630.[DOI]
-
240. Lapchak PA, Zhang JH, Noble-Haeusslein LJ. RIGOR guidelines: Escalating STAIR and STEPS for effective translational research. Transl Stroke Res. 2013;4(3):279-285.[DOI]
-
241. Percie du Sert N, Hurst V, Ahluwalia A, Alam S, Avey MT, Baker M, et al. The ARRIVE guidelines 2.0: Updated guidelines for reporting animal research. PLoS Biol. 2020;18(7):e3000410.[DOI]
-
242. Millán M, DeGregorio-Rocasolano N, Pérez de la Ossa N, Reverté S, Costa J, Giner P, et al. Targeting pro-oxidant iron with deferoxamine as a treatment for ischemic stroke: Safety and optimal dose selection in a randomized clinical trial. Antioxidants. 2021;10(8):1270.[DOI]
-
243. Tian R, Ma H, Ren J, Li Y, Zhang Z, Xu Z, et al. Selenium-containing nanoscale hydrogen-bonded organic framework nanozymes for multienzyme cascade antioxidant-targeted therapy of cerebral ischemia-reperfusion injury. ACS Nano. 2026;20(2):2435-2450.[DOI]
-
244. Wang W, Zhang Z, Liu Y, Kong L, Li W, Hu W, et al. Nano-integrated cascade antioxidases opsonized by albumin bypass the blood-brain barrier for treatment of ischemia-reperfusion injury. Biomater Sci. 2022;10(24):7103-7116.[DOI]
Copyright
© The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Publisher’s Note
Science Exploration remains a neutral stance on jurisdictional claims in published
maps
and institutional affiliations. The views expressed in this article are solely those
of
the author(s) and do not reflect the opinions of the Editors or the publisher.
Share And Cite



Science Exploration Style
Kalisvaart AC, Ratan RR. Cracking the neuronal ferroptosis code: In vitro insights into mechanisms and treatment of stroke. Ferroptosis Oxid Stress. 2026;2:202518. https://doi.org/10.70401/fos.2026.0017
Tips
Copy completed.
Submit a Manuscript
Author Instructions
Cite this Article
Article Metrics
0
View
0
Download
Cited
Article Updates

- Abstract
- Keywords
- 1. Introduction
- 2. The Emergence of Ferroptosis: Lessons from In Vitro Models
- 3. Redox Networks as Determinants of Ferroptotic Susceptibility
- 4. Translational Frontiers: Timing, Spatial Dynamics, and Cellular Crosstalk
- 5. Conclusion: The Path to Translation
- Supplementary materials
- Acknowledgements
- Authors contributions
- Conflicts of interest
- Ethical approval
- Consent to participate
- Consent for publication
- Availability of data and materials
- Funding
- Copyright
Science Exploration Style
Kalisvaart AC, Ratan RR. Cracking the neuronal ferroptosis code: In vitro insights into mechanisms and treatment of stroke. Ferroptosis Oxid Stress. 2026;2:202518. https://doi.org/10.70401/fos.2026.0017
copy
Share Link
copy