Metabolic reprogramming of amino acids dictates tumor susceptibility to ferroptosis
Guangyao Shan
1,#

,
Yunyi Bian
2,#
,
Shencheng Ren
1,#
,
Guoshu Bi
1,*
,
Cheng Zhan
1,*
*Correspondence to:
Guoshu Bi, Department of Thoracic Surgery, Zhongshan Hospital, Fudan University, Shanghai 200032, China.
E-mail: gsbi18@fudan.edu.cn
Cheng Zhan, Department of Thoracic Surgery, Zhongshan Hospital, Fudan University, Shanghai 200032, China. E-mail: czhan10@fudan.edu.cn
Cheng Zhan, Department of Thoracic Surgery, Zhongshan Hospital, Fudan University, Shanghai 200032, China. E-mail: czhan10@fudan.edu.cn
Ferroptosis Oxid Stress. 2026;2:202525. 10.70401/fos.2026.0023
Received: December 29, 2025Accepted: March 24, 2026Published: March 26, 2026
Abstract
Metabolic reprogramming fundamentally drives cancer progression, with aberrant amino acid metabolism serving as a critical nexus for maintaining redox homeostasis and dictating cell fate. Ferroptosis, an iron-dependent form of cell death driven by lipid peroxidation, is closely related to intracellular amino acid metabolic networks. Here, we systematically delineate how key amino acids function as multi-dimensional regulatory nodes that orchestrate tumor cell susceptibility to ferroptosis. We provide a focused analysis of the context-dependent mechanisms through which these metabolic pathways rewire cellular redox capacity, modulate central anti-ferroptotic defense nodes (e.g., GPX4), and reshape the tumor microenvironment. Finally, we highlight the profound metabolic plasticity and spatiotemporal heterogeneity of these networks, exposing the intrinsic vulnerabilities within the amino acid-ferroptosis axis that drive drug resistance and tumor evolution.
Keywords
Ferroptosis, amino acid metabolism, redox homeostasis, metabolic plasticity, therapeutic vulnerabilities
1. Introduction
Ferroptosis is an iron-dependent and lipid peroxidation-driven form of regulated cell death, which is distinct from traditional cell death pathways such as apoptosis, necrosis, and autophagy[1]. Operationally, it proceeds without the caspase activation and chromatin condensation hallmarks of apoptosis, lacks the massive cellular swelling and early plasma membrane rupture typical of necrosis, and occurs independently of the double-membrane vesicles characteristic of autophagy. Instead, the key features of ferroptosis include the generation of reactive oxygen species (ROS), elevated intracellular ferrous iron levels, inhibition of antioxidant pathways, and accumulation of lipid peroxides on cellular membranes. Morphologically, it is uniquely characterized by shrunken mitochondria with increased membrane density and reduced or absent cristae[2]. In recent years, ferroptosis has garnered significant attention due to its broad involvement in multiple pathophysiological processes, playing important roles in neurodegenerative diseases, ischemia-reperfusion injury, fibrosis, and more[3]. In the treatment of malignancies, inducing ferroptosis has emerged as a promising strategy to overcome resistance to conventional therapies, including chemotherapy, radiotherapy, and immunotherapy[4,5].
Metabolic reprogramming is a fundamental hallmark of cancer[6]. To meet the demands of rapid proliferation, tumor cells undergo metabolic remodeling to acquire energy and biosynthetic precursors. However, this hypermetabolic state also confers specific vulnerabilities[7]. Mounting evidence indicates that metabolic reprogramming is a central driver of the ferroptosis regulatory network, where the flux through carbohydrate, lipid, and amino acid pathways directly determines cellular susceptibility to lipid peroxidation[8,9].
Among various nutrients, profound alterations in amino acid metabolism provide multi-dimensional support for tumor cells spanning from energy supply to oxidative stress[10-12]. Amino acids are not only the fundamental blocks for protein synthesis but also serve as crucial metabolic intermediates. By supplying carbon to the tricarboxylic acid (TCA) cycle and nitrogen for nucleotide synthesis, they drive the continuous proliferation and biomass accumulation of tumor cells[10,13,14]. This metabolic dependency is particularly prominent in maintaining redox homeostasis[11]. Multiple studies have highlighted that tumor cells enhance the metabolism of glutamine, cysteine, and glycine to promote the synthesis of antioxidant molecules like glutathione (GSH)[10,11,15]. This mechanism allows tumor cells to neutralize ROS generated during metabolism, thereby sustaining survival under high oxidative stress. Concurrently, specific amino acids (e.g., glutamine) are involved in lipid synthesis and regulate oxidative phosphorylation to meet the energy demands of specific tumor cell populations, such as leukemia stem cells[14,16].
Tumor cells often exhibit “addiction” to specific amino acids due to the upregulated expression of amino acid transporters or enhanced activity of key catabolic enzymes[17]. Consequently, targeted disruption of amino acid supply or utilization represents a powerful strategy to sensitize cancer cells to ferroptosis. In light of this, this review systematically explores the regulatory landscape of amino acid metabolism in ferroptosis, elucidating the molecular mechanisms of ferroptotic evasion and the potential for leveraging these metabolic dependencies to enhance ferroptosis-based research and precision strategies.
2. The Canonical Redox Axis
2.1 Cysteine
Long before the formal conceptualization of ferroptosis, the absolute cellular dependency on specific amino acids for survival and oxidative defense was firmly established. In 1955, Harry Eagle demonstrated that cysteine is strictly essential for the proliferation of mouse L fibroblasts and human HeLa cells[18,19]. Building on this foundational observation, Shiro Bannai et al. later revealed that cultured cells exhibit a unique vulnerability to cystine depletion compared to other amino acids, laying the groundwork for the cystine-dependent ferroptosis paradigm[20].
Cysteine dictates the cellular threshold for lipid peroxidation and serves as the central node of the anti-ferroptotic regulatory network. As the rate-limiting precursor for GSH biosynthesis, cysteine determines the availability of the essential reductive cofactor for glutathione peroxidase 4 (GPX4)[4,21]. GPX4 relies on GSH to reduce toxic lipid peroxides on cell membranes into non-toxic alcohols, thereby preventing ferroptosis[22,23]. Intracellular cysteine is primarily sourced via the cystine/glutamate antiporter system xc- (an SLC7A11/SLC3A2 heterodimer), which imports extracellular cystine for subsequent cytosolic reduction. Consequently, the SLC7A11-cysteine-GSH-GPX4 axis constitutes the backbone of the cellular antioxidant defense system. In this system, the availability of cysteine is crucial for cell survival[24,25].
The profound reliance on this axis exposes a targetable metabolic vulnerability. Pharmacological inhibition of system xc- by classical ferroptosis inducers, such as erastin and imidazole ketone erastin, precipitates a rapid depletion of the intracellular GSH pool. This upstream substrate starvation collapses the antioxidant system and triggers ferroptosis, even when GPX4 remains functional[26]. Beyond pharmacological intervention, the tumor suppressor protein p53, interferon-γ secreted by immune cells, and radiotherapy can downregulate SLC7A11 expression, thereby inducing ferroptosis in tumor cells through deprivation of cysteine[27-29]. In contrast, drug-resistant cancer cells often remodel their metabolism to fortify this defense. For instance, the circadian regulator ARNTL2 directly enhances SLC7A11 transcription, sustaining high-capacity cysteine uptake. This metabolic adaptation establishes a robust shield against chemotherapeutic stress, as demonstrated in both in vitro and in vivo models of colorectal cancer[30]. Similarly, the stem cell factor SOX2 directly binds and activates the SLC7A11 promoter to confer ferroptosis resistance in lung cancer stem cells, which can be abrogated by SOX2 oxidation at Cys265[31]. Furthermore, lipid metabolism exhibits strict crosstalk with this axis. Inhibition of the lipid enzyme SCD1 blocks the AKT-NRF2 pathway to transcriptionally downregulate SLC7A11, priming lung adenocarcinoma for ferroptosis[32]. Post-transcriptionally, RNA-binding proteins exert context-dependent control: RBMS1 promotes SLC7A11 translation by bridging its UTRs in non-small cell lung cancer (NSCLC)[33], whereas RBMS2 destabilizes SLC7A11 mRNA to promote ferroptosis in colorectal cancer[34]. At the post-translational level, the lysosomal protein LAPTM4B suppresses the NEDD4L/ZRANB1-mediated ubiquitin-proteasomal degradation of SLC7A11 to counteract ferroptosis in NSCLC[35]. Concurrently, the E3 ligase ZNRF2 tunes the ubiquitination of the auxiliary subunit SLC3A2 at K147, driving its plasma membrane translocation to maximize cystine import in lung adenocarcinoma, which can be therapeutically blocked by the synthetic peptide K147[36].
Under extreme pressure from extracellular cysteine scarcity, cells exhibit complex compensatory logic. When external uptake is blocked, cells activate the GCN2-ATF4 signaling axis to drive the transsulfuration pathway, converting methionine to cysteine to sustain basic cell growth[37,38]. Concurrently, inhibition of cysteinyl-tRNA synthetase in the protein translation machinery leads to the accumulation of uncharged tRNA, triggering metabolic reprogramming. This includes upregulation of key enzymes like cystathionine β-synthase (CBS) to induce endogenous cysteine synthesis, thereby antagonizing ferroptosis[39]. Furthermore, broader amino acid starvation (such as arginase-induced arginine depletion) activates the GCN2-eIF2α pathway to suppress global protein translation. This translational arrest functions as a metabolic shunt, redirecting the critically limited intracellular cysteine pool away from protein synthesis and exclusively toward GSH biosynthesis to neutralize lipid peroxides[40,41]. Importantly, the restriction of non-cysteine amino acids in the tumor microenvironment (TME) also acts as a potent ferroptosis sensitizer. Such restriction triggers the GCN2-dependent integrated stress response and ATF4-driven mitochondrial respiration, which increases ROS leakage and drastically lowers the threshold for lipid peroxidation when GSH is depleted[42]. Beyond canonical GSH production, cancer cells could exhibit atypical metabolic adaptations to cysteine scarcity. Under cystine starvation, the catalytic subunit of glutamate-cysteine ligase catalytic subunit promiscuously utilizes alternative amino acids to synthesize non-canonical γ-glutamyl peptides. This atypical peptide generation buffers toxic glutamate accumulation, powerfully protecting NRF2-hyperactive NSCLC from ferroptosis[43].
Cysteine metabolism displays high spatial compartmentalization and functional diversity. For example, CHAC1-mediated GSH degradation may preferentially maintain mitochondrial cysteine levels to support iron-sulfur cluster synthesis, although such preservation of mitochondrial function may paradoxically promote lipid peroxidation under specific conditions in NSCLC[44]. Moreover, cysteine serves as a crucial precursor for hydrogen sulfide (H2S), which modulates antioxidant gene expression via protein S-sulfhydration[38,45].
The high cellular dependence on cysteine metabolism provides a clear point for clinical intervention. In non-malignant pathologies, such as diabetic wound healing, therapeutic efforts focus on mitigating ferroptosis by correcting cysteine deficiencies within hyperglycemic microenvironments[46]. Conversely, in pancreatic ductal adenocarcinoma, synthetic biology approaches have engineered tumor-colonizing bacteria that secrete cysteinase to locally deplete cysteine, thereby dismantling the anti-ferroptotic shield[47]. Crucially, the intersection of epigenetic modulation and amino acid metabolism constitutes a targetable vulnerability, as exemplified by the dual function of histone deacetylase (HDAC) inhibitors. In non-malignant tissues like neurons, specific HDAC inhibitors suppress ferroptosis by preserving redox homeostasis and upregulating neuroprotective cascades[48]. Conversely, in acute myeloid leukemia, HDAC inhibition transcriptionally represses SLC7A11, choking off cystine import and precipitating GSH depletion[49]. Nevertheless, safely balancing systemic amino acid requirements and identifying robust biomarkers for cysteine deprivation remain critical imperatives for future clinical translation[23,27].
2.2 Glycine
Glycine, as one of the three precursor amino acids for GSH synthesis, primarily exerts an inhibitory effect on ferroptosis. However, it may exhibit dual regulatory characteristics under specific pathological contexts[50,51].
Glycine mainly inhibits ferroptosis by participating in GSH biosynthesis. Together with cysteine and glutamate, glycine is used to synthesize GSH via the sequential actions of glutamate-cysteine ligase and glutathione synthetase. This process is crucial for clearing lipid peroxides during ferroptosis[52,53]. Therefore, glycine abundance directly limits the de novo synthesis rate of GSH. Depletion of intracellular glycine leads to decreased GSH reserves and, consequently, heightened sensitivity to ferroptosis.
Within the regulatory network of this core pathway, the serine-glycine conversion and its upstream transcription factors play pivotal roles. Serine hydroxymethyltransferase (SHMT) catalyzes the conversion of serine to glycine and 5,10-methylenetetrahydrofolate (5,10-CH2-THF). This reaction drives one-carbon metabolism, generating reduced coenzyme NAD(P)H to maintain cellular redox balance[54-56]. This reaction is the primary pathway for glycine synthesis. Previous research indicated that in hepatoblastoma models, the LATS2/YAP1 pathway activates ATF4, which further upregulates the expression of phosphoserine aminotransferase 1, a key enzyme in serine synthesis. This increases the synthesis of glycine and cysteine, thereby enhancing cellular antioxidant capacity and inhibiting ferroptosis[51].
Glycine uptake and exogenous supply are also important peripheral factors influencing ferroptosis. Dietary restriction of serine and glycine can effectively reduce the cellular capacity for GSH regeneration and exhibits significant synergistic effects with ferroptosis inducers to suppress tumor growth[50,57]. Furthermore, the impact of glycine on plasma membrane integrity has garnered attention. Although glycine is widely recognized for its ability to inhibit plasma membrane rupture mediated by NINJ1, recent evidence suggests that glycine cannot block ferroptosis-induced membrane damage[58]. This phenomenon reveals that the membrane damage mechanism in ferroptosis may possess unique properties independent of NINJ1 polymerization or may involve a glycine-insensitive, non-canonical pore formation process.
Beyond canonical GSH biosynthesis, the metabolic fate of glycine extends to profound epigenetic regulation. Although the effect, dominated by the GSH pathway, is anti-ferroptotic, prolonged or high-dose glycine exposure might instead promote ferroptosis in certain pathological states by inducing methylation reactions (e.g., GPX4 promoter methylation), which could downregulate the expression of antioxidant enzymes[57]. This mechanistic duality dictates that therapeutic interventions targeting amino acid metabolism must be rigorously calibrated to the precise tissue context and metabolic dosage.
2.3 GPX4-independent compensatory defense system
While the canonical SLC7A11-GSH-GPX4 axis is a primary barrier against lipid peroxidation, cancer cells can employ robust, GPX4-independent compensatory mechanisms to survive when GPX4 is deficient or when canonical amino acid supplies are compromised.
The most prominent parallel defense system is mediated by ferroptosis suppressor protein 1 (FSP1). Localized primarily at the plasma membrane, FSP1 acts as an oxidoreductase that uses NAD(P)H to reduce ubiquinone (Coenzyme Q10, CoQ10) to ubiquinol (CoQ10H2). Ubiquinol then functions as a potent lipophilic radical-trapping antioxidant, which directly halts the propagation of lipid peroxides independent of the GSH pool[59,60].
In addition to the FSP1-CoQ10 axis, other organelle-specific and metabolic pathways provide supplementary layers of defense. The GTP cyclohydrolase 1 (GCH1) pathway synthesizes tetrahydrobiopterin (BH4), another endogenous lipophilic antioxidant that protects cellular membranes from oxidative damage[61]. Furthermore, within the mitochondria, dihydroorotate dehydrogenase (DHODH) operates in parallel to mitochondrial GPX4 to reduce CoQ to CoQH2, specifically protecting the inner mitochondrial membrane from ferroptotic damage[62].
Crucially, the operation of these GPX4-independent surveillance systems is intrinsically coupled to amino acid metabolism. The generation of GTP, the obligate precursor for GCH1-mediated BH4 synthesis, demands a continuous influx of glutamine, glycine, and aspartate[63]. In parallel, DHODH-driven protection relies on de novo pyrimidine biosynthesis, which is fueled by glutamine and aspartate[64]. Consequently, even when circumventing the canonical cysteine-GSH axis, tumors remain metabolically tethered to the continuous flux of these specific amino acids to sustain alternative anti-ferroptotic shields.
3. The Mitochondrial Axis
3.1 Glutamine and glutamate
Glutamine metabolism exerts a profound, dichotomous influence on ferroptosis regulation. Following cellular uptake via the SLC1A5 transporter, the metabolic fate of this conditionally essential amino acid dictates whether it drives lethal lipid peroxidation or fortifies antioxidant defenses. In pro-ferroptotic contexts, especially during cystine deprivation, glutaminolysis acts as an indispensable driver. Glutamine is converted by glutaminase (GLS) to glutamate, which is subsequently transaminated to α-ketoglutarate (α-KG). The entry of α-KG into the mitochondrial TCA cycle fuels oxidative phosphorylation. However, this elevated mitochondrial respiration concomitantly generates substantial electron leakage, culminating in the burst accumulation of lipid-ROS that executes ferroptosis[65-68].
Conversely, glutamine-derived glutamate functions as a critical precursor for GSH biosynthesis, equipping cells with the antioxidant capacity to neutralize lipid peroxides[69-72]. This metabolic bifurcation is precisely governed by complex regulatory networks and subcellular contexts. For instance, the tumor suppressor p53 transcriptionally upregulates GLS2, directing glutamine flux toward α-KG and thereby sensitizing cells to mitochondria-dependent ferroptosis[73]. Notably, the requirement for mitochondrial metabolism varies by the ferroptotic trigger: while essential for cystine-deprivation-induced ferroptosis, its significance diminishes when ferroptosis is initiated by direct GPX4 inhibition, highlighting distinct metabolic dependencies[67,74].
Beyond the core pathway, upstream signaling and peripheral metabolic networks further reshape glutamine’s impact on ferroptosis. In hepatocellular carcinoma (HCC), loss of the tyrosine catabolic enzyme HPD forces cells into a state of strong glutamine addiction. This is mediated by activation of the AMPK/mTOR/p70S6K axis, which upregulates GLS activity to support rapid tumor proliferation, simultaneously exposing a metabolic vulnerability to ferroptosis[75]. In head and neck squamous cell carcinoma, blocking glutamine metabolism triggers autophagy and nutrient sensing, leading to compensatory accumulation of polyunsaturated fatty acids and, consequently, sensitizing cancer cells to lipid peroxidation[76]. Additionally, inhibiting GLS in lung adenocarcinoma can ameliorate nutrient competition in the TME, activating the anti-tumor immunity of CD8+ T cells[77]. Consequently, deciphering the contextual fate of glutamine is paramount for designing ferroptosis-targeted therapeutic interventions.
3.2 Aspartate and asparagine
Aspartate and its derivative, asparagine, primarily inhibit ferroptosis but exhibit dual regulatory characteristics under specific metabolic backgrounds. Aspartate mainly relies on the following three metabolic pathways to regulate ferroptosis: pyrimidine biosynthesis, urea cycle shunting, and transaminase-mediated TCA cycle anaplerosis.
Aspartate inhibits ferroptosis through a key mechanism involving its participation in pyrimidine biosynthesis to maintain mitochondrial antioxidant potential. Within the inner mitochondrial membrane, aspartate serves as a precursor for pyrimidine synthesis. Its metabolic product, dihydroorotate, is oxidized to orotate by DHODH, a process where CoQ is reduced to CoQH2[62]. CoQH2 is a potent lipophilic antioxidant capable of directly scavenging lipid peroxyl radicals. Therefore, the aspartate-DHODH axis constitutes an important ferroptosis defense system independent of GPX4 and FSP1, specifically responsible for clearing lipid peroxides within mitochondria. Furthermore, the dramatic consumption of N-carbamoyl-L-aspartate upon GPX4 system impairment confirms the compensatory protective role of this pathway under ferroptotic stress[62].
Beyond its role as a precursor for pyrimidine synthesis, aspartate can also inhibit ferroptosis through metabolic shunting into the urea cycle. Hu et al. revealed that in NSCLC, Argininosuccinate Synthase 1, a key urea cycle enzyme, promotes the catabolism of glutamine in the cytosol. This process channels glutamine-derived nitrogen into the urea cycle via aspartate, thereby preventing excessive glutamine entry into the TCA cycle and subsequently reducing ROS production[78].
Aspartate aminotransferase (GOT) primarily coordinates the interconversion between aspartate and TCA cycle intermediates (such as oxaloacetate and α-KG), playing an important role in maintaining mitochondrial membrane potential and redox homeostasis[78,79]. In certain tumor types, glutamine is converted via GOT to α-KG, which enters the TCA cycle, driving ROS production by enhancing oxidative phosphorylation and thereby promoting ferroptosis[79].
4. The Epigenetic and Translational Axis
4.1 Methionine
As an essential sulfur-containing amino acid, methionine exerts a context-dependent effect on ferroptosis, capable of both defending against it by maintaining antioxidant systems and, under specific metabolic backgrounds, exacerbating oxidative stress.
In pathways inhibiting ferroptosis, methionine primarily functions via the transsulfuration pathway and the one-carbon metabolism pathway. Methionine can be converted to homocysteine and subsequently to cysteine via the transsulfuration pathway. This metabolic flow is a crucial supply line for maintaining the intracellular GSH pool and GPX4 activity, especially when extracellular cystine uptake is impaired[3,80]. Besides, methionine can be converted to S-adenosylmethionine (SAM) by the key metabolic enzyme methionine adenosyltransferase 2A, participating in epigenetic regulation to inhibit ferroptosis. SAM mediates histone H3K4me3 modification, which upregulates the expression of ACSL3, promoting the production of monounsaturated fatty acids to resist lipid peroxidation in gastric cancer[81]. Methionine transmembrane transport is also an important factor influencing ferroptosis sensitivity. SLC43A2-mediated methionine uptake activates the NF-κB signaling pathway, thereby promoting the transcription of both SLC43A2 and GPX4. Concurrently, elevated methionine levels increase intracellular GSH content, forming a positive feedback loop[82].
However, the regulation of ferroptosis by methionine metabolism is not solely inhibitory. Recent studies have revealed its potential to promote a pro-oxidant environment under specific conditions. Xia et al. demonstrated that SAM, produced from methionine metabolism, is an essential methyl donor for CoQ10 synthesis, which plays a central role in lipid-ROS accumulation induced by cystine deprivation[83]. This finding implies that, in specific cellular contexts, depriving methionine might prevent lipid peroxidation by blocking ubiquinone synthesis.
Intervention strategies targeting methionine metabolism show great potential for clinical translation, but their efficacy is significantly influenced by the duration and frequency of intervention. Although long-term methionine deprivation may induce ferroptosis resistance by inhibiting CHAC1 protein synthesis, studies indicate that intermittent methionine starvation can significantly enhance tumor sensitivity to ferroptosis inducers and immunotherapy by stimulating CHAC1 transcription and accelerating GSH degradation[84]. Meanwhile, in glioma, methionine deprivation inhibits tumor proliferation and epithelial-mesenchymal transition via non-coding RNA networks such as TP53TG1/miR-96-5p/STK17B[85], further supporting the application value of dietary interventions in precision oncology. Additionally, while methionine shows preliminary value as a salivary biomarker for early cancer screening, the precise correlation between fluctuations in salivary amino acid concentrations and intratumoral metabolic states requires validation through larger-scale clinical data[86].
4.2 Serine
Serine functions as a potent endogenous suppressor of ferroptosis. Positioned at the nexus of glycolysis and one-carbon metabolism, it transcends its canonical role in macromolecular biosynthesis to serve as the central metabolic reservoir for antioxidant networks. By fueling these defense systems, serine dictates cellular redox homeostasis and robustly fortifies tumor cells against lethal lipid peroxidation[11,87].
Serine inhibits ferroptosis by driving the one-carbon metabolism unit to generate NADPH and GSH. Firstly, serine is converted to glycine by SHMT. This process accompanies the conversion of tetrahydrofolate to methylenetetrahydrofolate, ultimately driving the folate cycle to produce reduced NADPH, which is a key cellular reducing power[88]. NADPH serves as the cofactor for glutathione reductase to regenerate GSH and as the electron donor for the FSP1-CoQ10 antioxidant system, directly determining the cell’s capacity to clear lipid peroxides[11]. Secondly, serine is the direct carbon skeleton donor for cysteine synthesis, replenishing the cysteine pool via the transsulfuration pathway and thereby ensuring GSH synthesis[87].
Within this metabolic network, key enzymes CBS and phosphoglycerate dehydrogenase (PHGDH) play decisive roles. CBS is responsible for condensing serine and homocysteine to form cystathionine, the rate-limiting step of the transsulfuration pathway. Erdélyi et al. confirmed that under pressure from cysteine scarcity or ferroptosis induction, tumor cells heavily rely on CBS to utilize serine for maintaining cysteine levels. Additionally, CBS can drive persulfidation reactions, a specific post-translational modification mechanism that also significantly inhibits lipid peroxidation in breast cancer[89]. PHGDH is the rate-limiting enzyme for de novo serine synthesis, catalyzing the conversion of D-3-phosphoglycerate to 3-phosphohydroxypyruvate, the first critical step in serine biosynthesis[90,91]. In podocyte models of diabetic kidney disease, PHGDH deficiency exacerbates cellular damage (e.g., cytoskeleton disruption) and significantly promotes ferroptosis[92].
Moreover, serine metabolism indirectly regulates ferroptosis via “non-canonical” pathways such as epigenetic mechanisms. Serine is a major donor of one-carbon units, supporting SAM synthesis. SAM, as a universal methyl donor, directly influences the methylation status of histones and DNA, thereby modulating the transcriptional expression of ferroptosis-related genes[93]. This mechanism indicates that serine determines cell fate not only by directly providing antioxidant molecules but also by reshaping the epigenetic landscape of the genome.
4.3 Selenocysteine (Sec)
As a critical determinant of tumor ferroptosis susceptibility, Sec biosynthesis and incorporation fundamentally subvert canonical translation. Sec is uniquely synthesized in situ on its cognate tRNA[Ser]Sec via a sequential enzymatic cascade[94]. Its targeted integration into the anti-ferroptotic enzyme GPX4 requires reprogramming the UGA stop codon. This is orchestrated by the 3′-UTR SECIS element, which recruits SBP2 and eEFSec to circumvent translation termination and guide Sec-tRNA into the ribosomal A-site[95]. Evolutionary selection shapes that GPX4 strictly relies on a Sec residue, rather than a canonical cysteine, at its catalytic center. This requirement is essential for GPX4 to maintain its catalytic activity and resist irreversible inactivation when confronted with severe lipid hydroperoxide stress[96]. As demonstrated in engineered in vitro neurons and in vivo mouse models, GPX4 variants harboring a canonical cysteine are highly vulnerable to hydroperoxide-induced overoxidation, underscoring the absolute necessity of Sec for survival and central nervous system integrity[96]. To sustain the supply of this highly energy-intensive amino acid, tumor cells orchestrate multidimensional metabolic reprogramming and Sec-specific uptake mechanisms. Furthermore, selenium functions beyond acting as a mere translational building block; it can drive a robust adaptive transcriptional program via transcription factors such as TFAP2c and Sp1, which significantly upregulate the broader selenome, including GPX4, to confer enhanced transcriptional resistance against ferroptotic stress[97].
Therapeutically, the systemic dependency on exogenous selenium-containing amino acids exposes a critical metabolic vulnerability. In MYCN-amplified neuroblastoma, tumor cells are profoundly dependent on LRP8 receptor-mediated uptake of selenoprotein P to maintain Sec homeostasis. Therefore, genetic or pharmacological blockade of this influx precipitates robust ferroptosis[98]. Intracellularly, peroxiredoxin 6 (PRDX6) functions as a pivotal selenium receptor and chaperone, driving a highly efficient, SCLY-independent delivery system by covalently binding to selenoglutathione (GS-Se-SG). Consequently, PRDX6 is critical for sustaining GPX4 biosynthesis and anti-ferroptotic defense[99,100]. Furthermore, the incorporation of Sec during selenoprotein translation is deeply subjected to crosstalk regulation by other metabolic pathways. In HCC, the mevalonate pathway not only promotes CoQ10 synthesis but also directly facilitates Sec-tRNA modification. This crosstalk ensures the successful translation of Sec-dependent antioxidant enzymes, an adaptation that can be therapeutically dismantled using statins[101].
Recent paradigms have further expanded the metabolic repertoire of selenium-containing amino acids beyond classical selenoprotein synthesis. Notably, selenium-mediated antioxidant defense can bypass the rate-limiting translation of Sec-dependent enzymes. The Sec metabolic intermediate, hydrogen selenide, can directly and rapidly reduce ubiquinone within mitochondria via sulfide quinone oxidoreductase, establishing a GPX4-independent shield against lipid peroxidation[102]. Conversely, the incorporation of Sec does not universally confer ferroptosis resistance. The Sec residue within thioredoxin reductase 1 actively promotes the E3 ubiquitin ligase-mediated degradation of the KEAP-NRF2 axis. This cascade ultimately suppresses GPX4 expression, thereby exerting a paradoxical but potent pro-ferroptotic effect across both in vitro and in vivo models[103].
5. Emerging Amino Acids and Multi-level Crosstalk
5.1 Arginine
Arginine primarily acts as a “promoting factor” in ferroptosis regulation, but exhibits complex dual regulatory characteristics under specific conditions of metabolic deprivation or within distinct microenvironmental contexts[104,105]. As a semi-essential amino acid, arginine is not only a fundamental material for protein synthesis, but its catabolic products also serve as significant disruptors of intracellular redox balance, capable of directly driving lipid peroxidation through multiple metabolic branches.
In terms of core metabolic pathways, arginine promotes ferroptosis mainly via three routes: “polyamine metabolism”, the “urea cycle branch”, and “nitric oxide metabolism”. Firstly, intracellular arginine is converted to ornithine, which enters the polyamine synthesis pathway. During the catabolism of polyamines (such as spermidine and spermine), significant amounts of hydrogen peroxide (H2O2) are produced as byproducts. This high level of H2O2 directly provides the oxidant required for the Fenton reaction, thereby inducing lipid peroxidation[105]. Further research has found that this sensitization effect is modulated by KEAP1 status. SAT1 is the rate-limiting enzyme for polyamine catabolism. In tumor cells with a wild-type KEAP1 background, KRAS inhibitors activate the JNK/c-Jun/SAT1 axis, accelerating the conversion of arginine to polyamines and inducing ferroptosis. However, this metabolic sensitivity disappears in cells with KEAP1 mutations[104]. Secondly, metabolic intermediates generated from arginine in the urea cycle can also disrupt the antioxidant system. Fumarate, generated from arginine via argininosuccinate lyase, is electrophilic and can conjugate with GSH to form succinicGSH, directly depleting the intracellular antioxidant reserve and accelerating ferroptosis[106].
L-Arginine serves as the key precursor for nitric oxide (NO). Under catalysis by inducible nitric oxide synthase or oxidation by ROS, L-Arginine releases NO, which is rapidly converted to reactive nitrogen species (RNS)[107]. RNS not only directly oxidize and deplete GSH, but also downregulate SLC7A11 expression to restrict cystine uptake and inhibit glutathione reductase, thereby blocking GSH regeneration. Furthermore, NO can exacerbate mitochondrial damage by inhibiting the activity of aconitase activity[108,109].
The availability of arginine and its transport regulation constitute peripheral factors influencing ferroptosis. In colorectal cancer studies, Lin et al. found that due to the loss of ornithine transcarbamylase, colorectal cancer cells exhibit strong arginine dependence[110]. Upon deprivation of exogenous arginine, cells activate the AMPK-p53-p21 pathway and enter a reversible “quiescent state”. Notably, these quiescent cells show heightened sensitivity to ferroptosis inducers, providing a rationale for the clinical application of “arginine-restricted diets” combined with ferroptosis inducers[110]. Conversely, in tumors like liver cancer, where arginine concentration is abnormally elevated, arginine acts as an oncogenic signaling molecule. Mossmann et al. confirmed that arginine directly binds the RNA-binding protein RBM39, driving oncogenic metabolic reprogramming by regulating gene expression (e.g., ASNS) rather than inducing cell death[111].
Furthermore, the heterogeneity of arginine metabolism within the TME is a key determinant of ferroptosis sensitivity. Mbah et al. suggested that arginine accessibility in the TME not only affects cancer cell survival but also modulates the ferroptosis response and anti-tumor activity of infiltrating T cells[112]. In esophageal squamous cell carcinoma, dysregulation of the ISCU-p53 axis leads to upregulated expression of arginase 1 in macrophages. This not only promotes M2 polarization but also enhances macrophage resistance to ferroptosis via metabolic remodeling, ultimately contributing to resistance against PD-1 inhibitors[113]. Given that arginine deprivation therapies (e.g., ADI-PEG20) have entered clinical trials for various arginine auxotrophic tumors, future research needs to define how to precisely trigger ferroptosis in tumor cells via arginine metabolic pathways without damaging immune cell function[114].
5.2 Tryptophan
Tryptophan metabolism orchestrates ferroptotic resistance through a sophisticated defense network comprising direct radical-scavenging, receptor-mediated signaling, and co-factor biosynthesis.
Mechanistically, metabolites derived from the kynurenine and serotonin pathways, specifically 3-hydroxyanthranilic acid (3-HA) and 5-hydroxytryptamine (5-HT), function as potent radical-trapping antioxidants. These molecules possess unique chemical moieties enabling them to directly donate hydrogen atoms to lipid peroxyl radicals, thereby terminating the autocatalytic peroxidation chain[115]. This antioxidant protection holds significant clinical relevance for intestinal health. Multiple studies indicated that tryptophan derivatives produced by the gut microbiota (e.g., indoles, 5-HT) can protect the intestinal barrier by mitigating oxidative stress, preventing cell damage associated with inflammatory bowel disease[57,116]. The efficacy of this metabolic defense largely depends on the regulation of key enzymes. Among them, kynureninase promotes 3-HA generation to support cell survival, whereas monoamine oxidase A degrades 5-HT, weakening cellular resistance to ferroptosis[115].
Beyond direct antioxidation, the IL4i/tryptophan/aryl hydrocarbon receptor (AhR) axis constitutes a robust compensatory defense. The L-amino acid oxidase IL4i1 synergizes with IDO1 to convert tryptophan into indole-3-pyruvate, which dual-functions as a lipid-ROS scavenger and an endogenous ligand for the AhR. AhR activation transcriptionally upregulates a cohort of antioxidant genes, fortifying cellular tolerance to ferroptotic stress[117].
Furthermore, BH4, which is derived from tryptophan metabolism, serves as another potent antioxidant, assisting cancer cells in combating ferroptosis under oxidative stress[118].
5.3 Branched-chain amino acids (BCAA)
BCAAs (including leucine, isoleucine, and valine) exert bidirectional regulatory effects on ferroptosis. On one hand, BCAAs function as important carbon and nitrogen sources for glutamate, playing an inhibitory role by maintaining antioxidant system activity. On the other hand, abnormal accumulation of BCAAs can promote ferroptosis by altering lipid metabolism and signal transduction.
BCAAs exert their ferroptosis-inhibitory function primarily via the “branched-chain amino acid aminotransferases (BCAT)/glutamate/GSH” metabolic axis, a process dependent on BCAT1/BCAT2[119,120]. BCAT2 catalyzes the transamination of BCAAs to α-KG, generating branched-chain α-keto acids and glutamate. In liver, pancreatic, and prostate cancers, high expression of BCAT2 establishes resistance to ferroptosis in cancer cells[119,121]. Ferroptosis inducers such as sulfasalazine, erastin, and sorafenib can inhibit BCAT2 transcription by activating the AMPK/SREBP1 signaling pathway, leading to reduced intracellular glutamate levels and GSH depletion[119].
Conversely, impaired BCAA catabolism and the consequent intracellular accumulation of these amino acids actively promote ferroptosis. In cisplatin-induced chronic kidney disease models, miR-429-3p specifically inhibits key enzymes in the downstream BCAA catabolism pathway (e.g., ABAT, ALDH6A1) in renal proximal tubules, leading to abnormal intracellular BCAA accumulation[121]. This BCAA buildup does not act via the glutamate pathway but rather directly drives ferroptosis by upregulating pro-ferroptotic genes ACSL4 and TFRC[121]. Additionally, high concentrations of BCAAs can promote oxidative stress by activating the Akt-mTOR pathway, thereby damaging mitochondrial function[120].
5.4 Alanine
Alanine and its isomers act as potent metabolic suppressors of ferroptosis. Their protective mechanisms are multifaceted, encompassing the maintenance of mitochondrial redox homeostasis and the epigenetic silencing of pro-ferroptotic signaling cascades.
Alanine metabolism precisely regulates ferroptosis sensitivity through multiple pathways. In neuroblastoma, the OGT/FOXC1 signaling axis drives high expression of glutamate pyruvate transaminase 2, which converts glutamate and pyruvate to alanine and α-KG. This conversion not only provides anaplerotic substrates for the TCA cycle but, more critically, optimizes intracellular redox balance, conferring resistance to ferroptosis in neuroblastoma cells[122]. Consistently, endogenous alanine derivatives, such as γ-glutamyl-L-alanine, serve key defensive roles against oxidative damage, with their cellular depletion acting as a metabolic hallmark of heightened susceptibility to mitochondrial lipid peroxidation[123]. Furthermore, alanine participates in epigenetic regulation. Hong et al. found that lactyltransferase AARS1 can induce cellular ferroptosis in diabetic nephropathy through lactylation modification, and β-alanine, as a competitive substrate, can effectively block this pathway, demonstrating significant clinical intervention potential[124].
5.5 Sarcosine
Sarcosine exhibits context-dependent effects in regulating ferroptosis and oxidative stress. Multiple studies have shown that decreased sarcosine levels correlate with exacerbated oxidative stress and uncontrolled inflammation. Sarcosine supplementation can activate the macrophage GCN2 signaling axis to alleviate tissue damage, holding clinical significance in conditions like sarcopenia and diabetic immune stress[125,126]. Conversely, Shan et al. identified sarcosine as a potent ferroptosis inducer. It triggers mitochondrial ROS burst via the PDK4/PDHA1 pathway and meanwhile downregulates the iron export protein SLC40A1 to accumulate intracellular iron, thereby enhancing chemosensitivity in lung adenocarcinoma[127].
6. Challenges and Prospects
Although the central role of amino acid metabolism in regulating tumor ferroptosis is well-established (Figure 1 and Figure 2), translation from basic research to clinical therapies faces formidable challenges[128]. Current clinical efforts targeting these pathways highlight both the therapeutic potential and the inherent complexities of this approach (Table 1).
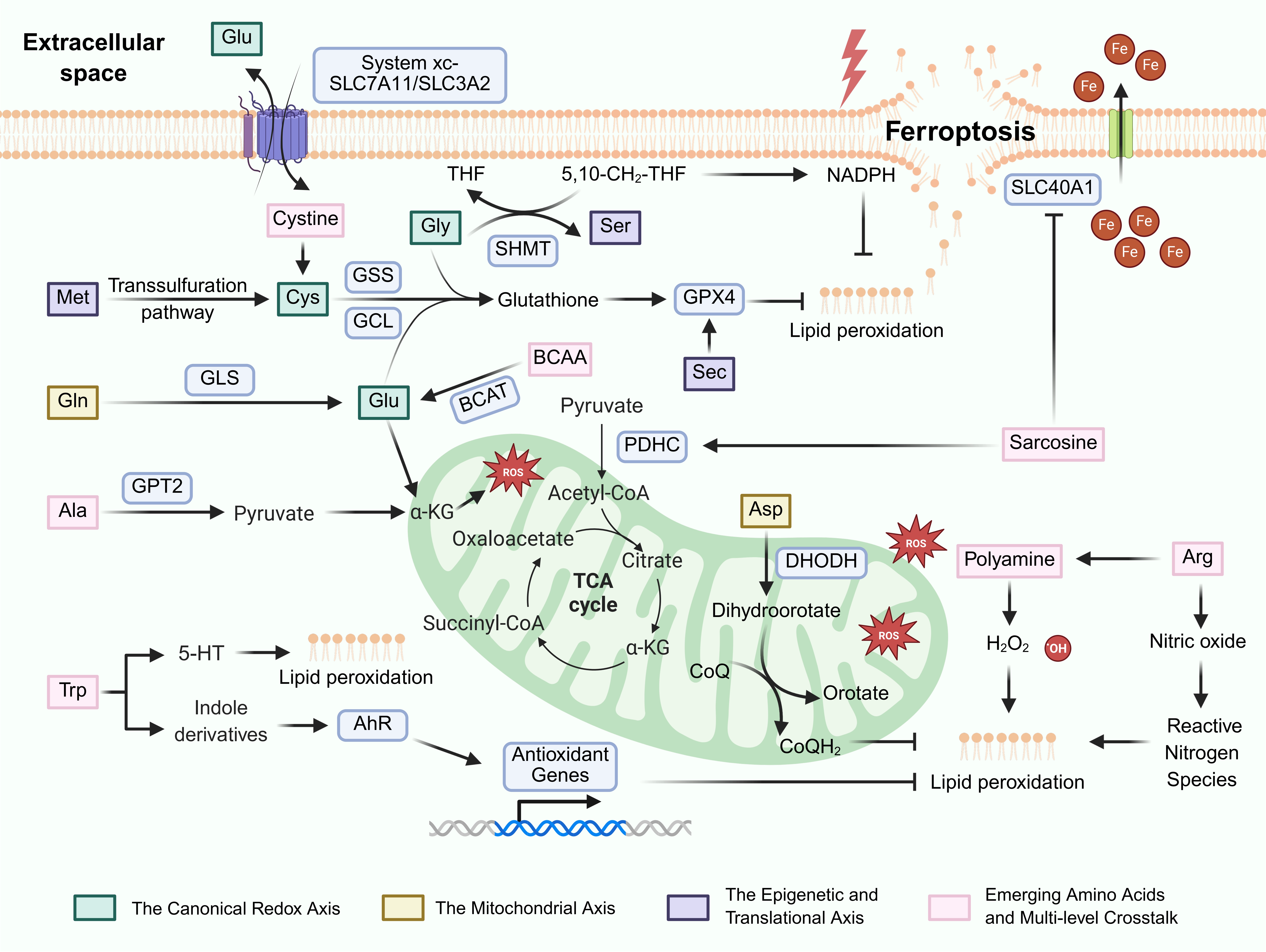
Figure 1. The regulatory landscape of amino acid metabolism in tumor ferroptosis. Created in BioRender.com. GCL: glutamate-cysteine ligase; GSS: glutathione synthetase; GPX4: glutathione peroxidase 4; GLS: glutaminase; BCAT: branched-chain amino acid aminotransferases; SHMT: serine hydroxymethyltransferase; GPT2: glutamate pyruvate transaminase 2; DHODH: dihydroorotate dehydrogenase; AhR: aryl hydrocarbon receptor; CoQ: coenzyme Q; THF: tetrahydrofolate; TCA: tricarboxylic acid; ROS: reactive oxygen species; BCAA: branched-chain amino acids.
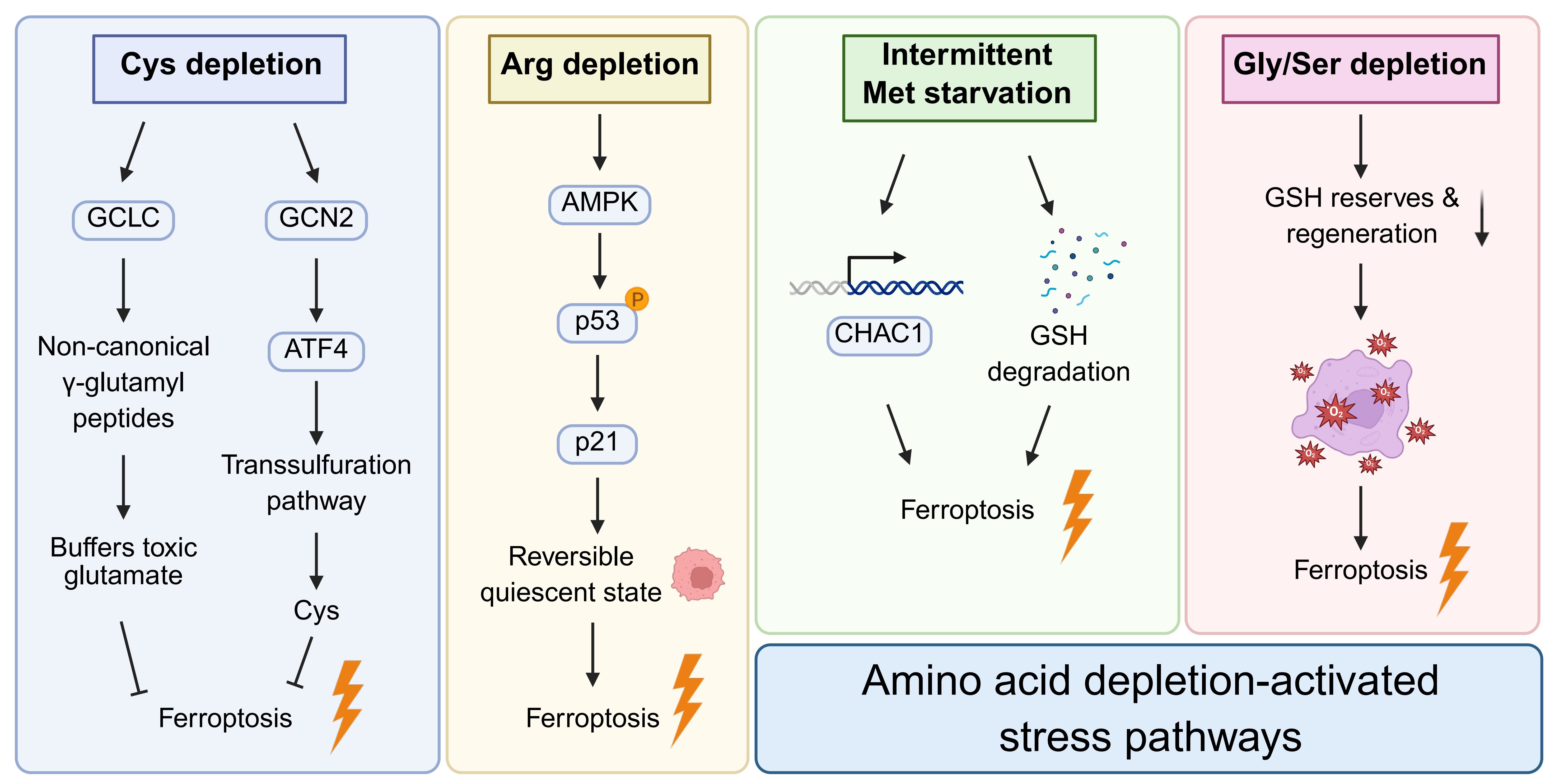
Figure 2. Context-dependent stress signaling pathways activated by specific amino acid deprivation. Created in BioRender.com. GCLC: glutamate-cysteine ligase catalytic subunit; GCN: general control nonderepressible; ATF: activating transcription factor; AMPK: AMP-activated protein kinase; GSH: glutathione.
Table 1. Clinical trials evaluating amino acid metabolism and ferroptosis-targeted therapies in cancer.
| Agent | Target | Tumor type | Primary aim | Status | Clinical trial ID |
| Sulfasalazine | SLC7A11 | Glioma | Determining optimal biological dose based on glutamate levels | Completed | NCT01577966 |
| Sulfasalazine | SLC7A11 | Glioblastoma | Evaluating safety as a radiosensitizer | Completed | NCT04205357 |
| Sulfasalazine | SLC7A11 | Glioblastoma | Evaluating efficacy in combination with chemoradiotherapy | Recruiting | NCT05664464 |
| Sulfasalazine | SLC7A11 | Acute myeloid leukemia | Evaluating safety, feasibility, and biomarkers | Recruiting | NCT05580861 |
| Sulfasalazine | SLC7A11 | Breast cancer | Assessing efficacy in reducing opioid use for bone pain | Completed | NCT03847311 |
| Sorafenib | SLC7A11 | Acute myeloid leukemia | Determining optimal dose in combination with chemotherapy | Active, not recruiting | NCT03247088 |
| CB-839 | GLS1 | Leukemia | Evaluating safety and dose in combination with azacitidine | Completed | NCT02071927 |
| ADI-PEG20 | Arginine | Hepatocellular carcinoma | Evaluating combination therapy with lenvatinib | Recruiting | NCT06034977 |
GLS: glutaminase.
A primary obstacle in the clinic is the high plasticity and compensatory capacity of tumor metabolic networks. For instance, pharmacological blockade of a single amino acid uptake pathway (e.g., System xc-) frequently triggers bypass mechanisms like the transsulfuration pathway, thereby diminishing the efficacy of monotherapies. Secondly, the regulation of ferroptosis by amino acids is highly context-dependent and spatiotemporally heterogeneous, which is dictated by the tissue of origin, specific genetic mutations, and the local TME. The same amino acid can exert opposite effects in different tumor types or even at different stages of the same tumor. Additionally, nutrient competition within the TME presents a non-negligible obstacle. Amino acid deprivation might induce ferroptosis in cancer cells while simultaneously impairing the function of infiltrating immune cells like T cells.
To overcome these translational barriers, future research must leverage single-cell and spatial omics technologies to comprehensively map amino acid metabolic profiles within the TME and identify robust biomarkers of ferroptosis susceptibility. Therapeutically, developing precise delivery systems, such as engineered bacteria or tumor-targeted nanocarriers, holds promise for maximizing ferroptosis and minimizing systemic side effects. Concurrently, combining amino acid deprivation therapies or inhibitors targeting non-canonical pathways (e.g., targeting DHODH or FSP1) with immune checkpoint inhibitors to reshape the metabolic state of the TME will be a crucial direction for overcoming drug resistance. Furthermore, the clinical value of non-invasive strategies like intermittent dietary intervention in ferroptosis modulation still requires validation through large-scale clinical trials.
7. Conclusion
Amino acid metabolism constitutes a core regulatory network of ferroptosis, which exhibits pathway dependency and circumstance-specific effects. Targeting key metabolic nodes can synergize with ferroptosis inducers to effectively kill tumor cells by disrupting redox homeostasis. Future studies should integrate spatiotemporal metabolic heterogeneity to develop precise strategies for clinical translation.
Authors contribution
Bi G, Zhan C: Conceptualization, funding acquisition, writing review & editing.
Shan G, Bian Y, Ren S: Data curation, writing-original draft, writing review & editing.
Conflicts of interest
The authors declare no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
This work was supported by the National Natural Science Foundation of China (Grant No. 82403100 to Guoshu Bi and Grant No. 82473184 to Cheng Zhan), the Natural Science Foundation of Shanghai (Grant No. 24ZR1409900 to Guoshu Bi), the Fellowship from the China Postdoctoral Science Foundation (Grant No. 2024M760555 to Guoshu Bi), and the 18th Special Fund (In-Station) from China Postdoctoral Science Foundation (Grant No. 2025T180549 to Guoshu Bi).
Copyright
© The Author(s) 2026.
References
-
1. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060-1072.[DOI]
-
2. Dos Santos AF, Fazeli G, da Silva TNX, Angeli JPF. Ferroptosis: Mechanisms and implications for cancer development and therapy response. Trends Cell Biol. 2023;33(12):1062-1076.[DOI]
-
3. Stockwell BR. Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications. Cell. 2022;185(14):2401-2421.[DOI]
-
4. Su Y, Zhao B, Zhou L, Zhang Z, Shen Y, Lv H, et al. Ferroptosis, a novel pharmacological mechanism of anti-cancer drugs. Cancer Lett. 2020;483:127-136.[DOI]
-
5. Zhang C, Liu X, Jin S, Chen Y, Guo R. Ferroptosis in cancer therapy: A novel approach to reversing drug resistance. Mol Cancer. 2022;21:47.[DOI]
-
6. Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell. 2011;144(5):646-674.[DOI]
-
7. Hassannia B, Vandenabeele P, Vanden Berghe T. Targeting ferroptosis to iron out cancer. Cancer Cell. 2019;35(6):830-849.[DOI]
-
8. Stockwell BR, Angeli JPF, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171(2):273-285.[DOI]
-
9. Zheng J, Conrad M. Ferroptosis: When metabolism meets cell death. Physiol Rev. 2025;105(2):651-706.[DOI]
-
10. Ren S, Zhou X, Wang Z, Yuan K. Amino acids metabolism: A potential target for cancer treatment. Mol Cancer. 2025;24:307.[DOI]
-
11. Liu X, Ren B, Ren J, Gu M, You L, Zhao Y. The significant role of amino acid metabolic reprogramming in cancer. Cell Commun Signal. 2024;22:380.[DOI]
-
12. Vettore L, Westbrook RL, Tennant DA. New aspects of amino acid metabolism in cancer. Br J Cancer. 2020;122(2):150-156.[DOI]
-
13. Tsun ZY, Possemato R. Amino acid management in cancer. Semin Cell Dev Biol. 2015;43:22-32.[DOI]
-
14. Jin J, Byun JK, Choi YK, Park KG. Targeting glutamine metabolism as a therapeutic strategy for cancer. Exp Mol Med. 2023;55(4):706-715.[DOI]
-
15. Tabe Y, Lorenzi PL, Konopleva M. Amino acid metabolism in hematologic malignancies and the era of targeted therapy. Blood. 2019;134(13):1014-1023.[DOI]
-
16. Jones CL, Stevens BM, D’Alessandro A, Reisz JA, Culp-Hill R, Nemkov T, et al. Inhibition of amino acid metabolism selectively targets human leukemia stem cells. Cancer Cell. 2018;34(5):724-740.[DOI]
-
17. Pathria G, Ronai ZA. Harnessing the co-vulnerabilities of amino acid-restricted cancers. Cell Metab. 2021;33(1):9-20.[DOI]
-
18. Eagle H. The specific amino acid requirements of a mammalian cell (strain L) in tissue culture. J Biol Chem. 1955;214(2):839-852.[DOI]
-
19. Eagle H. The specific amino acid requirements of a human carcinoma cell (strain HeLa) in tissue culture. J Exp Med. 1955;102(1):37-48.[DOI]
-
20. Bannai S, Tsukeda H, Okumura H. Effect of antioxidants on cultured human diploid fibroblasts exposed to cystine-free medium. Biochem Biophys Res Commun. 1977;74(4):1582-1588.[DOI]
-
21. Nakamura T, Conrad M. Exploiting ferroptosis vulnerabilities in cancer. Nat Cell Biol. 2024;26(9):1407-1419.[DOI]
-
22. Wu G, Lupton JR, Turner ND, Fang YZ, Yang S. Glutathione metabolism and its implications for health. J Nutr. 2004;134(3):489-492.[DOI]
-
23. Lee J, Roh JL. Cysteine metabolism at the crossroads of ferroptosis and cancer therapy. Crit Rev Oncol. 2025;215:104906.[DOI]
-
24. Xu T, Ding W, Ji X, Ao X, Liu Y, Yu W, et al. Molecular mechanisms of ferroptosis and its role in cancer therapy. J Cellular Molecular Medi. 2019;23(8):4900-4912.[DOI]
-
25. Chen X, Kang R, Kroemer G, Tang D. Broadening horizons: The role of ferroptosis in cancer. Nat Rev Clin Oncol. 2021;18(5):280-296.[DOI]
-
26. Sun S, Shen J, Jiang J, Wang F, Min J. Targeting ferroptosis opens new avenues for the development of novel therapeutics. Sig Transduct Target Ther. 2023;8:372.[DOI]
-
27. Zhou Q, Meng Y, Li D, Yao L, Le J, Liu Y, et al. Ferroptosis in cancer: From molecular mechanisms to therapeutic strategies. Sig Transduct Target Ther. 2024;9:55.[DOI]
-
28. Lang X, Green MD, Wang W, Yu J, Choi JE, Jiang L, et al. Radiotherapy and immunotherapy promote tumoral lipid oxidation and ferroptosis via synergistic repression of SLC7A11. Cancer Discov. 2019;9(12):1673-1685.[DOI]
-
29. Liang J, Bi G, Huang Y, Zhao G, Sui Q, Zhang H, et al. MAFF confers vulnerability to cisplatin-based and ionizing radiation treatments by modulating ferroptosis and cell cycle progression in lung adenocarcinoma. Drug Resist Updat. 2024;73:101057.[DOI]
-
30. Yang J, Lin D, Huang Y, Yin S, Chen M, Sun H, et al. Clock gene ARNTL2 enhances 5-fluorouracil resistance in colon cancer by upregulating SLC7A11 to suppress ferroptosis. Redox Biol. 2025;86:103798.[DOI]
-
31. Wang X, Chen Y, Wang X, Tian H, Wang Y, Jin J, et al. Stem cell factor SOX2 confers ferroptosis resistance in lung cancer via upregulation of SLC7A11. Cancer Res. 2021;81(20):5217-5229.[DOI]
-
32. Sen U, Coleman C, Gandhi N, Jethalia V, Demircioglu D, Elliott A, et al. SCD1 inhibition blocks the AKT–NRF2–SLC7A11 pathway to induce lipid metabolism remodeling and ferroptosis priming in lung adenocarcinoma. Cancer Res. 2025;85(13):2485-2503.[DOI]
-
33. Zhang W, Sun Y, Bai L, Zhi L, Yang Y, Zhao Q, et al. RBMS1 regulates lung cancer ferroptosis through translational control of SLC7A11. J Clin Investig. 2021;131(22):e152067.[DOI]
-
34. Wang X, Liu P, An Y, Hu Y, Qiao H, Miao H. RBMS2 mediates SLC7A11 transcription-translation to regulate ferroptosis in colorectal cancer. Free Radic Biol Med. 2025;240:504-513.[DOI]
-
35. Yan R, Liu D, Guo H, Liu M, Lv D, Björkblom B, et al. LAPTM4B counteracts ferroptosis via suppressing the ubiquitin-proteasome degradation of SLC7A11 in non-small cell lung cancer. Cell Death Dis. 2024;15(6):436.[DOI]
-
36. Zhang W, Li J, Zhao J, Lu D, Zhang M, Ma C, et al. Targeting ZNRF2-mediated SLC3A2 plasma membrane translocation enhances ferroptosis in lung adenocarcinoma. Oncogene. 2025;44(45):4339-4351.[DOI]
-
37. Zhu J, Berisa M, Schwörer S, Qin W, Cross JR, Thompson CB. Transsulfuration activity can support cell growth upon extracellular cysteine limitation. Cell Metab. 2019;30(5):865-876.[DOI]
-
38. Paul BD, Sbodio JI, Snyder SH. Cysteine metabolism in neuronal redox homeostasis. Trends Pharmacol Sci. 2018;39(5):513-524.[DOI]
-
39. Shimada K, Stockwell BR. tRNA synthase suppression activatesde novocysteine synthesis to compensate for cystine and glutathione deprivation during ferroptosis. Mol Cell Oncol. 2016;3(2):e1091059.[DOI]
-
40. Ye J, Kumanova M, Hart LS, Sloane K, Zhang H, De Panis DN, et al. The GCN2-ATF4 pathway is critical for tumour cell survival and proliferation in response to nutrient deprivation. EMBO J. 2010;29(12):2082-2096.[DOI]
-
41. Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell. 2003;11(3):619-633.[DOI]
-
42. Garellick VA, Gul N, Horrieh P, Mustafa D, Patel AAH, Dankis M, et al. Amino acid restriction sensitizes lung cancer cells to ferroptosis via GCN2-dependent activation of the integrated stress response. Redox Biol. 2026;90:103988.[DOI]
-
43. Kang YP, Mockabee-Macias A, Jiang C, Falzone A, Prieto-Farigua N, Stone E, et al. Non-canonical glutamate-cysteine ligase activity protects against ferroptosis. Cell Metab. 2021;33(1):174-189.[DOI]
-
44. Ward NP, Yoon SJ, Flynn T, Sherwood AM, Olley MA, Madej J, et al. Mitochondrial respiratory function is preserved under cysteine starvation via glutathione catabolism in NSCLC. Nat Commun. 2024;15:4244.[DOI]
-
45. Kimura H. Signalling by hydrogen sulfide and polysulfides via protein S-sulfuration. Br J Pharmacol. 2020;177(4):720-733.[DOI]
-
46. Huang Y, Ding Y, Wang B, Ji Q, Peng C, Tan Q. Neutrophils extracellular traps and ferroptosis in diabetic wounds. Int Wound J. 2023;20(9):3840-3854.[DOI]
-
47. Qiao C, Wang L, Huang C, Jia Q, Bao W, Guo P, et al. Engineered bacteria manipulate cysteine metabolism to boost ferroptosis-based pancreatic ductal adenocarcinoma therapy. Adv Mater. 2025;37(6):2412982.[DOI]
-
48. Zille M, Kumar A, Kundu N, Bourassa MW, Wong VSC, Willis D, et al. Ferroptosis in neurons and cancer cells is similar but differentially regulated by histone deacetylase inhibitors. eNeuro. 2019;6(1):ENEURO.0263-18.2019.[DOI]
-
50. Fujihara KM, Zhang BZ, Jackson TD, Ogunkola MO, Nijagal B, Milne JV, et al. Eprenetapopt triggers ferroptosis, inhibits NFS1 cysteine desulfurase, and synergizes with serine and glycine dietary restriction. Sci Adv. 2022;8(37):eabm9427.[DOI]
-
51. Zhu G, Xie Y, Bian Z, Ma J, Zhen N, Chen T, et al. N6-methyladenosine modification of LATS2 promotes hepatoblastoma progression by inhibiting ferroptosis through the YAP1/ATF4/PSAT1 axis. Int J Biol Sci. 2024;20(11):4146-4161.[DOI]
-
52. Lapenna D. Glutathione and glutathione-dependent enzymes: From biochemistry to gerontology and successful aging. Ageing Res Rev. 2023;92:102066.[DOI]
-
53. Liu X, Ma Q, Jia Z, Zhou Y, Zou C, Xiao Y, et al. ISG15 enhances the activity of γ-glutamate cysteine ligase to suppress apoptosis in high fat diet-promoted hepatocellular carcinoma. Adv Sci. 2025;12(19):2416401.[DOI]
-
54. Holeček M. Glycine as a conditionally essential amino acid and its relationship to l-serine. Metabolism. 2025;170:156330.[DOI]
-
55. Krupenko SA, Cole SA, Hou R, Haack K, Laston S, Mehta NR, et al. Genetic variants in ALDH1L1 and GLDC influence the serine-to-glycine ratio in Hispanic children. Am J Clin Nutr. 2022;116(2):500-510.[DOI]
-
56. Du C, Liu C, Yu K, Zhang S, Fu Z, Chen X, et al. Mitochondrial serine catabolism safeguards maintenance of the hematopoietic stem cell pool in homeostasis and injury. Cell Stem Cell. 2024;31(10):1484-1500.[DOI]
-
57. Yao H, Jiang W, Liao X, Wang D, Zhu H. Regulatory mechanisms of amino acids in ferroptosis. Life Sci. 2024;351:122803.[DOI]
-
58. Chen SY, Wu J, Chen Y, Wang YE, Setayeshpour Y, Federico C, et al. NINJ1 regulates ferroptosis via xCT antiporter interaction and CoA modulation. Cell Death Dis. 2024;15(10):755.[DOI]
-
59. Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. 2019;575(7784):688-692.[DOI]
-
60. Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature. 2019;575(7784):693-698.[DOI]
-
61. Kraft VAN, Bezjian CT, Pfeiffer S, Ringelstetter L, Müller C, Zandkarimi F, et al. GTP cyclohydrolase 1/tetrahydrobiopterin counteract ferroptosis through lipid remodeling. ACS Cent Sci. 2020;6(1):41-53.[DOI]
-
62. Mao C, Liu X, Zhang Y, Lei G, Yan Y, Lee H, et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature. 2021;593(7860):586-590.[DOI]
-
63. Zhu S, Xu R, Engel AL, Wang Y, McNeel R, Hurley JB, et al. Proline provides a nitrogen source in the retinal pigment epithelium to synthesize and export amino acids for the neural retina. J Biol Chem. 2023;299(11):105275.[DOI]
-
64. Ben-Sahra I, Howell JJ, Asara JM, Manning BD. Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science. 2013;339(6125):1323-1328.[DOI]
-
65. Berndt C, Alborzinia H, Amen VS, Ayton S, Barayeu U, Bartelt A, et al. Ferroptosis in health and disease. Redox Biol. 2024;75:103211.[DOI]
-
66. Dixon SJ, Olzmann JA. The cell biology of ferroptosis. Nat Rev Mol Cell Biol. 2024;25(6):424-442.[DOI]
-
67. Gao M, Yi J, Zhu J, Minikes AM, Monian P, Thompson CB, et al. Role of mitochondria in ferroptosis. Mol Cell. 2019;73(2):354-363.[DOI]
-
68. Jiang X, Peng Q, Peng M, Oyang L, Wang H, Liu Q, et al. Cellular metabolism: A key player in cancer ferroptosis. Cancer Commun. 2024;44(2):185-204.[DOI]
-
69. Wu Y, Li H, Yue K, Jing C, Duan Y. Ferroptosis in cancer: Metabolism, mechanisms and therapeutic prospects. Mol Cancer. 2025;24:303.[DOI]
-
70. Wang Z, Liu M, Yang Q. Glutamine and leukemia research: Progress and clinical prospects. Discov Onc. 2024;15:391.[DOI]
-
71. Xue X, Wang M, Cui J, Yang M, Ma L, Kang R, et al. Glutathione metabolism in ferroptosis and cancer therapy. Cancer Lett. 2025;621:217697.[DOI]
-
72. Koppula P, Zhuang L, Gan B. Cystine transporter SLC7A11/xCT in cancer: Ferroptosis, nutrient dependency, and cancer therapy. Protein Cell. 2021;12(8):599-620.[DOI]
-
73. Kang R, Kroemer G, Tang D. The tumor suppressor protein p53 and the ferroptosis network. Free Radic Biol Med. 2019;133:162-168.[DOI]
-
74. Jiang X, Stockwell BR, Conrad M. Ferroptosis: Mechanisms, biology and role in disease. Nat Rev Mol Cell Biol. 2021;22(4):266-282.[DOI]
-
75. Tong M, Wong TL, Zhao H, Zheng Y, Xie YN, Li CH, et al. Loss of tyrosine catabolic enzyme HPD promotes glutamine anaplerosis through mTOR signaling in liver cancer. Cell Rep. 2021;36(8):109617.[DOI]
-
76. Allevato MM, Trinh S, Koshizuka K, Nachmanson D, Nguyen TC, Yokoyama Y, et al. A genome-wide CRISPR screen reveals that antagonism of glutamine metabolism sensitizes head and neck squamous cell carcinoma to ferroptotic cell death. Cancer Lett. 2024;598:217089.[DOI]
-
77. Jiang P, Jiang Z, Li S, Li YX, Chen Y, Li X. The suppressive role of GLS in radiosensitivity and irradiation-induced immune response in LUAD: Integrating bioinformatics and experimental insights. Front Immunol. 2025;16:1582587.[DOI]
-
79. Yao X, Li W, Fang D, Xiao C, Wu X, Li M, et al. Emerging roles of energy metabolism in ferroptosis regulation of tumor cells. Adv Sci. 2021;8(22):2100997.[DOI]
-
80. Zhang HF, Klein Geltink RI, Parker SJ, Sorensen PH. Transsulfuration, minor player or crucial for cysteine homeostasis in cancer. Trends Cell Biol. 2022;32(9):800-814.[DOI]
-
81. Ma M, Kong P, Huang Y, Wang J, Liu X, Hu Y, et al. Activation of MAT2A-ACSL3 pathway protects cells from ferroptosis in gastric cancer. Free Radic Biol Med. 2022;181:288-299.[DOI]
-
82. Sun LL, He HY, Li W, Jin WL, Wei YJ. The solute carrier transporters (SLCs) family in nutrient metabolism and ferroptosis. Biomark Res. 2024;12:94.[DOI]
-
83. Xia C, Peng P, Zhang W, Xing X, Jin X, Du J, et al. Methionine-SAM metabolism-dependent ubiquinone synthesis is crucial for ROS accumulation in ferroptosis induction. Nat Commun. 2024;15:8971.[DOI]
-
84. Xue Y, Lu F, Chang Z, Li J, Gao Y, Zhou J, et al. Intermittent dietary methionine deprivation facilitates tumoral ferroptosis and synergizes with checkpoint blockade. Nat Commun. 2023;14:4758.[DOI]
-
85. Li J, Liu R, Hu H, Huang Y, Shi Y, Li H, et al. Methionine deprivation inhibits glioma proliferation and EMT via the TP53TG1/miR-96-5p/STK17B ceRNA pathway. npj Precis Onc. 2024;8:270.[DOI]
-
86. Bel’skaya LV, Sarf EA, Loginova AI. Diagnostic value of salivary amino acid levels in cancer. Metabolites. 2023;13(8):950.[DOI]
-
87. Wang H, Nicolay BN, Chick JM, Gao X, Geng Y, Ren H, et al. The metabolic function of cyclin D3–CDK6 kinase in cancer cell survival. Nature. 2017;546(7658):426-430.[DOI]
-
88. McBride MJ, Hunter CJ, Zhang Z, TeSlaa T, Xu X, Ducker GS, et al. Glycine homeostasis requires reverse SHMT flux. Cell Metab. 2024;36(1):103-115.[DOI]
-
89. Erdélyi K, Ditrói T, Johansson HJ, Czikora Á, Balog N, Silwal-Pandit L, et al. Reprogrammed transsulfuration promotes basal-like breast tumor progression via realigning cellular cysteine persulfidation. Proc Natl Acad Sci U S A. 2021;118(45):e2100050118.[DOI]
-
90. Zhao JY, Feng KR, Wang F, Zhang JW, Cheng JF, Lin GQ, et al. A retrospective overview of PHGDH and its inhibitors for regulating cancer metabolism. Eur J Med Chem. 2021;217:113379.[DOI]
-
91. Lee CM, Hwang Y, Kim M, Park YC, Kim H, Fang S. PHGDH: A novel therapeutic target in cancer. Exp Mol Med. 2024;56(7):1513-1522.[DOI]
-
92. Wang Y, Zhang Q, Lv S, Wang X, Liu Q, Liu X, et al. PHGDH alleviates DKD by regulating YB1/SLC7A11-mediated ferroptosis in podocytes. Transl Res. 2025;282:1-13.[DOI]
-
93. Ge T, Gu X, Jia R, Ge S, Chai P, Zhuang A, et al. Crosstalk between metabolic reprogramming and epigenetics in cancer: Updates on mechanisms and therapeutic opportunities. Cancer Commun. 2022;42(11):1049-1082.[DOI]
-
94. Stadtman TC. SELENOCYSTEINE. Annu Rev Biochem. 1996;65:83-100.[DOI]
-
95. Hilal T, Killam BY, Grozdanović M, Dobosz-Bartoszek M, Loerke J, Bürger J, et al. Structure of the mammalian ribosome as it decodes the selenocysteine UGA codon. Science. 2022;376(6599):1338-1343.[DOI]
-
96. Ingold I, Berndt C, Schmitt S, Doll S, Poschmann G, Buday K, et al. Selenium utilization by GPX4 is required to prevent hydroperoxide-induced ferroptosis. Cell. 2018;172(3):409-422.[DOI]
-
97. Alim I, Caulfield JT, Chen Y, Swarup V, Geschwind DH, Ivanova E, et al. Selenium drives a transcriptional adaptive program to block ferroptosis and treat stroke. Cell. 2019;177(5):1262-1279.[DOI]
-
98. Alborzinia H, Chen Z, Yildiz U, Freitas FP, Vogel FCE, Varga JP, et al. LRP8-mediated selenocysteine uptake is a targetable vulnerability in MYCN-amplified neuroblastoma. EMBO Mol Med. 2023;15(8):e18014.[DOI]
-
99. Chen Z, Inague A, Kaushal K, Fazeli G, Schilling D, da Silva TNX, et al. PRDX6 contributes to selenocysteine metabolism and ferroptosis resistance. Mol Cell. 2024;84(23):4645-4659.[DOI]
-
100. Ito J, Nakamura T, Toyama T, Chen D, Berndt C, Poschmann G, et al. PRDX6 dictates ferroptosis sensitivity by directing cellular selenium utilization. Mol Cell. 2024;84(23):4629-4644.[DOI]
-
101. Chen Y, Lee D, Kwan KK, Wu M, Wang G, Zhang MS, et al. Mevalonate pathway promotes liver cancer by suppressing ferroptosis through CoQ10 production and selenocysteine-tRNA modification. J Hepatol. 2025;83(6):1338-1352.[DOI]
-
102. Lee N, Park SJ, Lange M, Tseyang T, Doshi MB, Kim TY, et al. Selenium reduction of ubiquinone via SQOR suppresses ferroptosis. Nat Metab. 2024;6(2):343-358.[DOI]
-
103. Luan J, He J, Wu D, Duan Y, Hou G, Ma S, et al. Selenoprotein thioredoxin reductase 1 promotes cancer cells ferroptosis by suppressing GPX4 expression. Cell Death Differ. 2026.[DOI]
-
104. Bian Y, Shan G, Bi G, Xu Z, Liang J, Yan Y, et al. Targeting polyamine metabolism and ferroptosis enhances the efficacy of KRAS-targeted therapy depending on KEAP1 status. Nat Commun. 2025;16:9923.[DOI]
-
105. Bi G, Liang J, Bian Y, Shan G, Huang Y, Lu T, et al. Polyamine-mediated ferroptosis amplification acts as a targetable vulnerability in cancer. Nat Commun. 2024;15:2461.[DOI]
-
106. Guo X, Guo Y, Li J, Liu Q, Wu H. Arginine expedites erastin-induced ferroptosis through fumarate. Int J Mol Sci. 2023;24(19):14595.[DOI]
-
107. Fung TS, Ryu KW, Thompson CB. Arginine: At the crossroads of nitrogen metabolism. EMBO J. 2025;44(5):1275-1293.[DOI]
-
108. Mao G, Xin D, Wang Q, Lai D. Sodium molybdate inhibits the growth of ovarian cancer cells via inducing both ferroptosis and apoptosis. Free Radic Biol Med. 2022;182:79-92.[DOI]
-
109. Li W, Liu S, Ding H, Zhao R, Zang P, Li S, et al. Three-step depletion strategy of glutathione: Tunable metal–organic-framework-engineered nanozymes for driving oxidative/nitrative stress to maximize ferroptosis therapy. Nano Lett. 2024;24(6):2071-2080.[DOI]
-
110. Lin Y, Zhang Y, Huang T, Chen J, Li G, Zhang B, et al. Arginine deprivation induces quiescence and confers vulnerability to ferroptosis in colorectal cancer. Cancer Res. 2025;85(9):1663-1679.[DOI]
-
111. Mossmann D, Müller C, Park S, Ryback B, Colombi M, Ritter N, et al. Arginine reprograms metabolism in liver cancer via RBM39. Cell. 2023;186(23):5068-5083.[DOI]
-
112. Mbah NE, Lyssiotis CA. Metabolic regulation of ferroptosis in the tumor microenvironment. J Biol Chem. 2022;298(3):101617.[DOI]
-
113. Luo J, Zhang X, Liang Z, Zhuang W, Jiang M, Ma M, et al. ISCU-p53 axis orchestrates macrophage polarization to dictate immunotherapy response in esophageal squamous cell carcinoma. Cell Death Dis. 2025;16:462.[DOI]
-
114. Qiu F, Huang J, Sui M. Targeting arginine metabolism pathway to treat arginine-dependent cancers. Cancer Lett. 2015;364(1):1-7.[DOI]
-
115. Liu D, Liang CH, Huang B, Zhuang X, Cui W, Yang L, et al. Tryptophan metabolism acts as a new anti-ferroptotic pathway to mediate tumor growth. Adv Sci. 2023;10(6):2204006.[DOI]
-
116. Zhou J, Lu P, He H, Zhang R, Yang D, Liu Q, et al. The metabolites of gut microbiota: Their role in ferroptosis in inflammatory bowel disease. Eur J Med Res. 2025;30:248.[DOI]
-
117. Zeitler L, Murray PJ. IL4i1 and IDO1: Oxidases that control a tryptophan metabolic nexus in cancer. J Biol Chem. 2023;299(6):104827.[DOI]
-
118. Mao C, Wang M, Zhuang L, Gan B. Metabolic cell death in cancer: Ferroptosis, cuproptosis, disulfidptosis, and beyond. Protein Cell. 2024;15(9):642-660.[DOI]
-
119. Wang K, Zhang Z, Tsai HI, Liu Y, Gao J, Wang M, et al. Branched-chain amino acid aminotransferase 2 regulates ferroptotic cell death in cancer cells. Cell Death Differ. 2021;28(4):1222-1236.[DOI]
-
120. Liang J, Liao Y, Wang P, Yang K, Wang Y, Wang K, et al. Ferroptosis landscape in prostate cancer from molecular and metabolic perspective. Cell Death Discov. 2023;9:128.[DOI]
-
121. Sone H, Lee TJ, Lee BR, Heo D, Oh S, Kwon SH. microRNA-mediated attenuation of branched-chain amino acid catabolism promotes ferroptosis in chronic kidney disease. Nat Commun. 2023;14:7814.[DOI]
-
122. Li Q, Cheng Y, Yang C, Tian M, Wang X, Li D, et al. Targeting the exonic Circular OGT RNA/O-GlcNAc transferase/forkhead box C1 axis inhibits asparagine- and alanine-mediated ferroptosis repression in neuroblastoma progression. Research. 2025;8:703.[DOI]
-
123. Jiang M, Peng M, Meng A, Zhang W, Shi H, Han Q, et al. Multi-omics analysis reveals the toxic mechanism of tributyltin exposure causing digestive gland oxidative stress in cuttlefish (Sepia pharaonis). J Hazard Mater. 2025;489:137547.[DOI]
-
124. Hong J, Xu H, Yu L, Yu Z, Chen X, Meng Z, et al. AARS1-mediated lactylation of H3K18 and STAT1 promotes ferroptosis in diabetic nephropathy. Cell Death Differ. 2025.[DOI]
-
125. Liu Y, Ge M, Xiao X, Lu Y, Zhao W, Zheng K, et al. Sarcosine decreases in sarcopenia and enhances muscle regeneration and adipose thermogenesis by activating anti-inflammatory macrophages. Nat Aging. 2025;5(9):1810-1827.[DOI]
-
126. Anwardeen NR, Cyprian FS, Yassine HM, Al-Thani AA, Abdallah AM, Emara MM, et al. The retrospective study of the metabolic patterns of BCG-vaccination in type-2 diabetic individuals in COVID-19 infection. Front Immunol. 2023;14:1146443.[DOI]
-
127. Shan G, Bian Y, Ren S, Hu Z, Pan B, Zeng D, et al. Sarcosine sensitizes lung adenocarcinoma to chemotherapy by dual activation of ferroptosis via PDK4/PDHA1 signaling and NMDAR-mediated iron export. Exp Hematol Oncol. 2025;14:60.[DOI]
-
128. Linkermann A. Key questions in ferroptosis. Ferroptosis Oxid Stress. 2025;1:202503.[DOI]
Copyright
© The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Publisher’s Note
Science Exploration remains a neutral stance on jurisdictional claims in published
maps
and institutional affiliations. The views expressed in this article are solely those
of
the author(s) and do not reflect the opinions of the Editors or the publisher.
Share And Cite



Science Exploration Style
Shan G, Bian Y, Ren S, Bi G, Zhan C. Metabolic reprogramming of amino acids dictates tumor susceptibility to ferroptosis. Ferroptosis Oxid Stress. 2026;2:202525. https://doi.org/10.70401/fos.2026.0023
Tips
Copy completed.
Submit a Manuscript
Author Instructions
Cite this Article
Article Metrics
0
View
0
Download
Cited
Article Updates

- Abstract
- Keywords
- 1. Introduction
- 2. The Canonical Redox Axis
- 3. The Mitochondrial Axis
- 4. The Epigenetic and Translational Axis
- 5. Emerging Amino Acids and Multi-level Crosstalk
- 6. Challenges and Prospects
- 7. Conclusion
- Authors contribution
- Conflicts of interest
- Ethical approval
- Consent to participate
- Consent for publication
- Availability of data and materials
- Funding
- References
- Copyright
Science Exploration Style
Shan G, Bian Y, Ren S, Bi G, Zhan C. Metabolic reprogramming of amino acids dictates tumor susceptibility to ferroptosis. Ferroptosis Oxid Stress. 2026;2:202525. https://doi.org/10.70401/fos.2026.0023
copy
Share Link
copy