Synergistic regulation of lithium nucleation and anion-rich solvation structure via silver trifluoroacetate additive for stable lithium metal anodes Download PDF
Jing Bai

Wenjie Cheng
Tao Wang
Sheng Liu
*
*Correspondence to:
Sheng Liu, Institute of New Energy Material Chemistry, School of Materials Science and Engineering, Nankai University. Tianjin 300350, China.
E-mail: shengliu@nankai.edu.cn
Smart Mater Devices. 2025;1:202534. 10.70401/smd.2025.0016
Received: August 24, 2025Accepted: September 22, 2025Published: September 26, 2025
Abstract
Lithium (Li) metal, owing to its high theoretical specific capacity and low electrochemical potential, is considered one of the most promising anode materials for next-generation rechargeable batteries. However, interfacial instability severely hinders the practical application of Li anodes. Constructing a robust solid electrolyte interphase (SEI) with optimized chemical composition and structure has been recognized as an effective strategy to overcome this challenge. Here, we propose silver trifluoroacetate (AgTFA) as a multifunctional electrolyte additive that synergistically regulates Li nucleation and promotes the formation of an anion-rich solvation structure. Through a spontaneous in situ displacement reaction, uniformly distributed silver nanoparticles (Ag NPs) are generated on the Li surface, providing abundant lithiophilic nucleation sites to enable homogeneous Li deposition. Meanwhile, trifluoroacetate anions (TFA-) with an ultrahigh donor number, together with NO3- anions participating in Li+ solvation, markedly reduce the desolvation barrier and facilitate the formation of a LiF-Li3N-rich SEI. As a result, Li||Li symmetric cells exhibit remarkable cycling stability of up to 2,500 hours at 0.5 mA·cm-2/0.5 mAh·cm-2, while Li||LiFePO4 full cells deliver a high discharge capacity of 139.8 mAh·g-1 with an excellent capacity retention of 97.28% after 200 cycles at 1.0 C. This work demonstrates a feasible strategy for constructing durable SEI layers by coupling Li nucleation regulation with anion-rich solvation chemistry.
Graphical Abstract
Keywords
Li metal anodes, solid electrolyte interphases, Li nucleation, solvation structure, silver trifluoroacetate
1. Introduction
Rechargeable lithium (Li) metal batteries (LMBs) are widely regarded as leading candidates for next-generation energy storage systems owing to their use of Li metal anodes, which deliver an ultra-high theoretical specific capacity (3,860 mAh·g-1) and the lowest redox potential (-3.040 V vs. standard hydrogen electrode)[1-3]. Despite these advantages, the practical deployment of LMBs remains constrained by several critical challenges during cycling, including uncontrolled dendritic Li growth[4,5], unstable solid electrolyte interphases (SEIs)[6,7], and severe volume fluctuations[8,9]. The intrinsically high Fermi level and strong reactivity of Li metal trigger spontaneous and irreversible reactions with electrolyte components, giving rise to a fragile SEI layer. This layer often fails to accommodate the pronounced volume changes occurring during repeated Li plating/stripping cycles[10]. As a result, its mechanical breakdown leads to continuous electrolyte consumption, heterogeneous Li deposition, dendrite formation, and electrically isolated Li, ultimately causing low Coulombic efficiency (CE), poor cycling durability, and serious safety concerns[11,12].
Over the past decades, numerous strategies have been pursued to address these limitations, including anode structural design[13,14], construction of artificial interfacial layers[15,16], and the incorporation of electrolyte additives[17,18]. Among these, tailoring electrolyte composition through the introduction of functional additives, such as LiNO3, alkali cations, and perfluorinated surfactants, has emerged as a facile yet powerful approach to suppress Li dendrite formation and stabilize Li anodes[18,20]. By adjusting the type and concentration of additives, the solvation sheath of Li+ ions can be effectively engineered, which directly influences SEI properties and Li deposition behavior[21,22]. Thus, regulating the local solvation environment and promoting the preferential reduction of favorable electrolyte species are essential for building a stable and robust SEI.
Metal salt ions are among the most widely studied additives for constructing stable SEI through in situ chemical reactions[23]. The introduction of salts such as CuF2, InF3, and MgCl2 can promote the formation of metal-rich SEI layers containing metal particles or Li alloys, which serve as abundant Li-ion nucleation sites and facilitate uniform Li deposition[24-26]. However, the uneven distribution of these in situ generated particles or alloys may result in non-uniform SEI layers, thereby reducing interfacial stability. To overcome this issue, incorporating additional inorganic components with high mechanical and electrochemical stability into the SEI has been considered an effective strategy for reinforcing interfacial robustness. Among these components, lithium fluoride (LiF) has received particular attention due to its outstanding mechanical strength, wide band gap, high ionic conductivity, and large surface energy[27]. The presence of LiF within the SEI not only accelerates interfacial reaction kinetics but also promotes homogeneous Li+ flux, effectively suppressing dendrite growth[28]. Therefore, combining lithiophilic metal sites with LiF-rich SEI components through the rational design of multifunctional additives represents a promising approach for constructing hybrid SEIs. Such hybrid structures can reinforce the interfacial layer, homogenize current distribution, and enable uniform Li deposition, thereby laying a solid foundation for the commercialization of Li metal anodes[26,29].
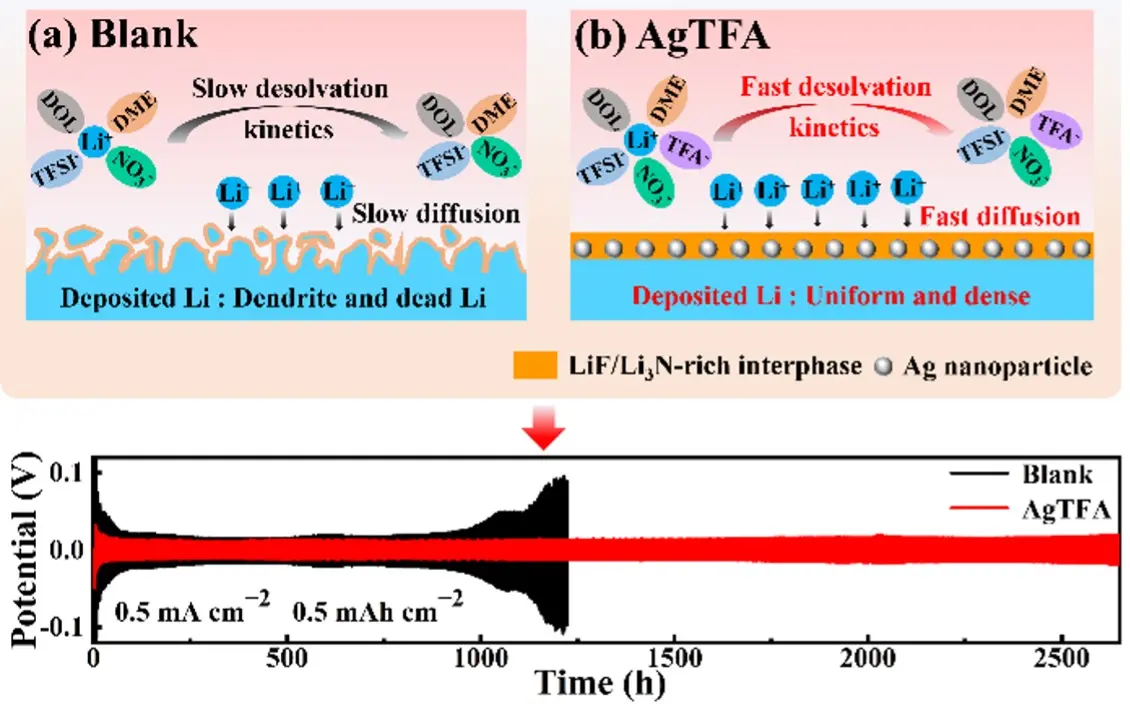
In this work, we introduce silver trifluoroacetate (AgCO2CF3, AgTFA) as a multifunctional electrolyte additive in ether-based electrolytes to stabilize Li metal anodes through the synergistic regulation of Li nucleation and anion-rich solvation structures. On one hand, are generated on the Li surface via the spontaneous in situ reduction reaction between AgTFA and Li metal. These uniformly distributed Ag NPs provide abundant lithiophilic nucleation sites, reduce interfacial resistance, and promote homogeneous Li deposition. Density functional theory (DFT) calculations further confirm that Li atoms exhibit much stronger binding to various Ag crystal facets than to Li surfaces, thereby enhancing nucleation in AgTFA-containing electrolytes. On the other hand, trifluoroacetate (TFA-) anions, characterized by an ultrahigh Gutmann donor number (DN, 34.0 kcal·mol-1) and low binding energy with Li+ ions, strongly coordinate with Li+ ions, participate in the solvation structure, and subsequently decompose into LiF[30]. Simultaneously, the presence of LiNO3 contributes to the formation of Li3N on the Li surface, leading to a composite SEI composed of Ag NPs, LiF, and Li3N. This hybrid SEI combines mechanical robustness with favorable ion transport properties. As illustrated in Figure 1, the synergistic effects of AgTFA result in accelerated interfacial kinetics, dendrite-free Li deposition, and enhanced interfacial stability. Consequently, Li||Li symmetric cells with AgTFA demonstrate stable cycling for over 2,500 hours at 0.5 mA·cm-2/0.5 mAh·cm-2, while Li||Cu cells maintain a high Coulombic efficiency of 98.28% after 300 cycles. Notably, Li||LiFePO4 full cells achieve a capacity retention of 97.28% after 200 cycles at 1 C.
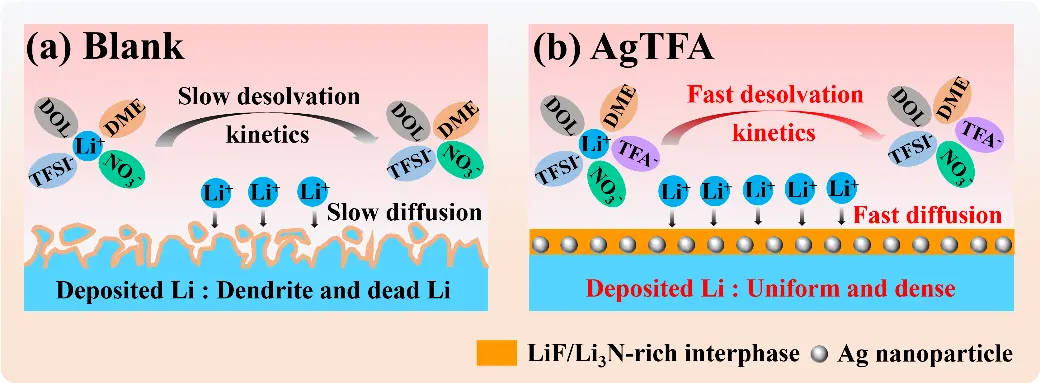
Figure 1. Schematic comparison of Li plating processes in the (a) blank and (b) AgTFA electrolyte. Li: lithium; AgTFA: silver trifluoroacetate; DOL: 1, 3-dioxopentane; TFA-: trifluoroacetate; DME: 1, 2-dimethoxyethane; TFSI-: bis(trifluoromethanesulfonyl)imide.
2. Experimental Section
2.1 Materials
AgTFA (AgCO2CF3, 97%) was purchased from Mindray Biochemical Technology Co. Ltd. (Shanghai, China). Lithium trifluoroacetate (LiCO2CF3, LiTFA, 97%) and deuterated acetone (C2D6SO, 99.8%) were obtained from Titan Technology Co. Ltd. (Shanghai, China). Lithium bis(trifluoromethylsulfonyl)imide (LiTFSI, 99.9%) and N-methyl-2-pyrrolidone (NMP, 99.9%) were supplied by Bailingwei Technology Co. Ltd. (Beijing, China). Anhydrous lithium nitrate (LiNO3, 99.99%) was purchased from Alfa Aesar (Shanghai, China) 1, 3-dioxopentane (DOL, 99.9%) and 1, 2-dimethoxyethane (DME, 99.9%) were supplied by Sigma-Aldrich (America). Li metal disks (14 mm diameter, 700 μm thickness) and Cu foils (35 μm thickness) were obtained from China Energy Lithium Co., Ltd. (Tianjin, China) and Jingliang Copper Industry Co., Ltd. (Shenzhen, China), respectively. Lithium iron phosphate (LiFePO4, LFP) powders were purchased from Darui Fine Chemicals Co. Ltd. (Shanghai, China). Conductive carbon (Super P) and poly(vinylidene) fluoride (PVDF) were obtained from Shenzhen Kejing Star Technology (Shenzhen, China). All raw materials were stored in an argon-filled glovebox and used without further purification.
2.2 Electrolyte preparation and electrode preparation
The blank electrolyte was prepared by dissolving 1 M LiTFSI and 0.2 M LiNO3 in a 1:1 (V/V) mixture of DOL and DME. AgTFA was then added to the blank electrolyte at concentrations of 5, 10, and 20 mM to obtain the AgTFA electrolytes. The formulation containing 10 mM AgTFA, which exhibited the best cycling performance, was selected for subsequent electrochemical and characterization tests. In addition, a series of electrolytes without LiNO3 was prepared to decouple the contributions of Ag⁺ ions in liquid nuclear magnetic resonance spectroscopy analysis.
The LFP cathodes were fabricated by blade-coating a slurry of LFP, Super P, and PVDF in a mass ratio of 8:1:1 onto aluminum foils, followed by drying at 110 °C for 12 h and punching into 10 mm-diameter round disks with an LFP loading of 1.5-2.0 mg·cm-2.
2.3 Coin cell assembly and electrochemical measurements
All batteries were assembled in CR2032-type coin cells inside an argon-filled glovebox, with Celgard-2300 membranes used as separators. Li||Li symmetric cells were assembled using two identical Li foils as electrodes. Li||Cu cells consisted of a Li disk anode paired with a Cu foil cathode, while Li||LFP cells were fabricated using a Li disk anode and an LFP cathode. The electrolyte volume was fixed at 40 μL for Li||Li and Li||Cu cells and 50 μL for Li||LFP cells.
Galvanostatic charge/discharge tests were carried out at 25 °C using a LAND-CT2001A battery testing system. The average CE was evaluated using Li||Cu half cells. For these measurements, the cathode was prepared by pre-depositing and stripping 3 mAh·cm-2 of Li on a Cu foil at 0.5 mA·cm-2, with stripping terminated at 1 V. Pre-depositing Li on the Cu electrode ensured a stable and clean Li surface, thereby eliminating the effects of oxide layers and defects, and providing a suitable surface for accurate CE evaluation. The Li||Cu cells were cycled for 10 plating/stripping cycles at 0.5 mA·cm-2 with a capacity of 0.5 mAh·cm-2 per cycle, with each stripping step terminated at 1 V. Li||Li symmetric cells were tested under different current densities and areal capacities (0.5 mA·cm-2/0.5 mAh·cm-2 and 1 mA·cm-2/1 mAh cm-2). Their rate performance was further evaluated at current densities ranging from 0.5 to 3.0 mA·cm-2. Li||LFP full cells were cycled galvanostatically between 2.5 and 4.0 V at 1 C (versus Li/Li+), with the theoretical capacity of LFP set at 170 mAh·g-1. The rate capability of the full cells was assessed by varying the C-rate from 0.2 to 5 C. Cyclic voltammetry (CV), Tafel plots, and linear sweep voltammetry were conducted on a CHI 600e workstation: Li||Cu cells were tested in the potential range of -0.1 to 0.5 V at a scan rate of 1 mV·s-1; Li||Li cells in the range of -0.2 to 0.2 V at 1 mV·s-1; and Li||LFP cells in the range of 2.5 to 4.0 V at 0.1 mV·s-1). Electrochemical impedance spectroscopy (EIS) was performed on a ZAHNER IM6ex workstation with a perturbation amplitude of 5 mV, over a frequency range of 100 kHz to 10 mHz.
2.4 Characterization
The morphologies of the treated and cycled Li electrodes were examined using a scanning electron microscope (SEM, JEO-LSM7800F) equipped with an energy dispersive spectrometry (EDS) detector. The phase composition of the bare and treated Li electrodes was analyzed by X-ray diffraction (XRD, Rigaku Mini Flex) with a 2θ scan range of 5 to 85 deg. X-ray photoelectron spectroscopy (XPS, ESCALAB 250Xi) was employed to investigate the elemental composition and chemical states of the treated and cycled Li electrodes. The 7Li and 1H NMR spectra of the liquid electrolytes were acquired using a 400 MHz Bruker Avance NEO spectrometer.
2.5 Computational methods
All periodic spin-polarized first-principles calculations were carried out using the Vienna Ab initio Simulation Package (VASP)[31-33]. The Perdew-Burke-Ernzerhof (PBE)[34] exchange-correlation functional with D3 dispersion correction[35] was employed to describe electronic interactions. The projected augmented wave (PAW) method[36] was applied with a plane-wave cutoff energy of 520 eV to model the interactions between nuclei and electrons. All Li and Ag slabs comprised five atomic layers, with the bottom two layers fixed during structural relaxation. A vacuum layer of 20 Å along the vertical direction was introduced to avoid spurious interactions between periodic images. The Brillouin zone was sampled using a Γ-centered 4 × 4 × 1 Monkhorst-Pack k-point grid. The energy and force convergence criteria were set to 10-6 eV and 0.02 eV/Å, respectively. Geometric structures were visualized using VESTA[37]. The adsorption energies (Eads) were calculated as:
where Etotal is the total energy of the slab with adsorbed molecules, and Eslab and Eadsorbate are the energies of the clean slab and isolated adsorbate, respectively.
All non-periodic quantum chemistry calculations were performed using Gaussian 16[38] at the M06-2X/def2-TZVP level[39,40] with D3 dispersion correction[35]. Frequency calculations were carried out to confirm the absence of imaginary frequencies. The binding energies between solvents or anions and Li+ were calculated as:
where Etotal is the total energy of the Li+-molecule complex, and Emolecule and ELi+ are the energies of the isolated solvent molecule (or anion) and Li+, respectively.
3. Results and Discussion
Owing to the strong reducibility of Li metal, Ag NPs are formed via a displacement chemical reaction upon contact between Li metal and the AgTFA electrolyte. The surface morphologies of the Li disk after immersion in AgTFA/DME solution for 4 h were examined using SEM coupled with EDS mapping. As shown in Figure 2a, a smooth and flat reaction layer is formed on the Li anode surface. The EDS mapping in the top-right inset of Figure 2a and the high-magnification SEM image in Figure 2b further reveal that the layer is composed of numerous evenly distributed Ag NPs. Meanwhile, the XRD pattern of the treated Li electrode (Figure 2c) exhibits additional Ag diffraction peaks compared with the bare Li electrode, confirming the formation of metallic Ag. To further verify the chemical composition of the treated surface, XPS analysis was conducted. In the Ag 3d spectra (Figure 2d), the peaks at 373.6 eV (Ag 3d3/2) and 367.5 eV (Ag 3d5/2) correspond to metallic Ag (0), providing direct evidence of the displacement reaction[26]. In the F 1s spectra (Figure 2e), peaks at 688.5 eV and 684.7 eV are assigned to the C-F bond and LiF, respectively, with LiF likely formed via reduction of TFA- by Li metal[41]. The C 1s spectra (Figure 2f) display signals corresponding to C-C and C-O bonds from the organic solvent and the C-F bond of AgTFA, possibly representing residual unreacted AgTFA, observed at 284.8, 286.3, and 292.6 eV, respectively. The presence of Ag NPs can accelerate Li nucleation dynamics and improve the stability of Li plating/stripping. DFT calculations (Figure 2d,e,f) show that, regardless of the surface facet, the Ag surface exhibits significantly higher Li adsorption binding energies than the Li surface. Therefore, Ag NPs act as lithiophilic sites, lowering the Li nucleation energy barrier and ensuring uniform Li deposition.
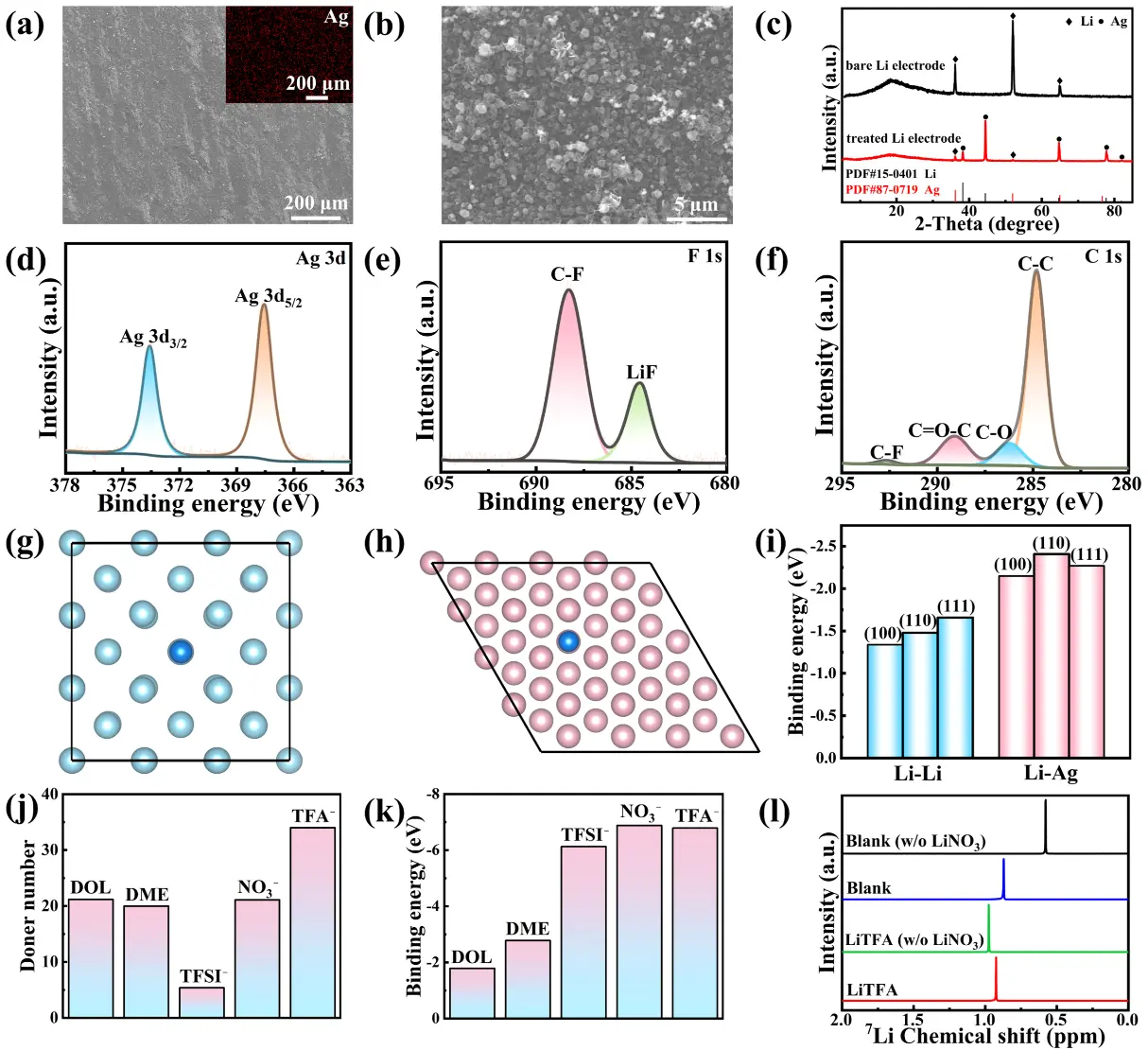
Figure 2. (a, b) SEM images and Ag distribution; (c) XRD patterns, and (d,e,f) XPS profiles (d: Ag 3d, e: F 1s; f: C 1s) of the Li electrode treated by AgTFA/DME solution; (g, h) Top views of Li atom adsorption on Li(100) and Ag(111) surfaces; (i) Binding energies of Li atom adsorption on different facets of Li and Ag surfaces; (j) Donor numbers of solvent molecules and anions in two electrolytes; (k) Binding energies of Li+ with solvent molecules and anions in two electrolytes; (l) 7Li NMR spectra of the different electrolytes. SEM: scanning electron microscope; XRD: X-ray diffraction; XPS: X-ray photoelectron spectroscopy; AgTFA: silver trifluoroacetate; DME: 1, 2-dimethoxyethane; NMR: nuclear magnetic resonance spectroscopy.
The solvation structure of Li+ ions with solvent molecules and anions plays a critical role in constructing a stable SEI. Gutmann DN, as a quantitative measure of Lewis basicity, is commonly used to evaluate the solvation capability of anions. It is generally accepted that solvent molecules and anions with high DN exhibit stronger coordination with Li+ ions and preferentially occupy the solvation sheath, thereby promoting Li+ desolvation[42]. As shown in Figure 2j, compared with the solvent molecules and anions in the blank electrolyte (DOL: 21.2 kcal·mol-1, DME: 20 kcal·mol-1, TFSI-: 5.4 kcal·mol-1, NO3-: 22 kcal·mol-1), the added TFA- anions possess an ultrahigh DN of 34.0 kcal·mol-1[30,43]. Moreover, the binding energies between solvent molecules or anions and Li+ ions reflect the desolvation energy barrier modulated by different components. DFT calculations (Figure 2k) indicate that NO3- and TFA- exhibit lower binding energies with Li+ ions (-6.88 eV and -6.79 eV, respectively), suggesting weak coordination and preferential decomposition, which explains the formation of a LiF-Li3N-rich SEI. Consequently, the ultrahigh DN and lower binding energy of TFA- enable it to compete with other solvents and anions, participate in the Li+ solvation sheath, and preferentially decompose to form a LiF-rich SEI. To experimentally investigate the impact of TFA- on the Li+ solvation shell, 7Li NMR tests were conducted using four selected electrolytes: blank ether electrolyte without LiNO3 (blank w/o LiNO3), blank ether electrolyte with LiNO3 (blank), LiTFA electrolyte without LiNO3 (LiTFA w/o LiNO3), and LiTFA electrolyte with LiNO3 (LiTFA). Different additives result in distinct coordination environments for Li+ ions. As shown in Figure 2l, compared with the blank electrolyte (0.578 ppm), the addition of LiNO3 shifts the 7Li NMR signal to a higher value (0.871 ppm), consistent with previous reports[44]. This shift arises because the electronegativity of O atoms in NO3- and TFSI- is lower than that of O atoms in DME and DOL, and thus, when NO3- participates in the Li+ solvation structure, it reduces the electron density around the Li+ nucleus[44]. In contrast, introducing TFA- into the electrolyte shifts the 7Li NMR signal even higher (0.976 ppm), consistent with observations in electrolytes containing LiTFA reported by Li et al.[45]. The strong coordination between the carbonyl group (C=O) of TFA- and Li+ allows TFA-, a strong electron-donating species, to enter the Li+ solvation shell and replace other solvent molecules or anions, further decreasing the electron density around the Li+ nucleus[46]. When both NO3- and TFA- are added to the blank electrolyte, the 7Li NMR signal shifts to 0.924 ppm, indicating that NO3- and TFA- collaboratively participate in the Li+ solvation structure and reduce Li+ binding to solvent molecules. Similarly, in carbonate-based electrolytes, the combined addition of NO3- and TFA- collectively results in an upward shift of the 7Li peak, confirming their strong coordination with Li+ ions[47]. Under the combined effect of NO3- and TFA-, Li+ desolvation is promoted, forming an anion-rich solvation structure that facilitates fast desolvation kinetics and establishes a foundation for a stable LiF-Li3N-rich SEI. Furthermore, 1H NMR spectra (Figure S1) show that signal peaks shift to lower values in the AgTFA electrolyte with LiNO3, further confirming the weakened interaction between the solvent and Li+ ions.
In addition, CV tests of the Li||Cu cells were conducted using 0.2 M LiTFA/DME as the electrolyte to further confirm the reductive decomposition of TFA- anions. As shown in Figure S2, distinct reduction onset potential appears at approximately 1.1 V (vs. Li/Li+) in the LiTFA/DME electrolyte compared with the DME electrolyte, which can be attributed to the reduction of TFA-[48]. This further demonstrates that TFA- participates in the Li+ solvation structure and decomposes to form a LiF-rich SEI on the electrode surface, thereby influencing interfacial charge transfer and interfacial energy.
To quantitatively evaluate the effect of the AgTFA additive on the reaction kinetics of LMBs, Li||Li and Li||Cu cells were assembled for electrochemical measurements. As shown in the CV curves of Li||Cu cells (Figure 3a), compared with the blank electrolyte, the AgTFA electrolyte exhibits a higher plating/stripping current (2.35 mA·cm-2) and a lower nucleation onset potential (12 mV), indicating enhanced Li deposition kinetics. The increased Li stripping capacity further confirms improved reversibility of lithium plating and stripping. Figure 3b presents the nucleation overpotentials of Li||Cu cells in different electrolytes. At the initial stage of Li nucleation, the nucleation and mass-transfer overpotentials in the AgTFA electrolyte are 34.02 mV and 9.3 mV, respectively, much lower than 64.82 mV and 25.1 mV in the blank electrolyte. This demonstrates that AgTFA contributes to the formation of a stable SEI, effectively reduces the energy barriers for Li nucleation and mass transfer, and promotes Li deposition kinetics. CV tests of Li||Li cells (Figure 3c) show that the current density increases from 2.4 mA·cm-2 in the blank electrolyte to 9.0 mA·cm-2 in the AgTFA electrolyte, indicating improved interfacial kinetics and rapid Li stripping/plating processes. Moreover, as shown in Figure 3d, the incorporation of AgTFA significantly enhances the exchange current density of the Li electrode from 0.39 mA·cm-2 in the blank electrolyte to 1.82 mA·cm-2 in the AgTFA electrolyte. This enhancement mainly arises from the formation of an Ag NPs-rich SEI, which accelerates interfacial charge transfer kinetics.
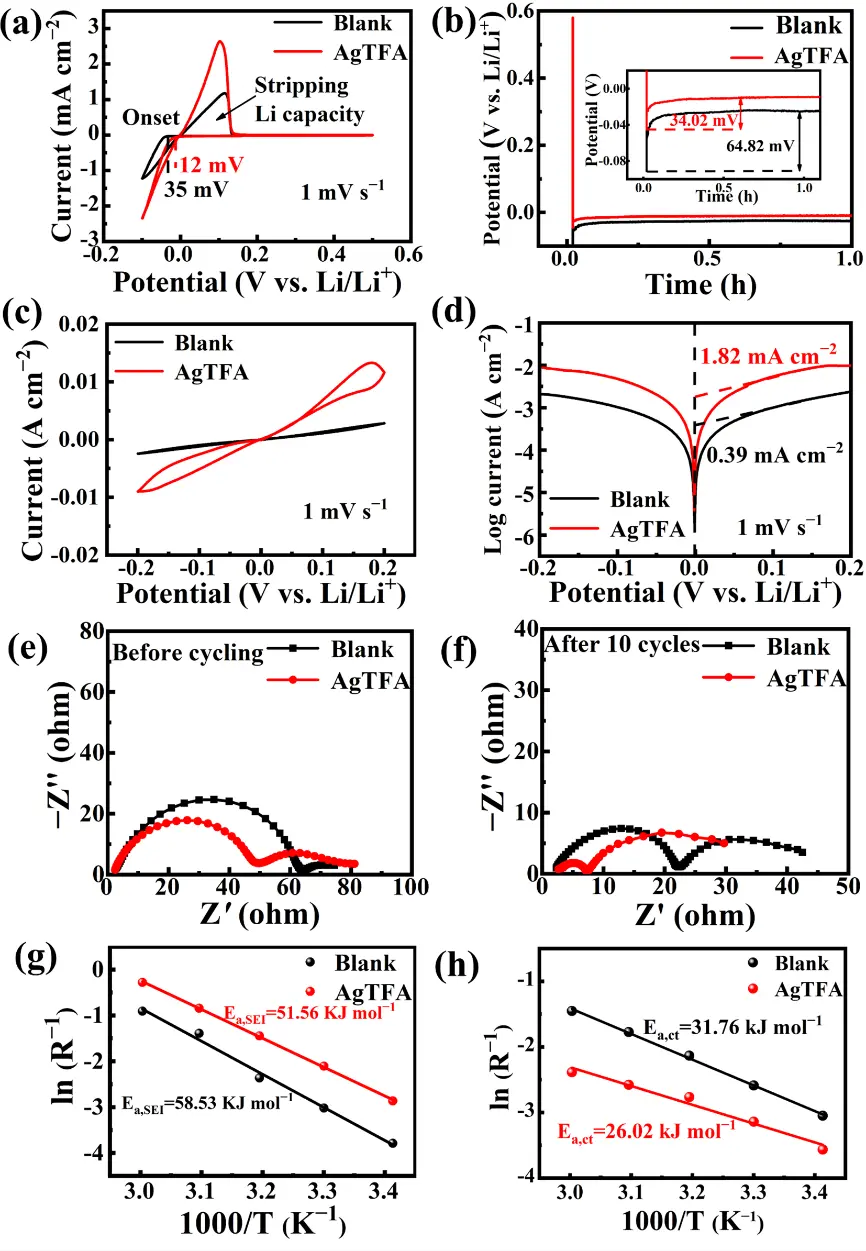
Figure 3. (a) CV curves and (b) the nucleation overpotentials of Li deposition of the Li||Cu cells in different electrolytes; (c) CV curves; (d) Tafel plots, electrochemical impedance spectra (e) before cycling and (f) after 10 cycles, and Arrhenius behavior of (g) the RSEI and (h) the Rct of Li||Li cells with different electrolytes. CV: Cyclic voltammetry; AgTFA: silver trifluoroacetate.
Furthermore, EIS analysis was conducted to evaluate the effect of AgTFA on interfacial stability. As shown in Figure 3e,f and Figure S3, both before and after cycling, cells using the AgTFA electrolyte exhibit smaller RSEI and Rct values compared with those using the blank electrolyte. The reduced interfacial resistance in the AgTFA electrolyte can be attributed to the presence of numerous Ag NPs with high conductivity and strong Li affinity, which accelerate interfacial charge transfer. Although the AgTFA electrolyte initially shows a relatively large RSEI, this value gradually decreases as the SEI forms during cycling, indicating that an Ag NPs-LiF-Li3N-rich stable SEI enables rapid Li+ transport and effectively lowers interfacial resistance. Based on EIS results at different temperatures (Figure S4), the diffusion activation energy (Ea, SEI) and desolvation activation energy (Ea, ct) were calculated according to the Arrhenius law[49]. As shown in Figure 3g,h, the Ea, SEI and Ea, ct values for the AgTFA electrolyte are 51.13 kJ·mol-1 and 26.02 kJ·mol-1, respectively, significantly lower than those of the blank electrolyte (58.53 kJ·mol-1 and 31.76 kJ·mol-1), indicating accelerated Li+ desolvation and transport.
Li||Cu cells with different electrolytes were further assembled to evaluate Li utilization during cycling. As shown in Figure 4a, the CE of Li||Cu cells using the blank electrolyte decreases rapidly from 96.95% in the initial cycle to 86.3%, mainly due to irreversible capacity loss caused by Li dendrites and dead Li. In contrast, the AgTFA electrolyte facilitates the formation of a stable SEI on the Li surface and promotes uniform Li plating/stripping. The initial CE is 99.01% and remains nearly 98.28% even after nearly 300 cycles. Moreover, the voltage-capacity profiles of Li||Cu cells with different electrolytes (Figure S5) show that cells with the AgTFA electrolyte maintain relatively low polarization voltage after 300 cycles, further demonstrating the critical role of AgTFA in stabilizing Li anodes. Figure 4b shows that the capacity retention increases from 99.15% in the blank electrolyte to 99.27% in the AgTFA electrolyte, while the average CE rises from 98.88% to 99.83%, confirming excellent reversibility and improved utilization of Li during deposition and stripping.
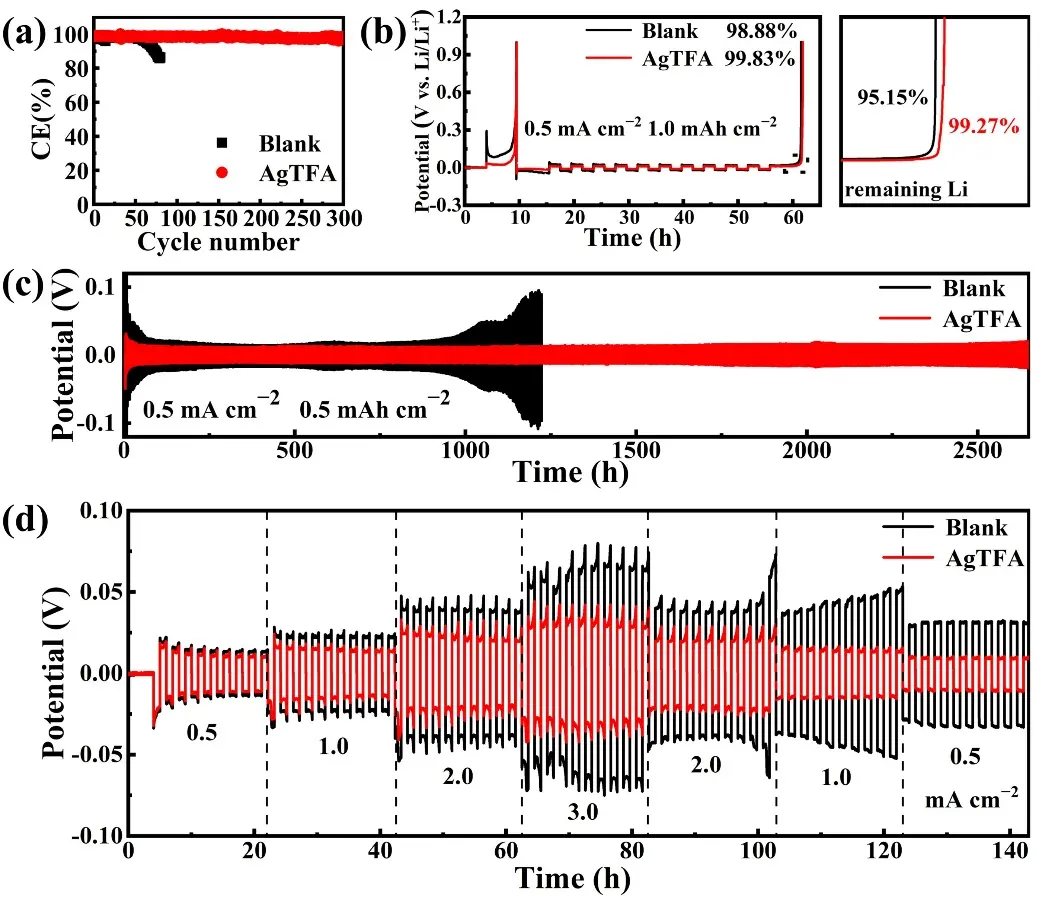
Figure 4. (a) Coulombic efficiency and (b) average coulombic efficiency of Li||Cu cells with different electrolytes; (c) Cycling performance and (d) rate performance of the Li||Li cells with different electrolytes at 0.5 mA·cm-2 for 0.5 mAh·cm-2. AgTFA: silver trifluoroacetate.
In addition, to further verify the advantages of an Ag NPs-LiF-Li3N stable SEI for LMBs, the cycling lifespan of Li||Li cells with different electrolytes was evaluated by monitoring voltage fluctuations. As shown in Figure 4c and Figure S6, at both 0.5 mA·cm-2 or 1.0 mA·cm-2, Li||Li cells with the blank electrolyte exhibit larger polarization voltages and poorer cycling stability. This is because the SEI is too fragile to suppress interfacial side reactions and continuously ruptures and reconstructs during cycling, leading to high interfacial resistance and increased polarization voltages[50]. In contrast, Li||Li cells with the AgTFA electrolyte exhibit remarkably stable cycling with low overpotentials for over 2,500 h at 0.5 mA·cm-2/0.5 mAh·cm-2, and they can still sustain stable cycling for 700 h even at a higher current density and areal capacity of 1.0 mA·cm-2/1.0 mAh·cm-2. This enhanced cycling stability arises from the Ag NPs-LiF-Li3N hybrid SEI, in which the lithiophilic Ag NPs promote uniform Li nucleation while the anion-rich solvation structure contributes to robust SEI formation. Furthermore, Li||Li cells with the AgTFA electrolyte exhibit superior rate performance, displaying consistently lower polarization voltages at different current densities (Figure 4d). This result demonstrates that the introduction of AgTFA significantly improves the stability of Li plating/stripping at high current densities by optimizing the SEI composition. In addition, the influence of AgTFA concentration (5, 10, 20 mM) on the cycling stability of Li anodes was investigated (Figure S7). The 10 mM concentration delivers the best stability, as a lower concentration is insufficient to generate a dense Ag NP interfacial layer and adequate LiF coverage, while an excessive concentration results in the formation of a thick, uneven Ag-rich layer. This leads to a coarse electrode surface, which is unfavorable for Li+ nucleation and transport.
Ex-situ SEM characterization was employed to investigate the influence of the AgTFA additive on the morphological evolution of Li in Li||Li symmetrical cells. After 10 cycles at 0.5 0.5 mA·cm-2/0.5 mAh·cm-2, the Li electrode cycled in the blank electrolyte exhibits dendritic deposition with non-uniform distribution, resulting in agglomerated surface structures (Figure 5a). In contrast, the Li electrode in the AgTFA electrolyte shows a smooth and flat surface with dense and uniform Li deposition (Figure 5b). EDS mapping analyses (Figure S8) indicate that O, C, F, and N elements are evenly distributed on the electrode surface in both electrolytes. Notably, in the AgTFA electrolyte, a uniform Ag signal is observed across the surface, accompanied by significantly enhanced F and N signals (Figure S9 and Figure S10), demonstrating that the AgTFA additive participates in SEI formation, resulting in a robust Ag NPs-LiF-Li3N-rich robust SEI. After 200 cycles, as shown in Figure S11a, Li electrodes in the blank electrolyte exhibit pronounced dendrites and numerous pores, indicating uneven Li deposition and increased surface roughness. By contrast, the electrode in the AgTFA-containing electrolyte displays uniform, island-like Li deposition with a dendrite-free morphology (Figure S11b). This improvement is attributed to the abundant Ag NPs providing numerous Li nucleation sites, combined with a LiF-Li3N-rich SEI that ensures homogeneous Li growth. Even under the harsh conditions of 2.0 mA·cm-2/2.0 mAh·cm-2, the cross-sectional view after 50 cycles in the AgTFA electrolyte shows a relatively intact structure and dense Li deposition without electrode pulverization or peeling, markedly superior to the blank electrolyte.
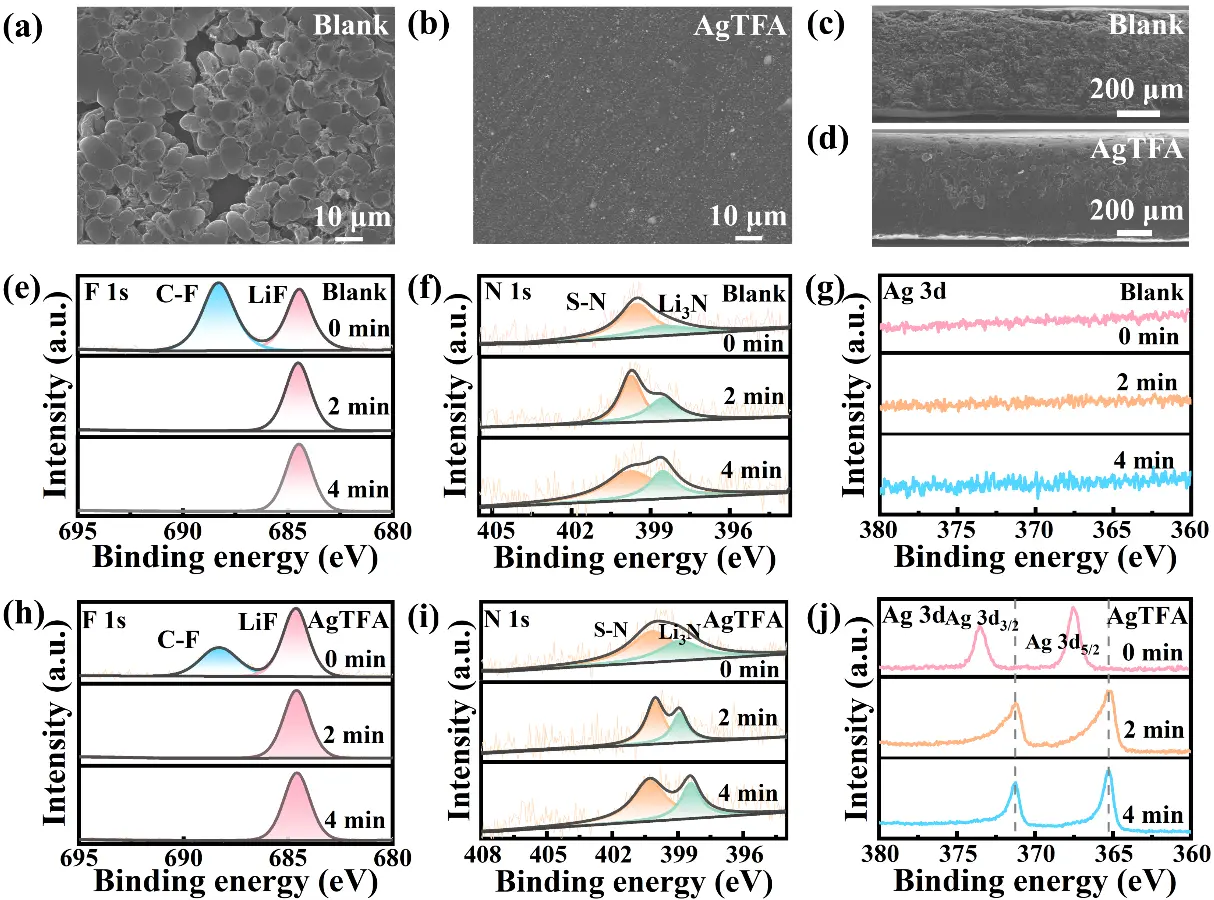
Figure 5. Top-view SEM images of Li metal anode after 10 cycles in (a) blank electrolyte and (b) AgTFA electrolyte; Cross-sectional SEM images of Li metal anode after 50 cycles in (c) blank electrolyte and (d) AgTFA electrolyte; (e, f, j) XPS spectra (e and h: F 1s, f and i: N 1s, g and j: Ag 3d) with etching depth profiles of the SEI on Li metal anode surface after 10 cycles in different electrolytes. SEM: scanning electron microscope; AgTFA: silver trifluoroacetate; XPS: X-ray photoelectron spectroscopy.
To elucidate how the AgTFA additive tailors the chemical composition of the SEI, XPS analysis was performed on Li||Li symmetrical cells after 10 cycles at 0.5 mA·cm-2/0.5 mAh·cm-2. In the F 1s spectra, C-F and LiF signals are observed at binding energies of 688.3 eV and 684.5 eV, respectively (Figure 5e,h). In the N 1s spectra, signals at 399.8 eV and 398.5 eV correspond to S-N and Li3N, respectively (Figure 5f,i)[51]. Notably, the contents of LiF and Li3N on the electrode surface in the AgTFA electrolyte account for 62.89% and 46.52% of F and N elements, respectively, significantly higher than 43.82% and 37.89% in the blank electrolyte, confirming that TFA- and NO3- participate in the Li+ solvation structure and decompose to form a LiF-Li3N-rich SEI. With increasing etching depth, the intensities of LiF and Li3N signals progressively increase, indicating the presence of a LiF-Li3N-rich SEI layer. Compared with the blank electrolyte, a metallic Ag signal appears at 367.5 eV and 373.5 eV in the AgTFA electrolyte (Figure 5j), which shifts toward lower binding energies with depth, attributed to the formation of Li-Ag alloys during Li deposition. The atomic percentages of F, N, and Ag at different depths are summarized in Table S1 and Table S2. The higher content of inorganic components in the AgTFA electrolyte underscores its role in stabilizing the Li anode. Therefore, the addition of AgTFA promotes the formation of an Ag NPs-LiF-Li3N-richstable SEI by modulating the Li+ nucleation and solvation structure, thereby regulating Li deposition and stripping behavior.
Li||LFP cells were assembled and tested to evaluate the practical application potential of the AgTFA electrolyte in LMBs. As shown in Figure 6a, full cells with the AgTFA electrolyte exhibit superior cycling stability compared with those using the blank electrolyte at 1 C. Specifically, the initial discharge capacity of the AgTFA cells is 143.7 mAh·g-1, which increases to 148.9 mAh·g-1 after activation. After 200 cycles, the discharge capacity remains at 139.8 mAh·g-1, corresponding to an outstanding capacity retention of 97.28% (Figure S12). In contrast, cells with the blank electrolyte show an initial discharge capacity of 142.7 mAh·g-1, which increases to 149 mAh·g-1 upon activation but declines rapidly to 122.4 mAh·g-1 after 200 cycles, corresponding to a capacity retention of only 85.77% (Figure S12). CV curves (Figure 6b) reveal that cells with the AgTFA electrolyte exhibit reduced overpotentials, evidenced by oxidation and reduction peaks shifting 30 mV and 25 mV earlier, respectively, along with a narrower potential gap between redox peaks. These results indicate high reaction reversibility and excellent charge transfer kinetics. The rate capability of the cells was further evaluated from 0.2 C to 5.0 C (Figure 6c). Clearly, full cells with the AgTFA electrolyte demonstrate superior rate performance, delivering high discharge capacities of 132.7 mAh·g-1 and 122.1 mAh·g-1 even at 3.0 C and 5.0 C, respectively, significantly outperforming cells with the blank electrolyte. The exceptional cycling stability and rate performance can be attributed to the formation of an Ag NPs-LiF-Li3N-rich stable SEI in the AgTFA electrolyte, which guides uniform Li plating/stripping and reduces interfacial resistance during cycling (Figure 6d,e). Before cycling, cells with both electrolytes show comparable resistance values. However, after 100 cycles, the resistance of cells with the AgTFA electrolyte (17.06 Ω) is markedly lower than that of cells with the blank electrolyte (23.52 Ω). Moreover, as shown in Figure S13, the Li metal surface in the blank electrolyte exhibits a rough and porous morphology, whereas the AgTFA electrolyte maintains a flat and uniform deposition. Therefore, the introduction of AgTFA effectively optimizes the chemical composition and structure of the SEI, regulates the Li plating/stripping process, and consequently enhances the discharge capacity and cycling stability of full cells.
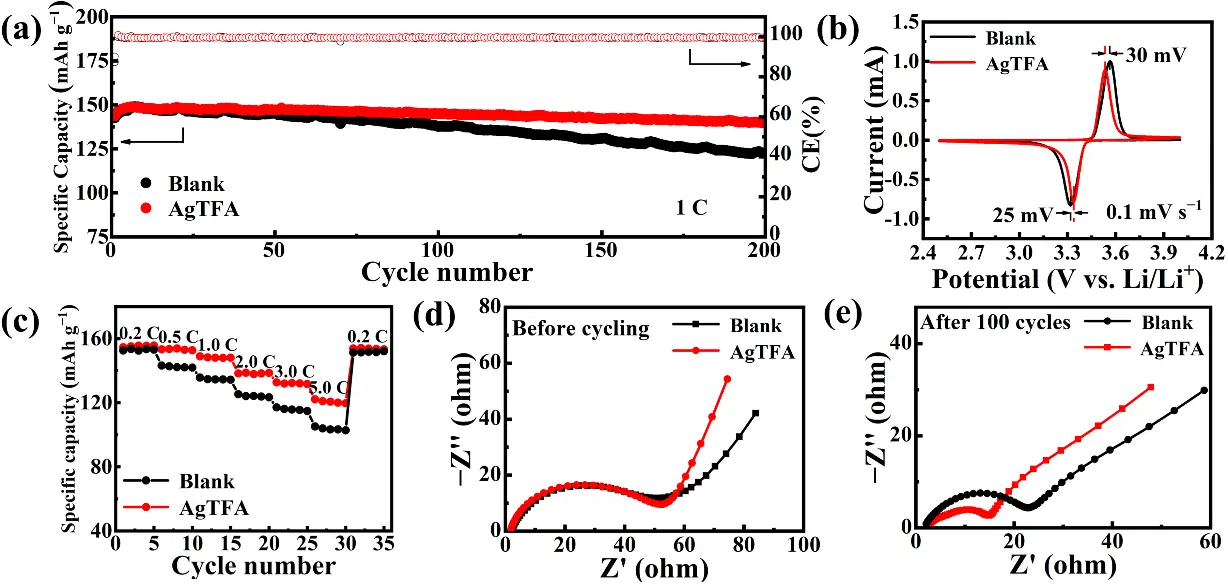
Figure 6. (a) Cycling performance of Li||LFP cells using different electrolytes at 1.0 C; (b) CV curves of Li||LFP cells; (c) Rate performance of Li||LFP cells using different electrolytes; and EIS plots of Li||LFP cells using different electrolytes (d) before cycling and (e) after 100 cycles at 1.0 C. Li: lithium; CV: Cyclic voltammetry; AgTFA: silver trifluoroacetate; LFP: lithium ferro-phosphate.
4. Conclusion
In summary, a stable Ag NPs-LiF-Li3N hybrid SEI was constructed via the synergistic regulation of Li nucleation and anion-rich solvation structure by introducing AgTFA as an electrolyte additive. Both experimental results and DFT calculations confirm that uniformly distributed Ag NPs, generated through an in situ displacement reaction between AgTFA and Li metal, serve as lithiophilic nucleation sites that substantially lower the energy barrier for Li nucleation and promote homogeneous Li deposition. Simultaneously, TFA- anions with an ultrahigh donor number, together with NO3- anions, strongly coordinate with Li+ ions, accelerate desolvation kinetics, and contribute to the formation of a LiF-Li3N-rich SEI layer. This hybrid interphase effectively suppresses Li dendrite growth and enhances the cycling stability of LMBs. Compared with other metal salts used as additives to construct metal-rich SEI layers, AgTFA uniquely provides both lithiophilic sites and an inorganic-rich SEI, synergistically reinforcing interfacial stability. Benefiting from these combined effects, Li||Li symmetrical cells with the AgTFA-containing electrolyte exhibit extended cycling stability, maintaining a low overpotential for over 2,500 h at 0.5 mA·cm-2/0.5 mAh·cm-2. Coupled with LFP cathodes, the full cells deliver a high discharge capacity of 139.8 mAh·g-1 and an impressive capacity retention of 97.28% at 1.0 C after 200 cycles. This work provides a feasible strategy for designing functional electrolytes through the tailored regulation of Li nucleation and solvation chemistry, offering a promising pathway for the development of high-performance LMBs.
Supplementary materials
The supplementary material for this article is available at: Supplementary materials.
Authors contribution
Bai J, Cheng W: Methodology, investigation, validation, writing-original draft.
Wang T: Methodology, validation.
Liu S: Conceptualization, supervision, funding acquisition, writing-review & editing.
Conflicts of interest
Sheng Liu is an Editorial Member of Smart Materials and Devices. The other authors declare no conflicts of interest.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
The data and materials could be obtained from the corresponding author upon request.
Funding
This work was supported by the National Natural Science Foundation of China (No. 22279064 and No. 22075151).
Copyright
© The Author(s) 2025.
References
-
1. Liu B, Zhang JG, Xu W. Advancing lithium metal batteries. Joule. 2018;2(5):833-845.[DOI]
-
2. Jagger B, Pasta M. Solid electrolyte interphases in lithium metal batteries. Joule. 2023;7(10):2228-2244.[DOI]
-
3. Xiao J, Shi F, Glossmann T, Burnett C, Liu Z. From laboratory innovations to materials manufacturing for lithium-based batteries. Nat Energy. 2023;8(4):329-339.[DOI]
-
4. Ning Z, Li G, Melvin DL, Chen Y, Bu J, Spencer-Jolly D, et al. Dendrite initiation and propagation in lithium metal solid-state batteries. Nature. 2023;618(7964):287-293.[DOI]
-
5. Harry KJ, Hallinan DT, Parkinson DY, MacDowell AA, Balsara NP. Detection of subsurface structures underneath dendrites formed on cycled lithium metal electrodes. Nature Mater. 2014;13(1):69-73.[DOI]
-
6. Li NW, Shi Y, Yin YX, Zeng XX, Li JY, Li CJ, et al. A flexible solid electrolyte interphase layer for long-life lithium metal anodes. Angewandte Chemie. 2018;57(6):1505-1509.[DOI]
-
7. Li B, Chao Y, Li M, Xiao Y, Li R, Yang K, et al. A review of solid electrolyte interphase (SEI) and dendrite formation in lithium batteries. Electrochem Energy Rev. 2023;6(1):7.[DOI]
-
8. Li Y, Li J, Xiao H, Xie T, Zheng W, He J, et al. A novel 3D Li/Li9Al4/Li-Mg alloy anode for superior lithium metal batteries. Adv Funct Mater. 2023;33(14):2213905.[DOI]
-
9. Yu J, Xiao J, Li A, Yang Z, Zeng L, Zhang Q, et al. Enhanced multiple anchoring and catalytic conversion of polysulfides by amorphous MoS3 nanoboxes for high-performance Li-S batteries. Angew Chem Int Ed. 2020;59(31):13071-13078.[DOI]
-
10. Lin D, Liu Y, Cui Y. Reviving the lithium metal anode for high-energy batteries. Nature Nanotech. 2017;12(3):194-206.[DOI]
-
11. Zheng X, Huang L, Luo W, Wang H, Dai Y, Liu X, et al. Tailoring electrolyte solvation chemistry toward an inorganic-rich solid-electrolyte interphase at a Li metal anode. ACS Energy Lett. 2021;6(6):2054-2063.[DOI]
-
12. Qin J, Pei F, Wang R, Wu L, Han Y, Xiao P, et al. Sulfur vacancies and 1T phase-rich MoS2 nanosheets as an artificial solid electrolyte interphase for 400 Wh kg-1 lithium metal batteries. Adv Mater. 2024;36(21):2312773.[DOI]
-
13. Bai J, Zhu Z, Liu X, Liu S. A dendrite growth-suppressing Li composite anode decorated with lithiophilic Li-Pb alloys for stable Li metal batteries. Chem Commun. 2025;61(40):7309-7312.[DOI]
-
14. Kong LL, Wang L, Ni ZC, Liu S, Li GR, Gao XP. Lithium-magnesium alloy as a stable anode for lithium-sulfur battery. Adv Funct Mater. 2019;29(13):1808756.[DOI]
-
15. Lu G, Nai J, Luan D, Tao X, Lou XW. Surface engineering toward stable lithium metal anodes. Sci Adv. 2023;9(14):eadf1550.[DOI]
-
16. Li S, Huang J, Cui Y, Liu S, Chen Z, Huang W, , et al. A robust all-organic protective layer towards ultrahigh-rate and large-capacity Li metal anodes. Nat Nanotechnol. 2022;17(6):613-621.[DOI]
-
17. Liu J, Zhou Y, Xiao Z, Xue M, Liu S, Yan T. Tailoring molecular structures for enhanced anchoring of polysulfides in lithium-sulfur batteries. Chem Eng J. 2024;484:149596.[DOI]
-
18. Wang YY, Gu JK, Zhang BH, Li GR, Liu S, Gao XP. Specific adsorption reinforced interface enabling stable lithium metal electrode. Adv Funct Mater. 2022;32(18):2112005.[DOI]
-
19. Duan H, You Y, Wang G, Ou X, Wen J, Huang Q, et al. Lithium-ion charged polymer channels flattening lithium metal anode. Nanomicro Lett. 2024;16(1):78.[DOI]
-
20. Ding F, Xu W, Graff GL, Zhang J, Sushko ML, Chen X, et al. Dendrite-free lithium deposition via self-healing electrostatic shield mechanism. J Am Chem Soc. 2013;135(11):4450-4456.[DOI]
-
21. Cheng H, Sun Q, Li L, Zou Y, Wang Y, Cai T, et al. Emerging era of electrolyte solvation structure and interfacial model in batteries. ACS Energy Lett. 2022;7(1):490-513.[DOI]
-
22. Mao M, Ji X, Wang Q, Lin Z, Li M, Liu T, et al. Anion-enrichment interface enables high-voltage anode-free lithium metal batteries. Nat Commun. 2023;14(1):1082.[DOI]
-
23. Shitaw KN, Tekaligne TM, Jiang SK, Huang CJ, Wu SH, Su WN, et al. Opportunities of liquid metals and liquid metal cations for Li-metal batteries. Chem Eng J. 2023;470:144062.[DOI]
-
24. Yan C, Cheng XB, Yao YX, Shen X, Li BQ, Li WJ, et al. An armored mixed conductor interphase on a dendrite-free lithium-metal anode. Adv Mater. 2018;30(45):1804461.[DOI]
-
25. Pang Q, Liang X, Kochetkov IR, Hartmann P, Nazar LF. Stabilizing lithium plating by a biphasic surface layer formed in situ. Angew Chem Int Ed. 2018;57(31):[DOI]
-
26. Zhong B, Wu J, Ren L, Zhou T, Zhang Z, Liu W, et al. Constructing a lithiophilic and mixed conductive interphase layer in electrolyte with dual-anion solvation sheath for stable lithium metal anode. Energy Storage Mater. 2022;50:792-801.[DOI]
Copyright
© The Author(s) 2025. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Publisher’s Note
Science Exploration remains a neutral stance on jurisdictional claims in published
maps
and institutional affiliations. The views expressed in this article are solely those
of
the author(s) and do not reflect the opinions of the Editors or the publisher.
Share And Cite



Science Exploration Style
Bai J, Cheng W, Wang T, Liu S. Synergistic regulation of lithium nucleation and anion-rich solvation structure via silver trifluoroacetate additive for stable lithium metal anodes. Smart Mater Devices. 2025;1:202534. https://doi.org/10.70401/smd.2025.0016
Tips
Copy completed.
Submit a Manuscript
Author Instructions
Cite this Article
Article Metrics
0
View
0
Download
Article Updates

Science Exploration Style
Bai J, Cheng W, Wang T, Liu S. Synergistic regulation of lithium nucleation and anion-rich solvation structure via silver trifluoroacetate additive for stable lithium metal anodes. Smart Mater Devices. 2025;1:202534. https://doi.org/10.70401/smd.2025.0016
copy