Electrocatalytic alcohol and aldehyde oxidation: advances in catalysts and reaction mechanisms for sustainable chemical synthesis Download PDF
Lei Chen
1
,
Zhong-Yong Yuan
2,*

*Correspondence to:
Zhong-Yong Yuan, School of Materials Science and Engineering, Nankai University, Tianjin 300050, China.
E-mail: zyyuan@nankai.edu.cn
Smart Mater Devices. 2026;2:202531. 10.70401/smd.2025.0017
Received: August 18, 2025Accepted: September 18, 2025Published: September 28, 2025
Abstract
Electrocatalytic oxidation of alcohols and aldehydes, known as alcohol oxidation reactions (AOR), provides a sustainable and efficient route for converting low-value feedstocks such as ethanol, glycerol, and 5-hydroxymethylfurfural into high-value chemicals, including organic acids and aldehydes, in line with the chemical industry’s transition toward carbon neutrality. This review synthesizes recent advancements in electrocatalytic AOR, emphasizing advances in catalyst design and detailed reaction mechanisms. A broad spectrum of catalysts is explored, ranging from noble metal-based (e.g., Pt, Pd, Au) to cost-effective non-noble metal-based (e.g., Ni, Cu, Co) materials, with attention to advanced strategies such as heteroatom doping, vacancy engineering, and alloying for fine-tuning electronic structures and optimizing intermediate adsorption. The review also delves into mechanistic insights, elucidating rate-determining steps, adsorption geometries, and electron-transfer pathways that govern AOR performance, supported by density functional theory analyses. Special emphasis is placed on the interplay between catalyst electronic structure and reaction kinetics, offering fresh perspectives for improving yield, selectivity, and Faradaic efficiency. Finally, current challenges, including catalyst stability, product selectivity, and scalability, are critically evaluated, and future directions such as in situ characterization and the development of non-noble metal catalysts are proposed to advance AOR toward large-scale, sustainable chemical synthesis.
Keywords
Electrocatalytic oxidation, alcohol oxidation, aldehyde oxidation, hydrogen evolution, catalyst design, sustainable synthesis
1. Introduction
The chemical industry is a cornerstone of modern society, supporting the production of essential goods such as fuels, fertilizers, pharmaceuticals, plastics, and specialty chemicals that underpin sectors ranging from agriculture to healthcare. However, its long-standing reliance on fossil-based processes has caused significant environmental issues, including greenhouse gas emissions, resource depletion, and pollution[1-3]. As global initiatives intensify to achieve carbon neutrality and mitigate climate change, there is an urgent need to transition toward sustainable and eco-friendly chemical manufacturing. Electrocatalytic organic synthesis (EOS) has emerged as a transformative approach, utilizing renewable electricity from wind, solar, and hydropower to drive chemical transformations under mild conditions (Figure 1). This provides a greener alternative to conventional approaches that depend on energy-intensive operations and hazardous reagents[4-6]. Within the EOS framework, electrocatalytic alcohol and aldehyde oxidation reactions (AOR) are particularly attractive because they can convert abundant, low-value feedstocks such as ethanol, glycerol, and biomass-derived 5-hydroxymethylfurfural (HMF) into high-value products including organic acids, aldehydes, and other functionalized molecules[7,8]. In contrast to traditional oxidation methods, which often require stoichiometric quantities of strong oxidants such as chromates or permanganates that generate toxic waste and operate under harsh conditions, AOR proceeds at ambient temperature and pressure while employing electrons as clean redox agents[9,10]. This approach minimizes environmental impact, enhances reaction selectivity, and improves controllability, positioning AOR as a cornerstone for sustainable chemical synthesis.
Figure 1. Sustainable production of fuels and chemicals via electrocatalytic oxidation.
AOR takes place at the electrode-electrolyte interface, where electrocatalysts facilitate electron transfer between reactants and the electrode surface, enabling the formation of reactive intermediates such as radicals, ions, or radical ions[11-13]. These intermediates undergo subsequent transformations to yield desired products with high efficiency and specificity. The performance of AOR is strongly dependent on the electrocatalyst’s electronic structure, which governs the adsorption and desorption of reactants and intermediates[14-16]. Key performance metrics, including yield, selectivity, and Faradaic efficiency (FE), defined as the percentage of electrical current contributing to the desired reaction, are directly influenced by the catalyst’s ability to modulate these interactions[17,18]. For example, strong adsorption of intermediates can lower reaction energy barriers, while optimized desorption prevents catalyst poisoning and ensures efficient product release[19,20]. Recent research in AOR has focused on developing catalysts that enhance these properties, with both noble-metal-based and non-noble metal-based materials showing considered potential. Noble metals are valued for their high catalytic activity and stability but are limited by high costs and scarcity, driving interest in cost-effective alternatives such as transition metal oxides and alloys[21,22]. Strategies including heteroatom doping, vacancy engineering, and alloying have been used to fine-tune catalyst electronic structures, optimizing interactions with key intermediates. These efforts have led to significant improvements in AOR performance, enabling the selective oxidation of alcohols such as glycerol to dihydroxyacetone or glyceraldehyde, and aldehydes such as HMF to 2,5-furandicarboxylic acid (FDCA), a key precursor for bioplastics[23,24]. By aligning AOR with renewable energy sources, these advancements contribute to global sustainability goals by reducing reliance on fossil fuels and minimizing waste. The lower oxidation potential of AOR compared with the oxygen evolution reaction (OER) makes it a promising alternative for H2 production. Wang et al. demonstrated the electrooxidation of HMF and furfural at an ultra-low potential of ~0.1 V vs. the reversible hydrogen electrode (RHE), significantly reducing electrolysis voltage[25]. This process not only lowered energy consumption but also produced H2 as a byproduct at the anode during aldehyde oxidation, unlike conventional aldehyde oxidation reactions that yield water. Notably, their system achieved a FE for H2 production of up to 200%, enabling H2 generation at both the anode and cathode simultaneously, representing a paradigm shift from traditional cathode-only H2 evolution. This approach enhances energy utilization efficiency and reflects the principles of green chemistry. To date, various substrates, including HMF, FF, and formaldehyde, have been investigated for coupling with the hydrogen evolution reaction (HER)[26-29]. As low-potential catalytic oxidation reactions continue to advance, they are expected to play a pivotal role in sustainable H2 production technologies, supporting the development of environmentally friendly energy systems.
Despite these advances, AOR faces several challenges that limit its widespread adoption. Catalyst stability under prolonged operational conditions, particularly for non-noble metal catalysts, remains a major barrier[30,31]. Achieving high selectivity in complex reaction mixtures requires a deeper understanding of intermediate adsorption and reaction pathways. In addition, scalability issues, such as the design of efficient reactors and integration into continuous-flow systems, are critical for translating laboratory successes into industrial applications[32,33]. To address these challenges, future research should focus on developing stable non-noble metal catalysts using advanced strategies such as vacancy engineering and heteroatom doping. In-situ characterization techniques, including Raman and infrared spectroscopy, can provide real-time insights into intermediate adsorption and catalyst dynamics, guiding the rational design of more effective systems[34-36]. Novel reactor designs, such as flow-based systems with high electrode area-to-electrolyte volume ratios, could enhance single-pass conversions and facilitate large-scale implementation[37,38].
This review presents a comprehensive overview of the electro-oxidation of low-carbon alcohols and aldehydes, including ethanol, glycerol, HMF, and furfural, with an emphasis on alkaline electrolytes and H-cell reactor configurations, focusing on catalyst development and reaction mechanisms. We begin by examining the design of advanced electrocatalysts, highlighting the role of electronic structure regulation in enhancing catalytic performance. Key strategies, including size effects, heteroatom doping, vacancy engineering, and alloying, are examined in the context of both noble and non-noble metal catalysts. Next, we explore the mechanistic foundations of AOR, detailing the pathways of alcohol and aldehyde oxidation, the influence of adsorption geometries, and the rate-determining steps that govern reaction kinetics. Insights from density functional theory (DFT) and in-situ characterization techniques are emphasized to clarity the interplay between catalyst properties and reaction outcomes. Emerging reactor technologies, such as flow cells and membrane-electrode assemblies, are also considered to illustrate pathways toward industrial application. The review further addresses critical challenges, including catalyst stability, product selectivity, and scalability, which continue to limit industrial implementation. By integrating findings from recent studies, we aim to provide a holistic understanding of how electronic structure regulation influences AOR performance, offering a roadmap for researchers and industry professionals to advance electrocatalytic AOR as a cornerstone of sustainable chemical synthesis.
2. History and Research Status of AOR
The historical development of electrocatalytic AOR is closely linked to the evolution of organic electrochemistry, which began in the early 19th century with pioneering experiments (Figure 2)[39]. Those early studies on the electrolysis of organic compounds established foundational principles for modern EOS. A pivotal milestone was the discovery of the Kolbe reaction in 1849, which demonstrated the electrochemical coupling of carboxylic acids to form hydrocarbons, representing one of the earliest examples of selective organic transformations via electrochemistry[40]. Despite this breakthrough, progress in organic electrochemistry remained limited until the mid-20th century due to constraints in electrode materials, electrolyte stability, and a lack of mechanistic understanding. Significant advancements during this period were driven by the development of electrode materials, including carbon-based and metal-based electrodes, and improvements in electrolyte formulations. The emergence of quantum mechanical theories further enabled a deeper understanding of electron transfer processes at the electrode-electrolyte interface, accelerating research in organic electrochemistry. A notable industrial success was the electro-hydrodimerization of acrylonitrile by Monsanto in the 1960s, producing adiponitrile, a key precursor for nylon, and demonstrating the scalability of electrochemical processes[41,42]. This achievement laid the groundwork for modern AOR research by highlighting the potential for large-scale, industrially relevant applications. In recent decades, AOR has emerged as a cornerstone of sustainable chemical synthesis, driven by global imperatives to address climate change, resource scarcity, and environmental degradation. The chemical industry, essential for producing fuels, fertilizers, pharmaceuticals, and specialty chemicals, has historically relied on fossil-based processes that contribute to greenhouse gas emissions and pollution[43,44]. Integrating AOR with renewable energy sources offers a pathway to decarbonize chemical manufacturing by harnessing electricity from wind, solar, and hydropower to drive reactions under mild conditions typically at ambient temperature and pressure. This approach significantly reduces energy consumption compared with traditional thermochemical processes, which often require high temperatures and pressures.
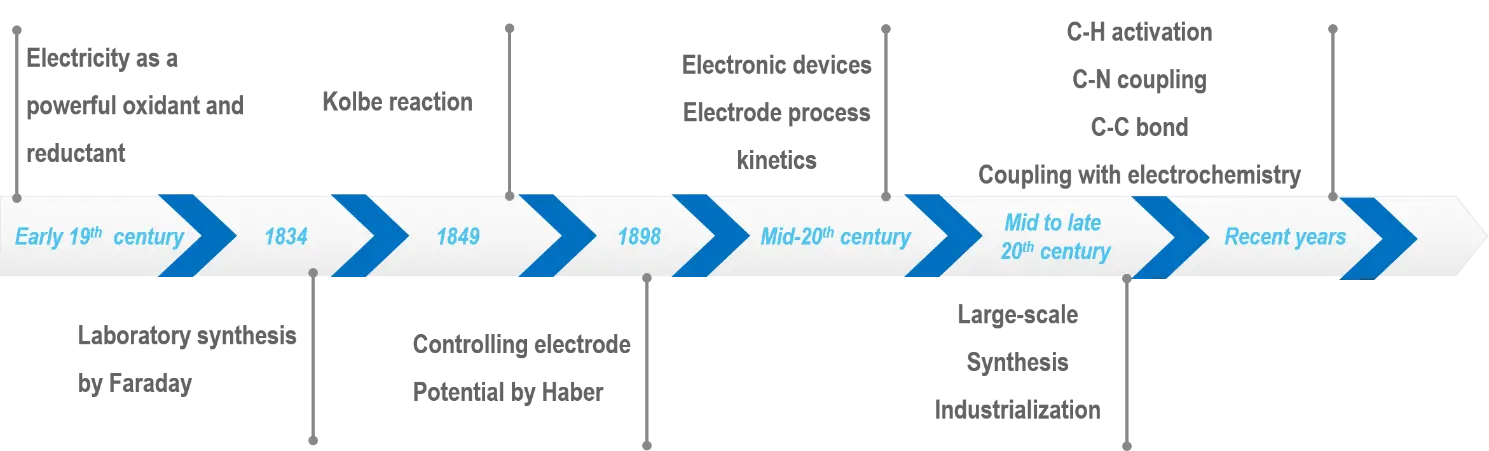
Figure 2. Timeline of key developments in EOS. EOS: electrocatalytic organic synthesis.
The contemporary significance of AORs is highlighted by their ability to convert biomass-derived feedstocks, such as ethanol, glycerol, and HMF, into high-value chemicals, including organic acids, aldehydes, and other functionalized molecules[45-47]. A notable example is the oxidation of HMF to FDCA, a key precursor for bio-based plastics. This process aligns AORs with the principles of a circular economy by utilizing renewable resources derived from lignocellulosic biomass, thereby enhancing sustainability in chemical production. Recent studies, such as those by Liu et al., have demonstrated the exceptional performance of CuxO supported on carbon foam (CuxO/CF) in catalyzing AORs, including the methanol oxidation reaction (MOR), ethylene glycol oxidation reaction, and glycerol oxidation reaction (GOR)[48]. In these reactions, formate emerges as the sole liquid product, achieving FE exceeding 80% across a broad potential range (1.3-1.7 V vs. RHE), with GOR reaching an impressive FE of up to 97%. The superior activity and selectivity of CuxO/CF can be attributed to its distinctive structural features: self-supported nanowire arrays that provide abundant active sites and efficient mass transport channels; a high density of interfaces between Cu2O and CuO, facilitate enhanced electron transfer; and the coexistence of multiple oxidation states (Cu0, Cu+, Cu2+), which optimizes the adsorption and desorption of reaction intermediates, thereby promoting formate as the dominant product with consistently high FEs above 80%.
AORs occur at the electrode-electrolyte interface, where electrocatalysts mediate electron transfer to generate reactive intermediates such as radicals, ions, or radical ions, which subsequently transform into the desired products. The efficiency of AORs depends on the electrocatalyst’s electronic structure, which governs the adsorption and desorption dynamics of reactants and intermediates, directly influencing yield, selectivity, and FE. Recent research has explored both noble-metal-based catalysts and non-noble metal-based alternatives, employing strategies such as heteroatom doping, vacancy engineering, and alloying to enhance catalytic performance. For example, Chen et al. developed an Au-Vo-NiO/CC electrocatalyst, where oxygen vacancies (Vo) in Ni-based nanostructures enhance the catalytic activity of Au, enabling efficient coupling of the formaldehyde oxidation reaction (FOR) with the HER[49]. At an ultralow potential of 0.47 V vs. RHE, this catalyst achieves bipolar hydrogen production with a FE approaching 100%, while simultaneously converting formaldehyde to high-value formate at the anode. Stability tests over 8 hours confirmed the electrode’s robustness, and DFT calculations revealed that the formation of the *CHO intermediate is the rate-determining step. Compared with conventional water electrolysis, this system reduces energy consumption by 40-80%. By replacing the energy-intensive OER with AOR at the anode, this paired electrolysis approach lowers the overall cell voltage, facilitating the simultaneous production of green hydrogen and valuable chemicals. Similarly, Chen et al. synthesized a Ni2P/NF catalyst for benzyl alcohol oxidation, achieving a potential of 1.32 V vs. RHE to drive a current density of 30 mA·cm-2, with benzyl alcohol conversion and FE reaching at least 96.0% and 92.4%, respectively[50]. DFT calculations indicate that the synergistic interaction between Ni and P optimizes the adsorption and desorption energies of intermediates, reducing the energy barrier of the rate-determining step for benzyl alcohol oxidation to 0.42 eV. These advancements highlight the potential of tailored electrocatalysts to enhance AOR efficiency, paving the way for sustainable chemical production and energy-efficient electrochemical systems.
Current research efforts are focused on addressing key challenges, including catalyst stability, scalability, and selectivity in complex reaction mixtures[51-53]. Non-noble metal catalysts, such as Ni- and Cu-based materials, are being developed to overcome the high cost and limited availability of noble metals. In situ characterization techniques, including Raman spectroscopy, X-ray absorption near-edge structure (XANES), and extended X-ray absorption fine structure (EXAFS), provide real-time insights into catalyst dynamics and intermediate adsorption, as demonstrated in studies of Ni-based catalysts for MOR[54]. Additionally, novel reactor designs, such as flow-based systems, are being explored to enhance single-pass conversions and enable industrial-scale implementation[55].
3. Reaction Mechanisms
Understanding the reaction mechanisms of electrocatalytic alcohol and aldehyde oxidation is crucial for designing efficient and selective catalysts[56]. This section explores the mechanisms underlying alcohol and aldehyde oxidation, emphasizing the fundamental chemical processes and reaction pathways involved. Key mechanisms, including radical-based, ionic, and catalytic processes, are discussed in the context of their roles in catalyst design and reaction optimization. By analyzing these mechanisms, this section aims to provide a comprehensive understanding of AOR function and recent research advancements. Specifically, it first explores the core oxidative processes of alcohol and aldehyde oxidation, followed by an examination of key mechanistic factors, such as adsorption behavior and microenvironment regulation, that govern reaction efficiency and inform catalyst design.
3.1 Mechanisms of alcohol oxidation
Alcohol oxidation proceeds via dehydrogenation or direct electron transfer mechanisms, influenced by the substrate, catalyst, and reaction environment (e.g., acidic or alkaline conditions)[57]. For primary alcohols like ethanol, the reaction involves sequential proton and electron removal, yielding aldehydes or carboxylic acids. Meng et al. employed 18O-labeled catalysts to elucidate the pivotal role of lattice oxygen in MOR, identifying the O 2p band center energy level as a robust descriptor for predicting formate yield and FE[58]. Experiments with seven catalysts (LaCoO3, SrCoO3-σ, FeOxHy, Fe2NiOxHy, FeNiOxHy, FeNi2OxHy, NiOxHy) confirmed a positive correlation between the O 2p band center energy level and both formate yield and FE. NiOxHy, with the highest O 2p band center energy level, achieved near 100% formate FE in membrane-electrode assembly (MEA) tests and maintained stability across a wide range of current densities. Gas chromatography-mass spectrometry analysis of 18O-labeled catalysts detected HC16O18OH (m/z = 48) in formate produced by SrCoO3-σ and NiOxHy, indicating lattice oxygen participation, whereas no such signal was observed for LaCoO3 and FeOxHy. The underlying mechanism suggests that when the O 2p band center is close to the Fermi level, lattice oxygen acts as a nucleophilic species, facilitating thermodynamically favorable reactions. Consequently, two reaction mechanisms were proposed: the surface adsorption mechanism and the lattice oxygen-mediated mechanism (LOM), which collectively explain the correlation between the O 2p band center and catalytic performance. Li et al. successfully constructed a Pd-Zn dual-site catalyst (PdZn/NC@ZnO) with uniformly exposed active sites on an N-doped carbon-coated ZnO (NC@ZnO) support, using Pd-Pd sites (Pdn/NC@ZnO) and single Pd sites (Pd1/NC@ZnO) as controls[59]. Aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (AC HAADF-STEM) revealed an ordered arrangement of Pd (bright) and Zn (dark) atoms in PdZn nanoparticles, with lattice spacings of 2.1 Å and 2.9 Å corresponding to the PdZn (200) and (110) planes, respectively (Figure 3a). High-resolution transmission electron microscopy further confirmed the PdZn (111) plane, verifying its intermetallic compound nature. In PdZn/NC@ZnO, PdZn nanoparticles were uniformly dispersed on the NC@ZnO surface, with an average particle size of 4.75 ± 0.77 nm (Figure 3b). In contrast, Pd nanoparticles in Pdn/NC@ZnO were smaller (2.75 ± 0.66 nm), while Pd1/NC@ZnO showed only isolated bright spots corresponding to single Pd atoms (AC HAADF-STEM, Figure 3c). The PdZn/NC@ZnO catalyst exhibited outstanding performance in the ethanol oxidation reaction (EOR), achieving a mass activity of 18.14 A·mgPd-1, 24 times higher than commercial Pd/C (0.76 A·mgPd-1), and a specific activity of 54.60 mA·cm-2, 37 times that of Pd/C. It also demonstrated excellent stability in a 24-hour chronoamperometry test. In situ characterization and DFT calculations revealed that the Pd-Zn dual sites facilitate the adsorption of ethanol and OH-, altering the initial dehydrogenation pathway in EOR from C-H bond cleavage at Pd-Pd sites to O-H bond cleavage (Figure 3d,e,f,g). This significantly lowers the reaction energy barrier (initial dehydrogenation free energy of -37.88 kcal·mol-1 compared to -35.38 kcal·mol-1 for Pd-Pd sites). Moreover, the reaction produces only acetate as the product, with no CO₂ formation.
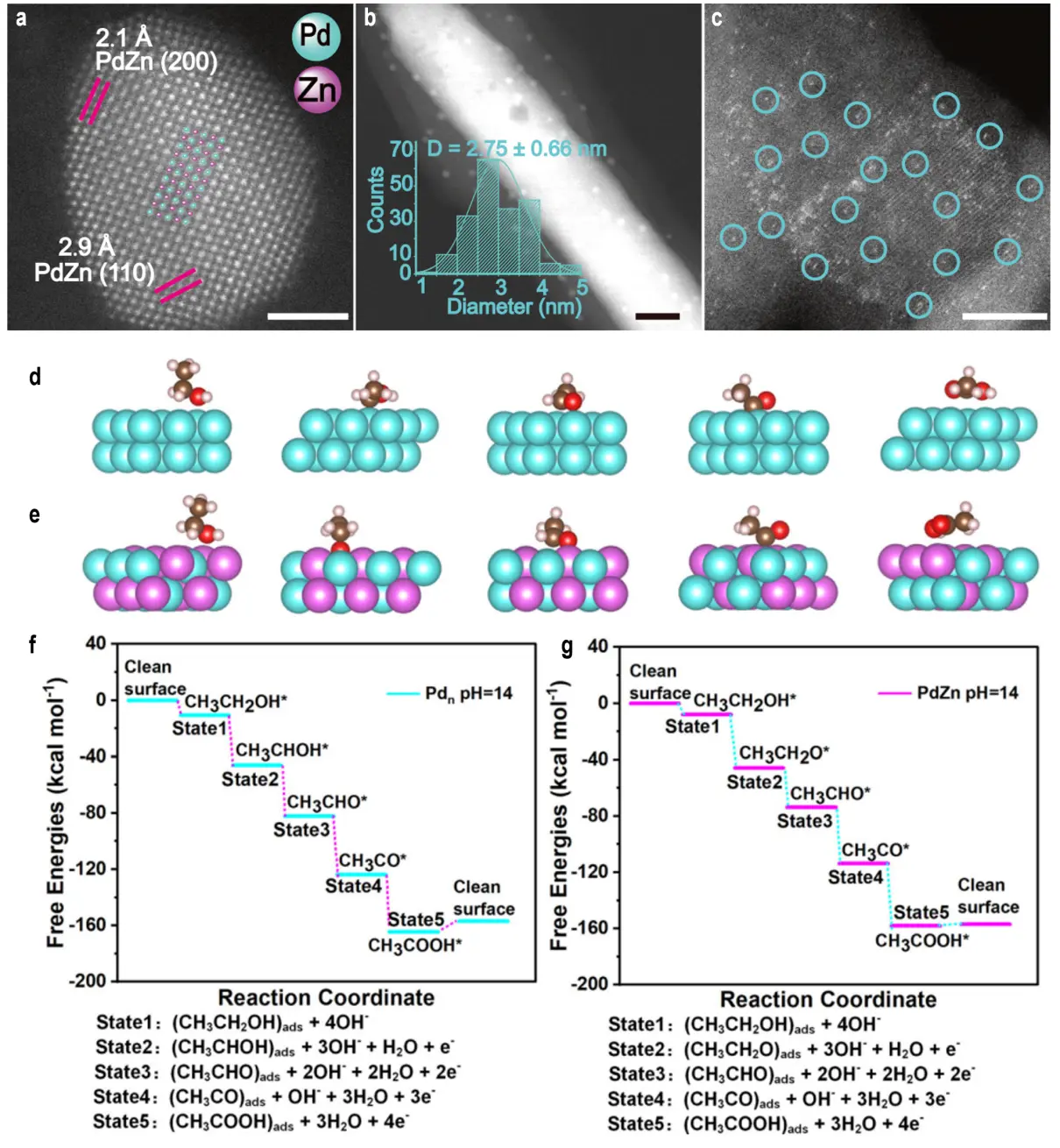
Figure 3. (a) AC HAADF-STEM elemental mappings of one PdZn nanoparticle in PdZn/NC@ZnO. Scale bar, 2 nm; (b) STEM image of Pdn/NC@ZnO, the inset is the size distribution histogram of Pd nanoparticles in Pdn/NC@ZnO. Scale bar, 20 nm; (c) AC HAADF-STEM image of Pd1/NC@ZnO. Scale bar, 5 nm; (d) DFT calculated models of Pd-Pd sites adsorbed with the reactive species from different EOR reaction states; (e) DFT calculated models of Pd-Zn dual sites adsorbed with the reactive species from different EOR reaction states; (f) DFT calculated free energy profiles of EOR over Pd-Pd sites (pH = 14, U = 0.82 V with respect to the RHE); (g) DFT calculated free energy profiles of EOR over Pd-Zn dual sites (pH = 14, U = 0.82 V with respect to the RHE)[59]. AC HAADF-STEM: aberration-corrected high-angle annular dark-field scanning transmission electron microscopy; DFT: density functional theory; EOR: ethanol oxidation reaction; RHE: reversible hydrogen electrode.
Recently, Wei et al. developed a nickel foam-supported NiCo2O4 (NiCo2O4/NF) catalyst via a facile hydrothermal/annealing process for anodic GOR[60]. This catalyst exhibited exceptional performance, requiring only 1.23 V vs. RHE to achieve a current density of 10 mA·cm-2 and delivering 152 mA·cm-2 at 1.6 V vs. RHE, with a formate FE exceeding 97%. The synergy between the NiCo2O4 nanostructure and the nickel foam substrate enabled a two-electrode electrolyzer (NiCo2O4/NF||Ni foam) to achieve 10 mA·cm-2 at a cell voltage of 1.35 V, which is 320 mV lower than conventional water electrolysis systems. Nuclear magnetic resonance analysis using 13C-labeled glycerol confirmed that NiCo2O4/NF cleaves all C-C bonds in glycerol, producing formate close to theoretical yields, with minor deviations attributed to glycolate formation. Characteristic peaks of reaction intermediates (glyceraldehyde, glyceric acid, glycolic acid) and products (formate, oxalate) were identified, supporting a proposed GOR pathway: glycerol → glyceraldehyde → glyceric acid → glycolic acid → formate. During GOR, the NiOOH active species on the NiCo2O4/NF surface dynamically regenerates after reacting with glycerol, sustaining catalytic activity. Under alkaline conditions, Feng et al. investigated methanol electrooxidation to formate on Ni-based layered double hydroxides (NiMn-LDH and NiFe-LDH)[61]. After normalization by electrochemical active surface area (ECSA), the MOR specific activity of NiMn-LDH (250.1 mA·cmECSA-2 at 1.45 V) was 3.3 times higher than that of NiFe-LDH (75.8 mA·cmECSA-2), while its OER specific activity is only 1/16 that of NiFe-LDH, indicating fundamental mechanistic differences between MOR and OER (Figure 4a,b). A two-electrode system was constructed with Pt/C as the cathode (for hydrogen evolution) and NiMn-LDH as the anode (for MOR), achieving cell voltages of 1.33 V and 1.43 V at current densities of 10 mA·cm-2 and 100 mA·cm-2, respectively, which are significantly lower than those of the commercial Pt/C//RuO2 system (1.40 V and 1.49 V) (Figure 4c). Figure 4d illustrates the overall MOR reaction framework, highlighting the reversible redox cycle of Ni2+(OH)2/Ni3+-OOH coupled with the catalytic MOR process. Blue and yellow arrows indicate proton transfer during precatalytic oxidation and NiM(-H) reduction in MOR, respectively. Figure 4e displays the Gibbs free energy for the precatalytic step (Ni2+ → Ni3+-OOH), showing that NiMn-LDH possesses a significantly lower ΔG (1.38 eV) than NiFe-LDH (2.04 eV), thereby facilitating the formation of active Ni3+-OOH species in NiMn-LDH. Figure 4f presents the free energy profile of the catalytic MOR pathway, where NiMn-LDH exhibits a modest energy barrier of 0.40 eV (non-spontaneous) at the *OCH2 dehydrogenation step, whereas all steps in NiFe-LDH are exothermic (spontaneous). Figure 4g,h show the optimized structures of MOR intermediates on NiMn-LDH and NiFe-LDH, respectively, visually demonstrating the synergistic interaction between Ni3+ and electrophilic oxygen species. Figure 4i proposes a bifunctional mechanism, in which Ni3+-OOH provides two types of active sites: Ni3+ for methanol adsorption and electrophilic oxygen atoms for hydrogen abstraction. These results directly explain the differences in catalytic activity: NiMn-LDH achieves superior MOR performance (100 mA·cm-2 at 1.41 V) compared with NiFe-LDH (100 mA·cm-2 at 1.45 V), owing to its lower energy barrier in the precatalytic step, more favorable formation of Ni3+-OOH, and higher synergistic efficiency of bifunctional sites. This also clarifies the intrinsic reasons behind NiMn-LDH’s non-spontaneous yet highly active behavior versus NiFe-LDH’s spontaneous but less active performance, namely that heteroatom doping modulates Ni2+/Ni3+ redox dynamics and intermediate adsorption energies, which ultimately dictate overall catalytic performance.
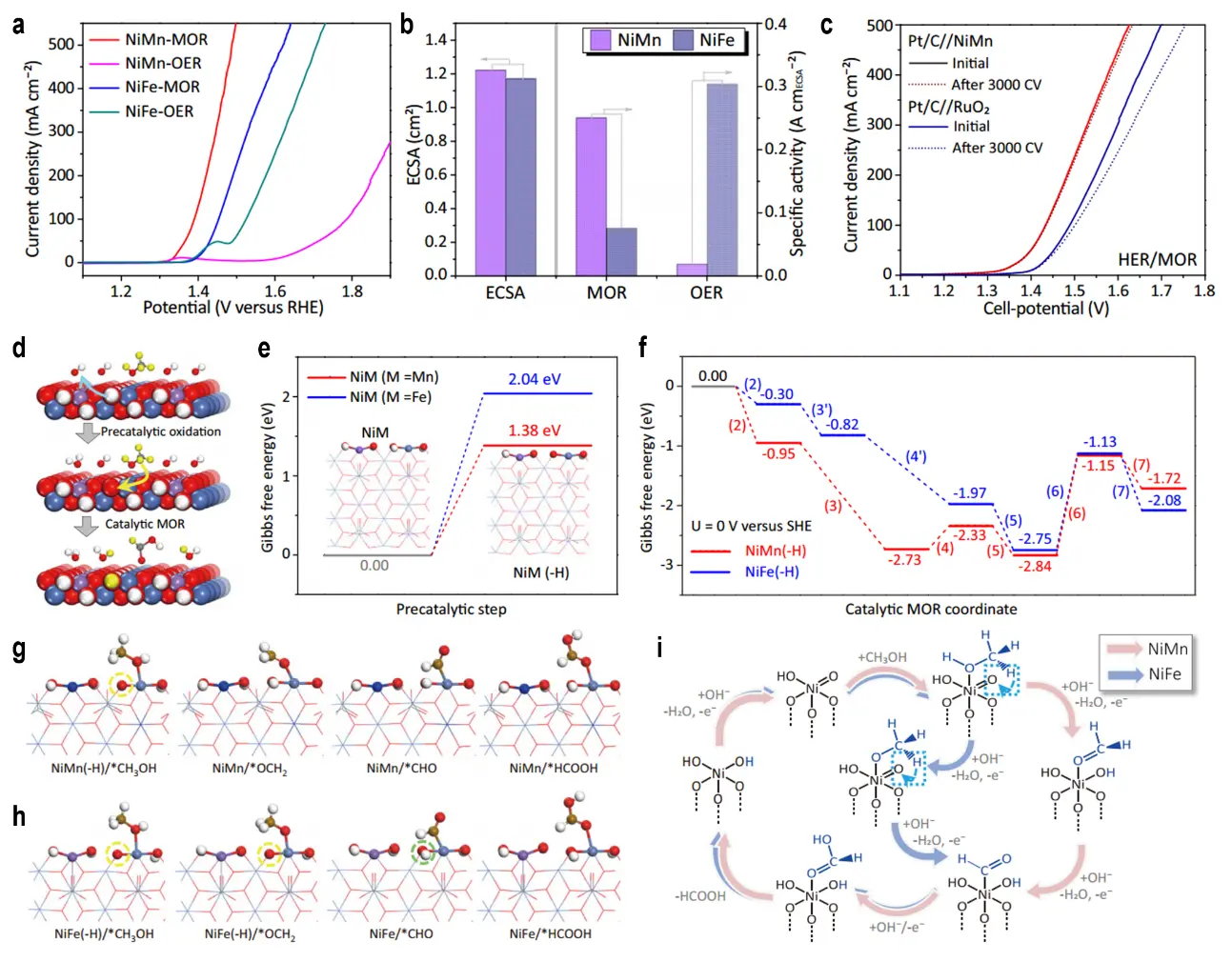
Figure 4. (a) IR-corrected LSV curves of NiMn and NiFe-LDHs both recorded in 1 M KOH without and with 3 M CH3OH; (b) ECSA normalized specific activities of NiMn and NiFe-LDHs for MOR (at 1.45 V RHE) and OER (at 1.65 V RHE); (c) Cell LSV curves (no iR correction) of HER/MOR electrolysis using an electrode pair of Pt/C//NiMn before and after 3,000 CV sweeps in a single chamber electrolyzer with 1 M KOH and 3 M CH3OH, using Pt/C//RuO2 as reference; (d) Reaction scheme for the overall MOR on NiM-LDHs (M = Mn, Fe). The blue and yellow arrows indicate the proton-transfers during the precatalytic oxidation process and that associated with the reduction of NiM(-H) to NiM during the catalytic MOR process. The hydrogens of methanol or derived from methanol are highlighted in yellow; Gibbs free energy diagrams for the (e) precatalytic process and (f) catalytic MOR process; Optimized structures of the intermediates in MOR process on (g) NiMn and (h) NiFe-LDHs, where, Ni: wathet, Fe: purple, Mn: blue, N: gray, O: red, H: white, C: brown. The yellow and green circles indicate the hydrogen-deficient and hydrogen-added oxygens in NiM(-H), respectively; (i) An unconventional bifunctional mechanism proposed for the overall MOR process[61]. IR:internal resistance; LSV: linear sweep voltammetry; ECSA: electrochemical active surface area; LDHs: layered double hydroxides; MOR: methanol oxidation reaction; RHE: reversible hydrogen electrode; HER: hydrogen evolution reaction.
3.2 Mechanisms of aldehyde oxidation
Aldehyde oxidation, such as the transformation of HMF to FDCA, is pivotal for valorizing biomass-derived feedstocks into high-value chemicals. These reactions typically proceed via sequential electron and proton transfers through multiple intermediates, with the reaction pathway and efficiency governed by the electrocatalyst’s electronic structure, surface properties, and interaction with reactants. Recent studies have elucidated detailed mechanisms and catalyst designs that enhance selectivity, FE, and energy efficiency in AORs, particularly under low-potential or reductive conditions. Wang et al. investigated the HMF oxidation reaction (HMFOR) using a Co4N@CeO2 heterostructure, where CeO2 functions as an electron pump to optimize the interfacial electronic structure[62]. The reaction mechanism involves the initial adsorption of HMF onto the catalyst surface, followed by its oxidation to FDCA via *OH-mediated pathways. At the Co4N@CeO2 heterointerface, electron transfer from Co4N to CeO2 increases the density of electronic states near the Fermi level, thereby improving charge transfer kinetics. DFT calculations reveal that CeO2 strengthens OH- adsorption, generating reactive *OH species that attack the aldehyde group of HMF, facilitating sequential C-H and O-H bond cleavage to form FDCA. The moderate HMF adsorption energy, being neither too strong nor too weak, ensures efficient substrate binding and product desorption, minimizing over-oxidation. This mechanism underscores the critical role of interfacial electronic modulation in promoting nucleophilic attack by *OH, which is essential for achieving high FDCA selectivity.
For low-potential aldehyde oxidation reactions (LP-AOR), Fu et al. developed a Cu nanosheet array on carbon foam (CF@Cu-NS) to catalyze FOR to formate, coupled with simultaneous H2 production at -0.1 to 0.3 V vs. RHE[63]. The reaction proceeds via a one-electron oxidation pathway, initiated by formaldehyde hydration to methanediol (CH2(OH)2) in aqueous media. DFT calculations, using Cu(111) as a model indicate that methanediol deprotonates to form the methanediol anion (CH2(OH)O-), which undergoes C-H bond cleavage to yield HCOO- and an adsorbed *H. The low energy barrier for *H recombination to H2 (0.66 eV), compared to its reaction with OH- to form H2O (0.96 eV), favors selective H2 production. Further oxidation of formate to CO2 is hindered by a high energy barrier (0.66 eV), ensuring negligible CO2 formation and high formate selectivity. This mechanism highlights the role of Cu surface properties in stabilizing MDA intermediates and promoting H2 evolution over competing pathways, achieving near-100% FE for both formate and H2. In furfural electrocatalytic reduction, Wan et al. engineered a Cu/Ce catalyst to shift the reaction mechanism from PCET to electrochemical HAT, enabling efficient conversion of furfural to furfuryl alcohol[64]. The mechanistic shift is driven by oxygen-vacancy-rich CeO2, which weakens hydrogen adsorption on Cu, facilitating a hydrogen formation-diffusion-hydrogenation chain. At the Cu/ceria interface, furfural molecules are locally enriched, lowering the energy barrier for the rate-determining HAT step. The reaction pathway involves initial furfural adsorption, followed by hydrogen atom addition to the carbonyl group, forming FA. DFT calculations indicate that oxygen vacancies in CeO2 stabilize partially reduced intermediates, while the Cu/CeO2 interface enhances local furfural concentration, promoting HAT over PCET. This results in a FE of 97 ± 1% and a production rate of 19.1 ± 0.4 mol·h-1·m-2 at current densities above 0.1 A·cm-2. The synergy between Cu and ceria highlights the importance of interfacial engineering in controlling reaction pathways and optimizing product selectivity. These mechanistic insights demonstrate how tailored catalyst designs, through electronic structure modulation, vacancy engineering, and interfacial synergy, govern the reaction pathways in aldehyde oxidation and reduction. By elucidating key intermediates and rate-determining steps, these studies provide a foundation for developing efficient, selective, and sustainable electrocatalytic systems for biomass valorization.
3.3 Adsorption mechanisms in AOR
Building on the oxidative pathways discussed, the following subsection examines adsorption behavior, which plays a pivotal role in facilitating these reactions on catalyst surfaces. The adsorption geometry of reactants and intermediates is a key determinant of AOR performance, as it directly influences reaction kinetics, selectivity, and FE. The orientation of functional groups, such as the C=O bond in HMF or furfural, on catalyst surfaces significantly affects the energy barriers for critical steps, including dehydrogenation. For instance, vertical adsorption of the C=O group on catalysts, such as MoO2-FeP@C, aligns the reactive site with the catalyst’s active centers, lowering the energy barrier for bond activation and enhancing reaction efficiency[65]. This precise alignment facilitates optimal interaction between the substrate’s reactive bonds and the catalyst’s active sites, promoting efficient electron and proton transfer. Designing efficient hybrid electrolysis systems requires a thorough understanding of how adsorption behavior governs catalytic performance. The adsorption mode of substrate molecules is a prerequisite for effective catalysis, with the distance between active sites and reactive bonds playing a pivotal role in bond activation. Modulating surface atom arrangements and oxidation states is a common strategy to tune adsorption behaviors. However, complex organic molecules with substituent groups often adopt inert adsorption configurations, which hinder catalytic activity by misaligning reactive bonds or sterically blocking active sites. Consequently, achieving precise control over adsorption modes to optimize electrocatalytic performance remains a significant challenge. Zhu et al. addressed this challenge by developing a ligand-confined pyrolysis strategy to synthesize a ruthenium single-atom catalyst anchored on N,S-co-doped carbon (Ru-SA/NSC), which enhances electrocatalytic oxidation by tailoring the adsorption modes of alcohol substrates[66]. Their study revealed that benzyl alcohol (BA) adsorbs horizontally via its oxygen atom onto Ru sites, enabling efficient oxidation to benzaldehyde at a low potential of 0.97 V vs. RHE, achieving a current density of 10 mA·cm-2. This configuration yielded a product formation rate of 96%, selectivity of nearly 99%, and an FE of approximately 100%. In contrast, pyridine methanol (PM), which adsorbs vertically through its nitrogen atom, exhibited no reaction activity due to unfavorable alignment with the Ru active site. The horizontal adsorption of BA minimizes the distance between the reactive C-O bond and the Ru center, facilitating dehydrogenation and electron transfer, whereas the vertical adsorption of PM misaligns the reactive group, suppressing catalysis. By engineering catalysts to favor specific adsorption modes, researchers can lower energy barriers, enhance selectivity, and maximize FE. The challenge of mitigating inert adsorption configurations, particularly for complex molecules, necessitates advanced strategies to precisely control surface interactions, paving the way for more efficient and selective electrocatalytic systems for biomass valorization.
3.4 Microenvironment regulation in AOR
Recent advances in electrocatalysis have focused on engineering the electrode-electrolyte interface and catalyst microarchitecture to create tailored microenvironments that enhance reactant adsorption, reaction kinetics, and product selectivity. By modulating local conditions, such as electrolyte composition, salting-out effects, or nanostructured catalyst designs, researchers have overcome these barriers, achieving high FE, selectivity, and scalability. This section explores key strategies for microenvironment regulation, highlighting their mechanistic underpinnings and potential for industrial applications in biomass valorization. The electrocatalytic oxidation of long-chain fatty alcohols, such as octanol, is often hindered by poor water solubility, resulting in low reactant concentrations at the electrode surface, limited mass transfer, and reduced reaction rates. To address this, Cheng et al. developed an electrolyte engineering strategy by incorporating cetyltrimethylammonium hydroxide (CTAOH) to modulate the hydrophobic microenvironment at the electrode-electrolyte interface, significantly enhancing the electrocatalytic oxidation performance of Au-catalysts[67]. Electrocatalytic oxidation of fatty alcohols, driven by renewable electricity, can be performed under mild aqueous conditions and coupled with the HER at the cathode to enhance hydrogen production efficiency, making it an ideal green process. However, long-chain fatty alcohols, owing to their strong hydrophobicity and low water solubility, result in insufficient reactant concentration at the electrode surface and limited mass transfer, severely restricting reaction rates (Figure 5a). CTAOH, an amphiphilic surfactant containing a hydrophobic long alkyl chain and a hydrophilic quaternary ammonium group, can preferentially adsorb on the electrode surface to create a hydrophobic microenvironment. This arrangement repels interfacial water molecules and enriches fatty alcohols at the interface (Figure 5b). Using nickel foam (NF) as the substrate and electrodeposited gold (ED-Au/NF) as the active component, the addition of CTAOH to 1 M KOH significantly enhanced the electro-oxidation activity of octanol on ED-Au/NF, as shown in Figure 5c. Molecular dynamics simulations were employed to model the Au(111) electrode in a 1 M KOH system, elucidating CTAOH’s regulatory role at the interface. In the pure KOH system, the electrode surface exhibited the highest water density (Figure 5d,e). Upon adding CTAOH, the long alkyl chains of CTA+ repelled water molecules, reducing the interfacial water density by 40% (Figure 5f) and establishing a hydrophobic microenvironment. In pure KOH, octanol aggregated in the bulk solution due to its hydrophobicity, with minimal adsorption on the electrode. In contrast, in the CTAOH, octanol was driven toward the electrode by hydrophobic interactions, increasing the local interfacial concentration by 2.5 times (Figure 5g,h) and significantly improving mass transfer efficiency. Therefore, CTAOH enhances mass transfer by repelling interfacial water molecules and enriching fatty alcohols at the electrode surface. This strategy is also applicable to other fatty alcohols, such as heptanol, nonanol, and decanol, providing a general solution for the aqueous electrocatalytic conversion of water-insoluble organic compounds.
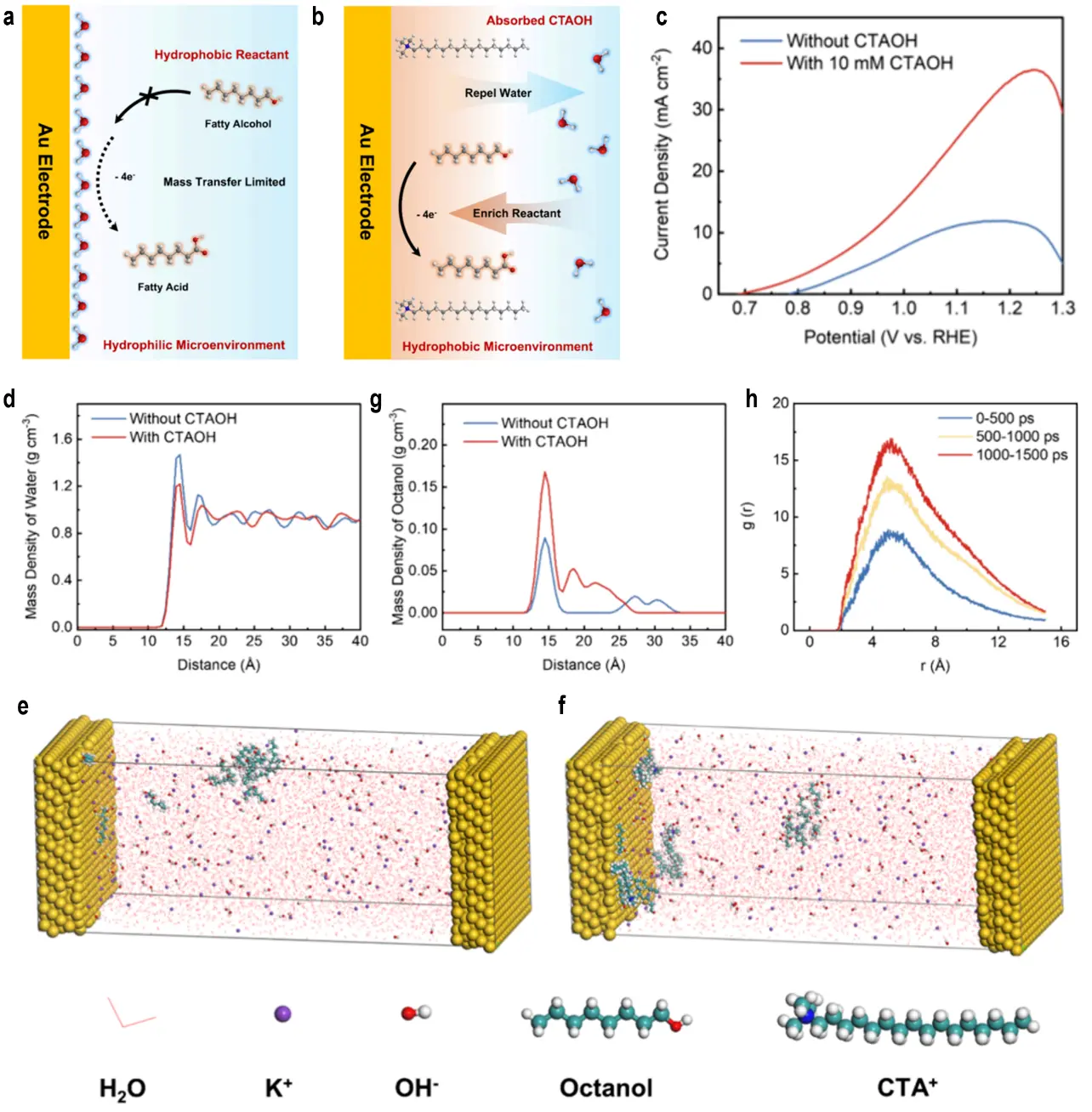
Figure 5. Schematic illustration of (a) the challenges associated with the electrocatalytic oxidation of fatty alcohols and (b) the electrolyte engineering strategy proposed in this study to address these challenges; (c) LSV curves for the electrocatalytic oxidation of octanol in 1 M KOH with and without 10 mM CTAOH; Snapshots of the final state for the (d) pure KOH system and the (e) CTAOH-containing system; (f) Density profiles of water calculated from MD simulations in the pure KOH system and CTAOH-containing system; (g) Density profiles of octanol calculated from MD simulations in pure KOH system and CTAOH-containing system; (h) Radial distribution functions of CTAOH-octanol obtained from MD simulations[67]. LSV: linear sweep voltammetry; CTAOH: cetyltrimethylammonium hydroxide; MD: molecular dynamics.
In acidic or neutral conditions, AOR kinetics are slow and typically require noble metal catalysts, whereas in alkaline conditions, high oxidation efficiency can be achieved; however, aldehydes are prone to over-oxidation to acids due to their high reactivity, resulting in low selectivity. Wang et al. proposed a microenvironment regulation strategy to induce a salting-out effect, enabling selective alcohol oxidation to aldehydes with 100% selectivity on NiO catalysts[68]. By reducing electrolyte alkalinity and increasing cation and substrate concentrations, this approach suppresses aldehyde hydration, promoting salting-out at the gas-liquid interface to prevent over-oxidation to acids. The mechanism relies on disrupting aldehyde-water hydrogen bonds, favoring aldehyde precipitation and stabilizing the aldehyde product. This strategy is versatile, extending to reactions such as HMF oxidation to 2,5-furandicarboxaldehyde and amine oxidation to imines, demonstrating broad applicability and industrial potential.
Selective coupling of different alcohols remains challenging due to the difficulty in designing catalysts and electrolytes that promote specific reaction pathways. Wang et al. developed a paired electrolysis strategy for anodic oxidative coupling of ethanol and benzyl alcohol to produce cinnamaldehyde (CAL), coupled with cathodic nitrate reduction (NO3-RR) for ammonia production, replacing the HER to prevent product hydrogenation[69]. A Pt-loaded Ni(OH)2 catalyst (Pt-Ni(OH)2) accelerates intermediate coupling kinetics, while a salting-out effect, induced by high K+ concentrations, balances the selective oxidation and coupling rates of ethanol and benzyl alcohol. The mechanism involves K+ ions disrupting aldehyde-water hydrogen bonds, promoting aldehyde-aldehyde interactions and enriching acetaldehyde around benzaldehyde to accelerate coupling. K+, with its moderate ionic radius and hydration energy, outperforms Na+ and Li+, which form thicker hydration shells that weaken aldehyde interactions. This system achieved 85% coupling selectivity and an ammonia production rate of 278 μmol·h-1 at 100 mA·cm-2, with a low cell voltage of 1.63 V, offering a membrane-free, energy-efficient, and scalable approach with broad substrate applicability. Catalyst microarchitecture can also be exploited to engineer localized microenvironments that enhance AOR performance. Molecular electrocatalysts such as 2,2,6,6-tetramethylpiperidinyl-N-oxyl (TEMPO) and 4-acetamido-TEMPO (ACT) are effective mediators for alcohol oxidation, but their turnover often relies on diffusion-limited contact with the electrode, necessitating high concentrations of costly aminoxyl radicals. To address this, Wang et al. developed a carbon-based nanovolcano (CNV) structure grown on 3D graphite felt, where the cavity functions as a nanoreactor, creating a localized microenvironment enriched with intermediates[70]. The CNV prolongs the residence time of reactants, while surface oxygen-containing functional groups form hydrogen bonds with ACT, anchoring it to the catalyst surface. This design achieved a high turnover frequency of 31,818 h-1 with only 0.5 mol% ACT, efficiently catalyzing alcohol oxidation under mild conditions. The system successfully oxidized benzyl and heterocyclic substrates and enabled scale-up for the oxidation of a chiral primary alcohol precursor to produce levetiracetam, a pharmaceutical intermediate, demonstrating its considerable industrial potential.
In summary, the reaction mechanisms of AOR involve complex electron and proton transfer processes that are finely modulated by catalyst composition, electronic structure, and adsorption behavior. Advances in catalyst design, including heteroatom doping, vacancy engineering, and nanostructuring, have enabled precise control over reaction pathways, thereby improving both selectivity and efficiency. The integration of AOR with HER presents a promising strategy for sustainable chemical synthesis, with ongoing research aimed at elucidating intermediate dynamics and optimizing catalyst performance for potential industrial applications.
4. Catalyst Design for AOR
Electrocatalysts are pivotal to the performance of AOR, as they govern reaction kinetics, selectivity, and FE. Both noble metal-based and non-noble metal-based catalysts have been extensively studied, with strategies such as alloying, heteroatom doping, vacancy engineering, and nanostructuring employed to optimize their electronic structures and surface properties[71]. This section provides an overview of key catalyst types and the strategies used to fine-tune their electronic structures for enhanced AOR performance.
4.1 Noble metal-based catalysts
Noble metal catalysts, including Pt- and Au-based materials and their alloys, play a central role in AORs owing to their high electrocatalytic activity and tunable electronic properties. These catalysts facilitate the selective transformation of biomass-derived alcohols into valuable chemicals, such as aldehydes, acids, and coupled products, often paired with hydrogen evolution to improve energy efficiency. Nevertheless, challenges such as catalyst poisoning, over-oxidation, and limited substrate scope necessitate innovative designs, including alloying, surface engineering, and tandem reaction strategies. This section reviews recent advances in Pt- and Au-based catalysts, as well as alloy-based systems, highlighting mechanistic insights and their contributions to sustainable chemical production.
4.1.1 Pt-based catalysts
Pt catalysts exhibit high activity for AORs but are prone to CO poisoning during methanol oxidation, which diminishes their performance. To overcome this limitation, Chen et al. synthesized two-dimensional ultrathin PtTe alloy metallenes (PtTe A-ML) via liquid-phase chemical reduction, exploiting high atomic utilization and alloy effects to enhance formic acid oxidation reaction (FAOR) performance[72]. The PtTe A-ML catalyst proceeds exclusively through the direct oxidation pathway for FAOR, effectively suppressing the formation of toxic CO intermediates. In acidic media, its FAOR activity is 43 times higher than that of commercial Pt nanocrystals and 5.6 times higher than Pd nanocrystals. This enhanced activity is attributed to the oxophilicity of Te atoms and electron transfer from Pt to Te, which optimizes the electronic structure of Pt sites and reduces CO adsorption strength. Furthermore, PtTe A-ML shows superior CO oxidation activity, improving FAOR durability by mitigating poisoning effects. This mechanism underscores the synergy between alloying and electronic modulation in achieving high selectivity and stability. Gao et al. empolyed in situ Raman spectroscopy, online HPLC, and DFT calculations to elucidate the relationship between various Pt surface oxides (PtOHads, PtOads, α-PtO2) and electrocatalytic activity[73]. Only PtOHads and α-PtO2 were identified as catalytically active centers, whereas PtOads exhibited no measurable activity. In the potential windows of 0.6-0.95 V (RHE, associated with PtOHads) and > 1.15 V (RHE, associated with α-PtO2), two distinct current peaks, M and N, were observed, with α-PtO2 displaying superior activity. These surface species govern product selectivity by facilitating the dehydrogenation of the gem-diol intermediate (CH3CH(OH)2): PtOHads preferentially yields acetaldehyde, whereas α-PtO2 favors acetate formation. DFT calculations (Figure 6a,b,c,d) further revealed that the activation barrier for ethanol dehydrogenation to acetaldehyde is 0.37 eV on α-PtO2 compared with 0.42 eV on PtOHads, while the barrier for gem-diol dehydrogenation to acetate, it is 0.19 eV on α-PtO2 versus 0.39 eV on PtOHads. The substantially lower activation energies accelerate reaction kinetics, which is reflected in the higher current intensity (peak N > peak M). This mechanistic relationship was confirmed on Pt(111), Pt(110), and Pt(100) single-crystal electrodes, providing a universal benchmark for activity modulation in Pt-based catalysts and related oxidation processes.
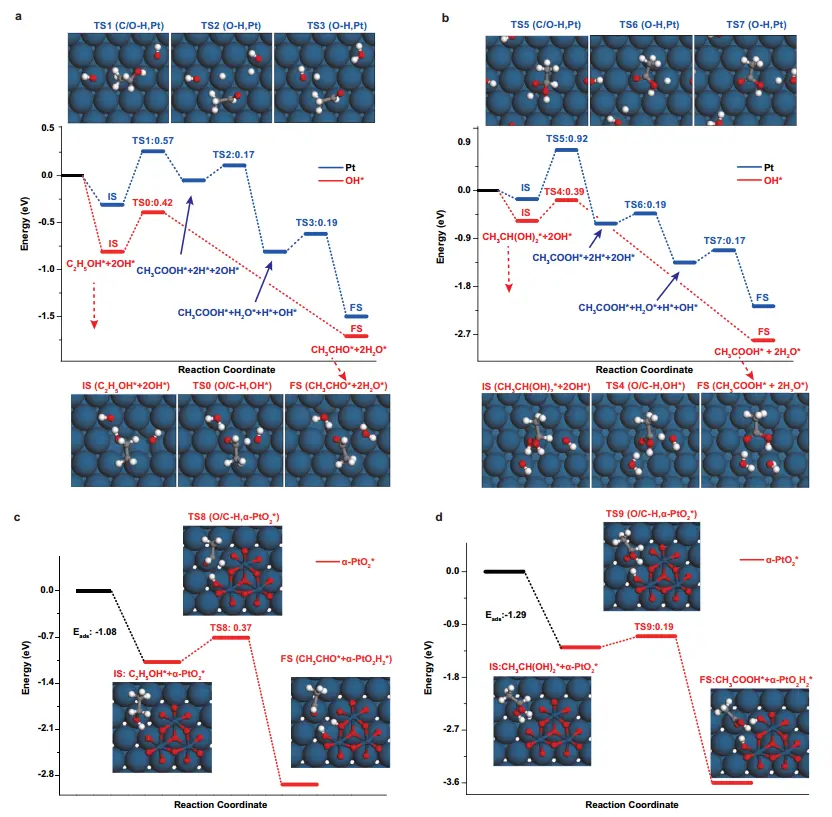
Figure 6. Energy profiles of the ethanol oxidation. (a,b) The comparisons of energy profiles for the formation of the acetaldehyde and acetic acid on Pt with (red path) and without (blue path) co-adsorbed OH; (c,d) Energy profiles for the formation of acetaldehyde and acetic acid on Pt with α-PtO2*, respectively. Surface structures of transition states (TS1-9) can be found in the above plots. Blue, gray, red, and white balls stand for Pt, C, O, and H atoms, respectively[73].
4.1.2 Au-based catalysts
Au catalysts supported on carbon-based materials, such as Au/CGOOH, exhibit excellent performance in the selective oxidation of HMF to FDCA, a key precursor for bio-based polymers. Duan et al. developed a tandem electrocatalytic strategy employing Au/CuO catalysts to selectively oxidize benzyl alcohol to benzaldehyde, followed by spontaneous aldol condensation with ketones (e.g., acetone or cyclohexanone) to yield α,β-unsaturated ketones in a single step[74]. The Au/CuO interface facilitates OH* generation and benzyl alcohol adsorption, thereby promoting selective oxidation. In a flow electrolyzer, this strategy achieved a benzalacetone production rate of 9.5 mmol·h-1, with a co-production of 0.4 L·h-1 of H2, delivering an atom economy of 89% and an E-factor of 0.71, consistent with green chemistry principles. The high efficiency was attributed to the synergistic interaction at the Au/CuO interface, which stabilizes reactive intermediates and facilitates condensation. Shi et al. reported an electrocatalytic cross-coupling strategy using Au catalysts to realize carbon-chain elongation by coupling ethylene glycol (EG) with C1-C4 primary alcohols, thereby producing C3-C6 α-hydroxy carboxylic acids (α-HCAs)[75]. For instance, coupling EG with methanol to form lactic acid (LA) under optimized conditions (low-roughness Au foil, 4 M KOH, 100 mA·cm-2, 60 °C) stabilized aldehyde intermediates (glycolaldehyde and formaldehyde) and suppressed overoxidation, affording 268.1 μmol cm-2·h-1 of LA with a 28.7% FE. This approach is versatile and applicable to a broad range of primary alcohols, and notable enabled the conversion of waste PET-derived EG into LA (224.5 μmol·cm-2). The underlying mechanism involves controlled aldehyde generation and cross-coupling, with high alkalinity and temperature enhancing intermediate stability. Building on this strategy, Duan et al. developed an Au/Ni(OH)2 catalyst for the efficient conversion of biodiesel-derived glycerol to lactic acid (77% selectivity, 317.7 mA·cm-2 at 0.95 V vs. RHE) and PET-derived ethylene glycol to glycolic acid (91% selectivity, 326.2 mA·cm-2 at 1.15 V vs. RHE)[76]. Utilizing a membrane-free flow electrolyzer, this strategy produced 11.2 g of lactic acid (accompanied by 9.3 L H2) from triglycerides and 13.7 g of glycolic acid (with 9.4 L H2) from PET waste, offering a sustainable pathway for upgrading waste into biodegradable polymer monomers while co-generating hydrogen. The energy consumption for H2 production(3.1-3.2 kWh·m-3) is lower than that of advanced water electrolysis systems (~4.2 kWh·m-3), owing to the Au/Ni(OH)2 interfacial effect, which enhances selective oxidation while suppressing side reactions.
4.1.3 Alloy-based catalysts
Alloying noble metals with other elements modulates the d-band center, thereby optimizing the adsorption energies of reactants and intermediates to enhance AOR performance. For instance, PtRh nanosponges improve AOR activity by tailoring the electronic structure[77]. Sun et al. synthesized a Cu3Ag7 catalyst supported on carbon foam (Cu3Ag7/CF) for FOR, achieving current densities of 100 mA·cm-2 and 500 mA·cm-2 at low potentials of 0.13 V and 0.36 V vs. RHE, respectively, in a 10.0 g·L-1 paraformaldehyde solution (Figure 7a,b)[78]. After five cycles at these current densities, no significant voltage decay was observed, confirming excellent stability (Figure 7c). DFT calculations were conducted to elucidate the underlying catalytic mechanism. Figure 7d illustrates the proposed oxidation pathway of HCHO under alkaline conditions, showing that HCHO first hydrates to form the H2C(OH)O- intermediate, which then adsorbs on the catalyst surface and undergoes dehydrogenation to yield HCOOH*, eventually converting to formate with concomitant H2 release. Figure 7e compares the adsorption energies of H2C(OH) on Cu, Ag, and Cu3Ag7, revealing that Cu3Ag7 exhibits the lowest adsorption energy (-0.7 eV), markedly outperforming pure Cu (-0.61 eV) and pure Ag (-0.42 eV) in stabilizing the intermediate. Figure 7f presents the optimized adsorption configuration of H2C(OH)O on the Cu3Ag7 surface, where the intermediate is anchored via its O atom at a Cu2Ag1 vacancy, with the H2COH group adopting a staggered conformation. This unique adsorption mode originates from the synergistic interaction between Cu 3d and Ag 4d orbitals. Figure 7g depicts the initial state (IS), transition state (TS), and final state (FS) of the rate-determining dehydrogenation step (C-H bond cleavage) of H2C(OH)O, clearly demonstrating that the TS on the Cu3Ag7 surface is more stable and associated with a lower energy barrier. Figure 7h further compares the energy profiles for H2 formation via the Tafel step on the three catalysts, confirming that Cu3Ag7 exhibits a remarkably low barrier of 0.66 eV, thereby providing a significant thermodynamic advantage and theoretical basis for efficient anodic hydrogen evolution. Wang et al. subsequently designed a bifunctional CuxAgy/CF catalyst for the co-electrolysis of furfural and formaldehyde, enabling the simultaneous production of FA, formate (FM), and H2[79]. The Cu7Ag3/CF catalyst achieved a 96.2% FE for the electrochemical hydrogenation of furfural to FA at the cathode, while Cu3Ag7/CF delivered a 100% FE for FOR to FM at the anode, with both electrodes concurrently producing H2. At a cell voltage of 0.50 V, the electrolyzer attained a current density of 500 mA cm-2. DFT calculations combined with in situ spectroscopy confirmed that the catalyst optimizes intermediate adsorption and reduces the energy barrier of the rate-determining step, thereby boosting overall efficiency. The Cu-Ag synergistic effect enables selective hydrogenation and oxidation, rendering this system highly effective for biomass valorization.
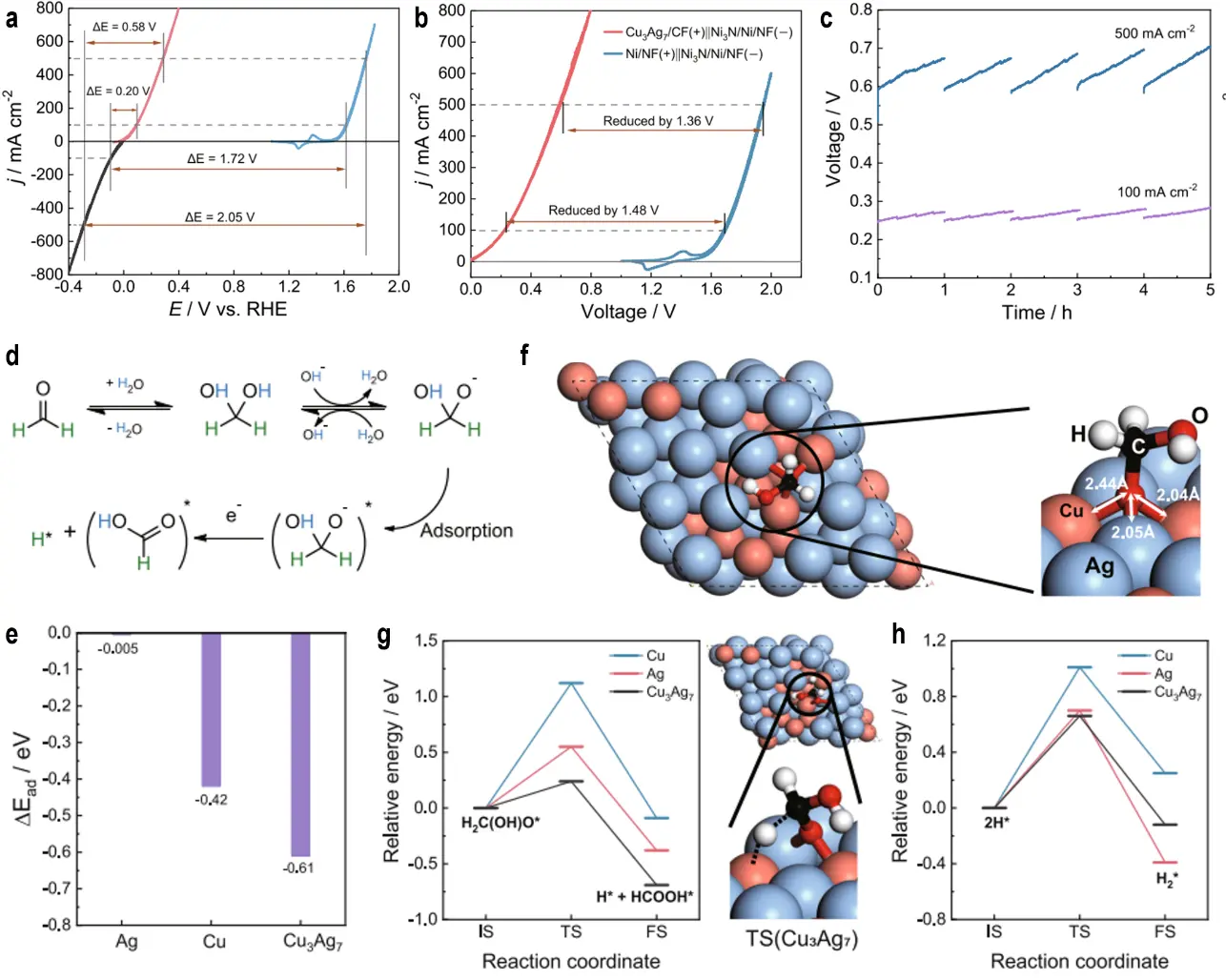
Figure 7. (a) CV curves of Cu3Ag7/CF for HCHO oxidation (red) in 1.0 M KOH with 0.6 M HCHO, Ni3N/Ni/NF for HER (black), and Ni/NF for OER (blue) in 1.0 M KOH. Voltage gaps to reach 100 and 500 mA·cm-2 are indicated; (b) The two-electrode CV curves of HER/FOR (red) and HER/OER (blue) collected at 10 mV·s-1, in which Cu3Ag7/CF and Ni3N/Ni/NF were employed as the anode and cathode for the former while Ni/NF and Ni3N/Ni/NF for the latter; (c) Chronopotentiometric curves forfive consecutive controlled-current electrolysis cycles; (d) Proposed mechanism of HCHO oxidation to HCOOH; (e) Computed adsorption energy of the H2C(OH)O intermediate on the three model surfaces; (f) Optimized adsorption geometry of H2C(OH)O on Cu3Ag7; (g) Initial (IS), transition (TS), and final (FS) states of H2C(OH)O dehydrogenation on the three model surfaces, together with the TS structure on Cu3Ag7; (h) H2 formation via the Tafel step on the three surfaces[78]. CV: cyclic voltammetry; CF: carbon felt; HER: hydrogen evolution reaction; OER: oxygen evolution reaction; NF: nickel foam; FOR: formaldehyde oxidation reaction; IS: initial state; TS: transition state; FS: final state.
4.2 Non-noble metal-based catalysts
Non-noble metal catalysts, particularly those based on Ni, Cu, and Co, have emerged as cost-effective and earth-abundant alternatives to noble metal catalysts for AORs. Their competitive performance in biomass valorization and energy-efficient electrocatalytic systems renders them highly attractive for sustainable chemical production. Although challenges such as lower intrinsic activity and susceptibility to deactivation remain, recent advances in structural engineering, electronic modulation, and heteroatom doping have markedly improved their catalytic activity, selectivity, and stability. This section examines mechanistic insights and innovative design strategies for Ni-, Cu-, and Co-based catalysts, emphasizing their pivotal roles in advancing AORs for applications ranging from aldehyde oxidation to coupled hydrogen production.
4.2.1 Ni-based catalysts
Ni-based catalysts are highly effective for AORs owing to their tunable electronic structures and high density of active sites. For instance, NiOOH-CuO supported on Cu foam (NiOOH-CuO) demonstrates outstanding performance in the EOR, delivering a current density of 200 mA cm-2 at a low potential of 1.347 V vs. RHE, surpassing all previously reported 3d transition metal-based EOR catalysts[80]. DFT calculations reveal that the synergy between CuO and NiOOH originates from interfacial charge redistribution, where electron transfer from Cu to Ni optimizes the adsorption energies of key intermediates and reduces the energy barrier of the rate-determining step (CH3CHO* → CH3CO*). This accelerates EOR kinetics and enhances selectivity toward acetaldehyde. Hausmann et al. developed a liquid-phase plasma modification strategy to construct hierarchical nanoflower architectures on nickel foam (NF-Plasma), markedly boosting its electrocatalytic activity[81]. Plasma treatment increased the density of redox-active Ni sites by 39-fold and enhanced the double-layer capacitance by sixfold, thereby improving charge transfer and expanding the reservoir of accessible active sites. In the selective oxidation of benzyl alcohol and HMF, NF-Plasma achieved current densities of up to 800 mA·cm-2 with FE exceeding 95%. The improved performance is attributed to the hierarchical morphology, which mitigates diffusion limitations, as evidenced by increased current densities under stirring and at elevated substrate concentrations. Moreover, the catalytic activity exhibits a strong correlation with double-layer capacitance, underscoring the critical role of the expanded electrode-electrolyte interface area. While Ni-based catalysts are effective for AORs, they typically favor acid formation over C-C bond coupling. To overcome this limitation, Wang et al. synthesized a Pt-Ni(OH)2 catalyst, in which Pt accelerates ethanol oxidation kinetics, promoting acetaldehyde (CH3CHO) generation and its subsequent coupling with benzaldehyde (PhCHO)[69]. Characterization confirmed a uniform Pt distribution and strong electronic interactions with Ni(OH)2. Furthermore, in situ Raman spectroscopy and electrochemical impedance spectroscopy (EIS) revealed accelerated electron transfer and facilitated formation of NiOOH active species, thereby enhancing coupling efficiency.
Glycerol, a major byproduct of biodiesel production, can be valorized through oxidation to generate a variety of high-value chemicals, most of which are more valuable than glycerol itself. These products range from C1 to C3 molecules, including formic acid (FA), glycolic acid, glyceric acid, and the target product 1,3-dihydroxyacetone (DHA) (Figure 8a). Among these, DHA is particularly attractive due to its high economic value; however, its selective formation requires the oxidation of glycerol’s secondary alcohol group, a process hindered by significant steric effects. Electrochemical glycerol oxidation typically employs noble metal catalysts (e.g., Pt, Pd), with only a limited number of modified catalysts (e.g., Bi/Pt) capable of producing DHA. For non-noble metal catalysts, Ni-based systems (with NiOOH as the active phase) have traditionally been applied only under strongly alkaline conditions (pH ≥ 13), where they predominantly yield FA through extensive C-C bond cleavage, thereby preventing the formation of C3/C2 products (Figure 8b). Alcohol oxidation on NiOOH proceeds via two distinct dehydrogenation pathways that govern selectivity: hydrogen atom transfer (HAT, an indirect mechanism) and hydride transfer (PD, a potential-dependent mechanism). Goetz et al.[82] investigated the selective oxidation of bio-based polyols, using glycerol as a model, and clarified the contributions of these two mechanisms. By carefully tuning the electrolyte pH (9-13) and applied potential (1.48-1.62 V vs. RHE), they achieved selective oxidation of the secondary alcohol group in glycerol to produce DHA[82]. As shown in Figure 8c,d,e, at low pH (9-10) and moderate potentials, the HAT pathway dominates (accounting for up to 75% at pH 9), suppressing the PD mechanism and minimizing C-C bond cleavage, thereby yielding DHA with a relative selectivity above 70% (with a FE up to 71%). In contrast, at high pH (13) or under strongly positive potentials, the PD pathway and extensive C-C bond cleavage prevail, producing mainly FA. This study establishes a direct correlation between reaction mechanism, operating conditions, and product selectivity, providing a generalizable strategy for the targeted conversion of bio-based polyols.
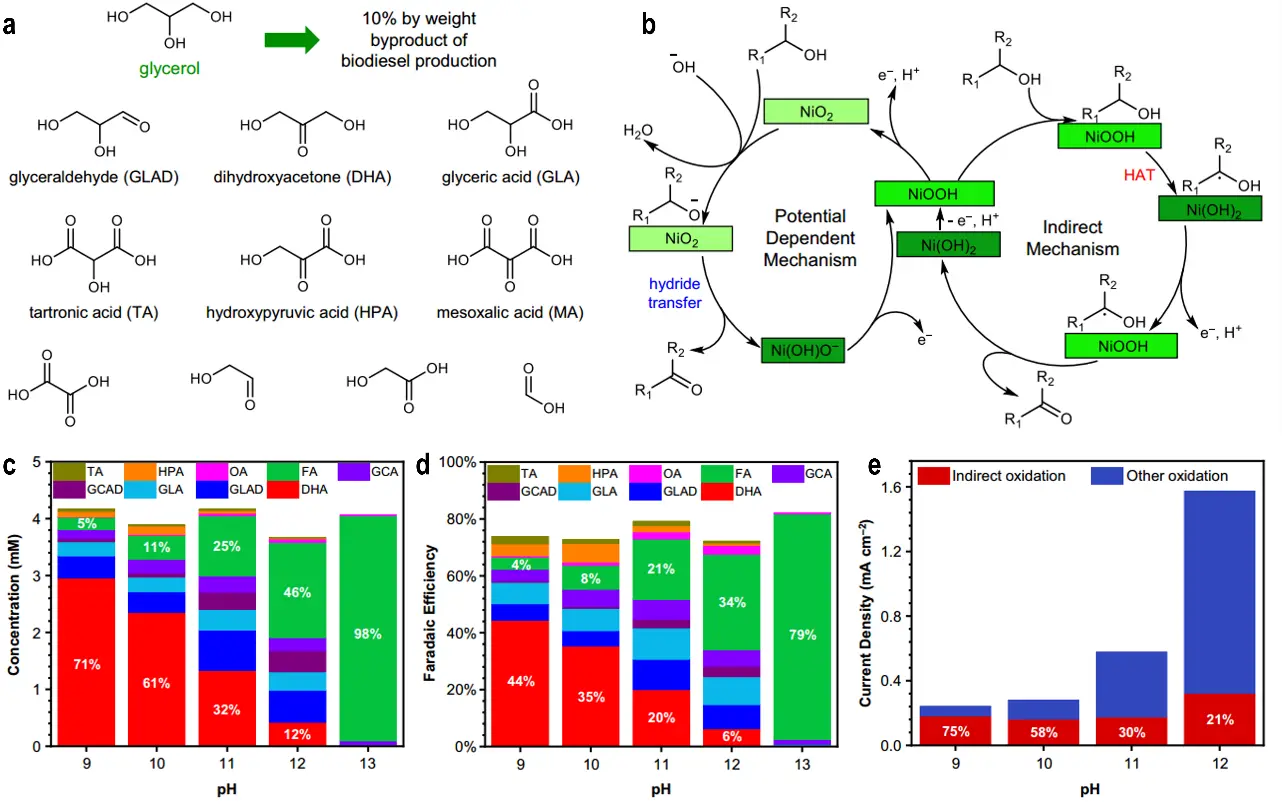
Figure 8. (a) Selected products from glycerol oxidation, C1-C3 molecules that can be obtained from oxidative valorization of glycerol; (b) Simplified representations of two possible alcohol dehydrogenation mechanisms on NiOOH electrodes; (c,e) Glycerol oxidation results after electrolysis at a constant potential of 1.52 VRHE under various pH conditions[82].
4.2.2 Cu-based catalysts
Cu-based catalysts show great promise for LP-AORs, which enable simultaneous aldehyde oxidation and hydrogen production near 0 V vs. RHE. Wang et al. designed RuCe-co-doped Cu2O nanotubes supported on Cu foam (RuCe-Cu2O/CF) as a dual hydrogen production catalyst, replacing the OER with FOR[83]. At a current density of 100 mA·cm-2, this catalyst delivered a low HER overpotential of 172 mV and initiated FOR at -0.018 V vs. RHE, thereby achieving efficient H2 generation. In the coupled HER-FOR system, an industrial-grade current density of 800 mA·cm-2 was obtained at a cell voltage of 0.55 V, with stable H2 production sustained at both electrodes. DFT calculations further revealed that Ru and Ce doping lowers the C-H bond cleavage barrier in FOR while optimizing H* adsorption energy (ΔGh ≈ -0.1 eV) for HER, thus enhancing overall efficiency. Despite these advantages, Cu-based LP-AOR catalysts often suffer from deactivation due to the oxidation of Cu(0) to Cu(I) (Cu(OH)ads), coupled with slow non-electrochemical reduction rates. Zou et al. investigated the deactivation-reactivation cycle of Cu catalysts and developed a self-reactivating PdCu system with markedly improved stability[84]. For Cu NP, the Cu valence state increased from 0.2 to 1.0 after 10 minutes of reaction, with EXAFS analysis showing the disappearance of Cu-Cu bonds and the emergence of stronger Cu-O bonds, resulting in complete deactivation. By contrast, PdCu NP maintained s stable Cu valence of 0.3 over 60 minutes, with no notable changes in Cu-Cu/Pd or Cu-O bond signals, thereby preserving activity (Figure 9a,b,c,d). DFT calculations were further employed to elucidate the molecular-level origin of PdCu’s superior performance over pure Cu. As shown in Figure 9e, for the non-electrochemical reduction of Cu(I) by formaldehyde, the reaction energy barrier on pure Cu is 0.75 eV, whereas Pd incorporation lowers this barrier significantly to 0.45 eV. This demonstrates that Pd accelerates the reduction of Cu(I) to Cu(0), providing an energetic basis for catalyst self-regeneration and sustained performance. Figure 9f compares the free-energy changes associated with H-H2 conversion on the surfaces of pure Cu and PdCu catalysts. For pure Cu, this process requires overcoming a non-spontaneous energy difference of 0.15 eV, whereas for PdCu, the free-energy difference for H recombination into H2 is -0.08 eV (spontaneous). This result indicates that Pd not only facilitates catalyst reactivation but also establishes thermodynamically favorable conditions for anodic H2 production, thereby supporting a bipolar hydrogen production mechanism. Figure 9g presents the complete free-energy profile for formaldehyde oxidation to formic acid, identifying formate desorption as the rate-determining step for both catalysts. The free-energy barrier for this step is 0.77 eV on pure Cu but decreases significantly to 0.49 eV on PdCu. Furthermore, PdCu exhibits lower adsorption free energies for reaction intermediates (e.g., *H2C(OH)O, *HCOO), confirming that Pd incorporation optimizes the kinetics of the overall reaction pathway and enhances LP-AOR. activity. Benefiting from these improvements, the bipolar hydrogen production device achieved a current density of 400 mA·cm-2 at 0.42 V and operated continuously for 120 hours, demonstrating robust stability and efficiency.
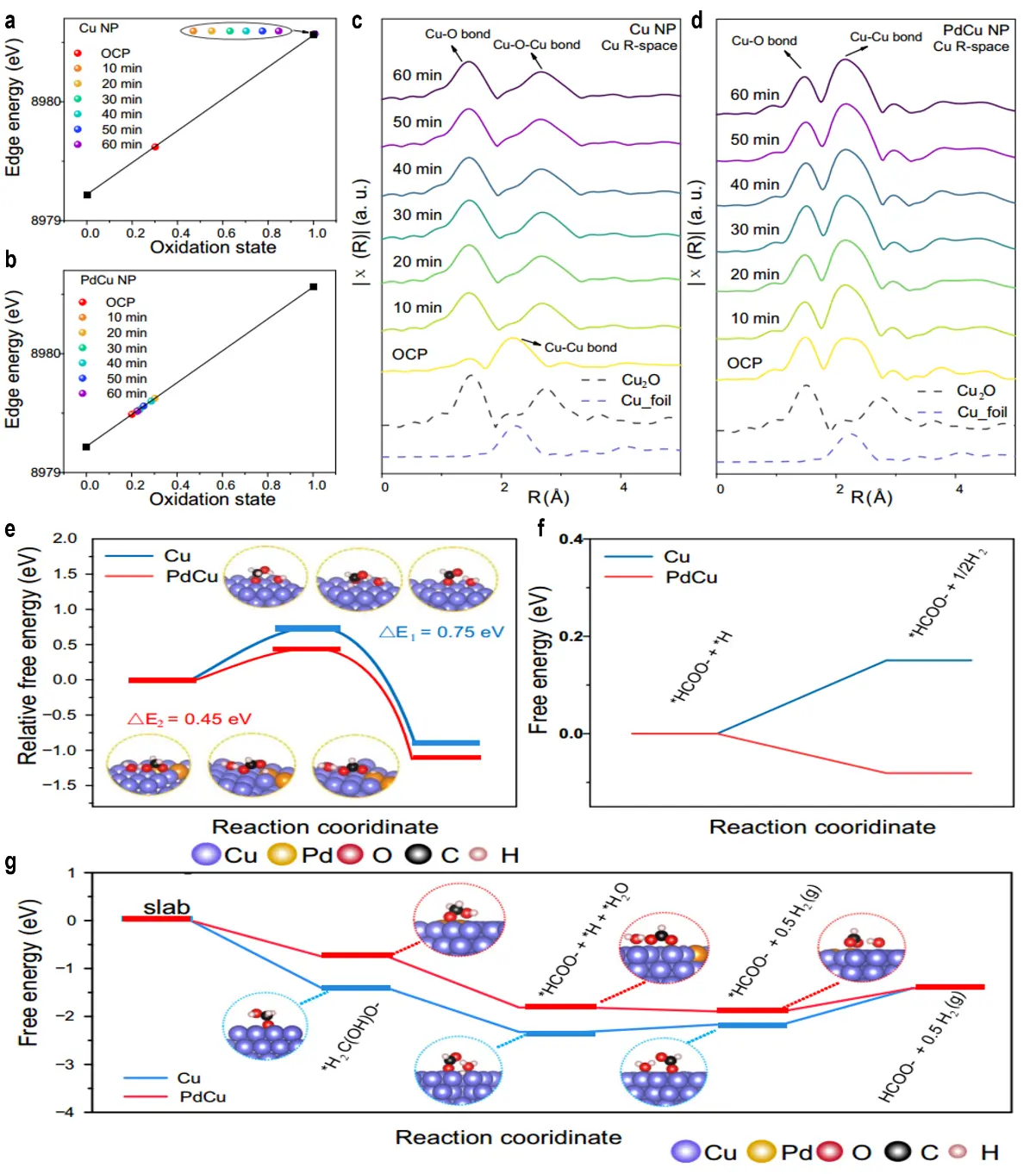
Figure 9. The valence fitting of Cu based on Cu K-edge XANES in (a) Cu NP and (b) PdCu NP; Corresponding FT EXAFS spectra of (c) Cu NP and (d) PdCu NP; (e) Energy barrier of the transition state of the non-electrochemical reaction on Cu and PdCu; (f) The Gibbs free energy difference map of hydrogen released on Cu and PdCu; (g) The Gibbs free energy difference map of LP-AOR process on Cu and PdCu[84]. NP: nanoparticle; XANES: X-ray absorption near-edge structure; FT EXAFS: Fourier transform extended X-ray absorption fine structure; LP-AOR: low-potential alcohol oxidation reaction.
The production of enols has traditionally relied on thermocatalytic semi-hydrogenation of alkynols, but this approach suffers from several inherent limitations. In contrast, electrocatalytic processes, powered by renewable electricity, operate under mild conditions (ambient temperature and pressure) and employ water as a hydrogen source, thereby circumventing the drawbacks of thermocatalysis (Figure 10a,b). Despite these advantages, progress in the electrocatalytic semi-hydrogenation of alkynols has been hindered by the lack of efficient catalysts. Zhang et al. developed an electrocatalytic strategy employing Cu nanoarrays (Cu NAs) as the cathode catalyst, enabling highly selective conversion of alkynols to enols under ambient conditions with water as the hydrogen source[85]. Using 2-methyl-3-butyn-2-ol (MBY) as a representative substrate, Cu NAs in alkaline solution at -0.88 V vs. RHE achieved a partial current density of 750 mA·cm-2 with 97% selectivity. In a 25 cm2 dual-electrode flow electrolyzer (Figure 10c), single-pass MBY conversion reached 93%, with a continuous 2-methyl-3-buten-2-ol (MBE) production rate of 169 g·gCu-1·h-1. As MBY concentration increased from 0.1 M to 1.5 M, the current density at -0.7 V vs. RHE decreased from 0.88 A·cm-2 to 0.18 A·cm-2, while the FE for MBE at 1.3 A·cm-2 increased from 20% to 64%, accompanied by significant HER suppression. DFT calculations (Figure 10d) revealed that MBY adsorption on the Cu(111) surface exhibits a free energy of -3.20 eV, markedly lower than that of water (-0.33 eV), indicating preferential MBY adsorption that suppresses water binding. In situ attenuated total reflection Fourier-transform infrared (ATR-FTIR) spectroscopy (Figure 10e) further confirmed this behaviour: the C≡C stretching peak of MBY shifted from 2,118 cm-1 to 1,672 cm-1 (indicative of σ-π adsorption), while the C=C peak of MBE appeared at 1,594 cm-1 (lower than the 1,649 cm-1 of free MBE), signifying weak adsorption that facilitates desorption. These theoretical and in situ spectroscopic analyses demonstrate that the strong, exothermic adsorption of alkynols coupled with the facile desorption of enols on the Cu surface enhances semi-hydrogenation performance. This strategy is applicable to a wide range of alkynol substrates, and techno-economic analysis estimates a production cost of < 1,500 USD per ton, significantly lower than that of conventional thermocatalytic processes, underscoring its potential as a viable and scalable alternative.
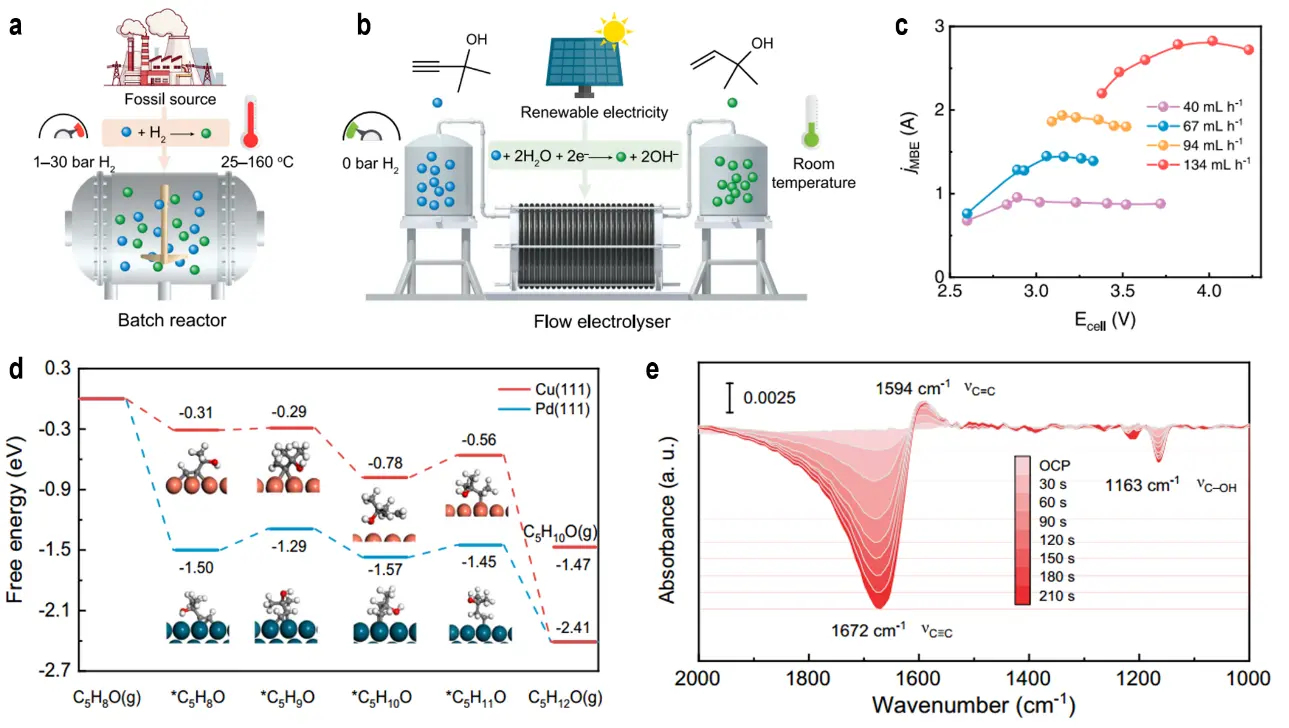
Figure 10. Schematic illustrations of (a) the industrial thermocatalytic process and (b)the proposed electrocatalytic process; (c) Partial current density of MBE at various cell voltages and electrolyte flow rates; (d) Free energy diagrams of the MBY hydrogenation process on Cu(111) and Pd(111) surfaces; (e) In situ electrochemical ATR-FTIR spectra obtained during the electrocatalytic semi-hydrogenation of MBY on the Cu NAs[85]. MBE: 2-methyl-3-buten-2-ol; MBY: 2-methyl-3-butyn-2-ol; ATR-FTIR: attenuated total reflection Fourier-transform infrared.
4.2.3 Co-based catalysts
Co-based catalysts exhibit remarkable versatility in AORs through structural and electronic engineering. Guan et al. synthesized N-doped CoFeP nanorods on nickel foam (N-CoFeP/NF) via a combination of hydrothermal synthesis, nitridation, and phosphorization, yielding a highly crystalline nanorod morphology[86]. This catalyst required only 1.38-1.42 V vs. RHE to drive AORs, thereby reducing energy consumption. For alcohols containing secondary hydroxyl groups in the carbon chain (e.g., glycerol, isopropanol), C-C bond cleavage occurred, producing formic acid (74% FE) and acetic acid, whereas primary alcohols without bond cleavage exhibited lower activity due to less favorable reaction kinetics. Luo et al. prepared flower-like NiCo2S4 nanosheet arrays on carbon cloth (CC@NiCo2S4) via electrodeposition, serving as a bifunctional catalyst for water/methanol co-electrolysis[87]. The array structure and tuned surface magnetism preferentially exposed the (01-1) facet, enhancing both catalytic activity and stability at high current densities. DFT and X-ray photoelectron spectroscopy (XPS) revealed that Ni and Co atoms on the (01-1) surface undergo a transition from low-spin to high-spin states upon OH adsorption, improving surface stability and boosting methanol oxidation and HER activity. Peng et al. developed a Re-modulated Co(OH)2 catalyst on carbon cloth (Re-Co(OH)2/CC) for the furfural oxidation reaction[88]. Rhenium doping reduced Co binding energy, increased the Co3+/Co2+ ratio, and introduced oxygen vacancies (Ov), as confirmed by XPS and electron paramagnetic resonance (EPR). The presence of Ov facilitated OH- adsorption, promoting the dehydrogenation of Co(OH)2 to the active Co(OH)O species and lowering the FOR dehydrogenation barrier from 0.07 eV to 0.02 eV. Additionally, Ov enhanced furfural chemisorption (open-circuit potential reduced by 0.24 V, C=O bond redshifted), thereby accelerating nucleophilic dehydrogenation and reducing the FOR overpotential to 171 mV at 10 mA·cm-2.
4.3 Electronic structure regulation strategies
The performance of AORs is fundamentally governed by the electronic structure of electrocatalysts, which dictates reaction kinetics, product selectivity, and FE. To precisely tailor these electronic properties, a variety of strategies have been developed, including heteroatom doping, vacancy engineering, alloying, surface functionalization, support engineering, and oxidation-state regulation. These approaches optimize the adsorption of reactants and intermediates, reduce reaction energy barriers, and improve catalyst stability, thereby enabling efficient and selective AORs for both biomass valorization and coupled hydrogen production. This section highlights these strategies, with a particular focus on their mechanistic roles and contributions to sustainable electrocatalytic systems.
4.3.1 Heteroatom doping
Heteroatom doping with metals (e.g., Co, Fe) or non-metals (e.g., N, P, S) can modulate the local charge distribution and shift the d-band center of catalysts, thereby enhancing the adsorption of reactants and intermediates. Tao et al. developed CoMoP nanosheets by introducing Mo into CoP, leveraging d-electron complementarity to optimize the electronic structure for coupled HMFOR and HER[89]. In 1 M KOH, CoMoP exhibited a low HMFOR onset potential of 1.03 V and a favorable HER overpotential, delivering high current densities in a dual-electrode configuration at 1.36 V with stable operation for 55 hours. In situ characterization and DFT calculations revealed that Mo doping induces lattice expansion, upshifts the Co d-band center, and promotes the generation of CoOOH and MoOx active species, which synergistically enhance hydrogen adsorption for HER and hydroxyl-mediated oxidation for HMFOR. Cai et al. further modified NiFe-LDH with F doping to enhance EOR activity[90]. Fluorine incorporation lowers the dehydrogenation activation barrier, accelerates NiFe-LDH deprotonation, and reduces the EOR onset potential. Moreover, F doping weakens the adsorption of OH- and EOR products, enabling high current densities (10-250 mA·cm-2) with FE > 95% and excellent stability for the optimized NiFe-0.10F catalyst at 1.386 V.
4.3.2 Vacancy engineering
The deliberate introduction of lattice defects, such as oxygen vacancies, into catalyst structures represents an effective strategy to lower adsorption energy barriers and enhance catalytic activity. These defects generate electron-deficient sites that facilitate reactant binding and accelerate key catalytic steps. Despite increasing attention to defect chemistry in heterogeneous catalysis, the specific roles of vacancies in AORs, particularly their influence on reaction pathways and product distributions, remain insufficiently understood. To address this knowledge gap, Wang et al. utilized β-Ni(OH)2 as a model system to systematically investigate the catalytic roles of Ni vacancies (VNi) and oxygen vacancies (VO) in AORs, including primary alcohol and vicinal diol oxidation[91]. They demonstrated that oxygen-vacancy-deficient β-Ni(OH)2 drives alcohol dehydrogenation exclusively via the LOM, converting primary alcohols into carboxylic acids without C-C bonds cleavage. By contrast, oxygen-vacancy-rich VR-β-Ni(OH)2 activates an additional vacancy-induced adsorbed oxygen-mediated mechanism (VO-AOM), enabling vicinal diol oxidation to both carboxylic and formic acids, accompanied by C-C bond scission. While Ni vacancies enhance dehydrogenation activity, they do not contribute to C-C bond cleavage. Building on these insights, Tian et al. designed a vacancy-rich IrRuOx catalyst to replace anodic OER with ethanol electrooxidation, thereby enabling paired electrosynthesis of H2 and acetic acid[92]. This catalyst displayed outstanding activity in acidic electrolytes, achieving an acetic acid production rate of 30 mmol cm-2·h-1 and a partial current density of 3 A·cm-2, tenfold higher than the previously reported highest activity. DFT calculations combined with in situ FTIR spectroscopy revealed that the reaction proceeds through the coupling of ethanol-derived CH3CO* intermediates with hydroxyl radicals (·OH) generated from water dissociation. Notably, the catalyst maintained stable operation at 1 A cm-2 for 40 hours, with an average acetate FE of 62%.
4.3.3 Alloying
Alloying noble or non-noble metals with other elements is an effective strategy to enhance electrocatalytic performance by improving electrical conductivity, reducing overpotentials, and increasing the density of active sites through modulation of electronic structures and surface properties[93]. Liu et al. synthesized Pd3Pb@Pt core-shell nanocubes with tunable Pt shell thickness via epitaxial growth, exploiting the lattice mismatch between the Pd3Pb core and Pt shell to induce tensile strain[94]. This strain, combined with the synergistic effects of the core-shell configuration, resulted in markedly enhanced EOR activity. In particular, the Pd3Pb@2.9% Pt catalyst with a three-atomic-layer Pt shell delivered an outstanding mass activity of 8.60 A·mg-1(Pd+Pt), underscoring the impact of precise structural engineering. Beyond conventional metal alloys, multimetallic oxides and hydroxides also harness synergistic interactions to boost catalytic efficiency. For instance, Xie et al. developed ternary NiVRu LDH nanosheet arrays (NiVRu-LDHs NAs/NF) for selective GOR coupled with hydrogen evolution[95]. At an industrially relevant current density of 1 A·cm-2, the catalyst achieved efficient co-production of formic acid and H2 at a low voltage of 1.933 V, with yields of 12.5 and 17.9 mmol·cm-2·h-1, respectively, and FE approaching 80% for HCOOH and 96% for H2. Experimental and theoretical analyses revealed that Ru incorporation modulates the local electronic structure of Ni-based LDHs, lowers the energy barrier for Ni2+ oxidation to the GOR-active Ni3+ species, and facilitates C-C bond cleavage, thereby enhancing formic acid selectivity.
4.3.4 Surface functionalization
Surface functionalization of catalysts with groups such as hydroxyl and carboxyl enhances interactions with polar molecules like alcohols and aldehydes, thereby improving catalytic selectivity and efficiency[96,97]. Glycerol, a C3 platform molecule, serves as a key substrate in electroreforming, where it can undergo direct oxidation to C3 products or oxidation-coupled C-C bond cleavage to generate C1/C2 products. While C3 products are economically more valuable, C-C cleavage enables the transfer of more electrons, thereby promoting H2 production. Consequently, balancing the reaction network is critical to optimizing electroreforming efficiency. A major challenge arises from the high solubility of oxidation products (e.g., formate and glycerate), which, together with downstream over-oxidation pathways, leads to low yields and complicates product separation. Oxalate represents a desirable product due to its resistance to over-oxidation and limited solubility, offering potential for solid-phase isolation alongside pure H2 production. However, precisely regulating oxidative strength and C-C cleavage to favor oxalate formation remains a significant challenge. To address this, Gong et al. developed a Ni(OH)2 catalyst functionalized with chelating phenanthroline ligands (Ni-phen), tuning its electronic structure through functional group modifications to achieve adjustable selectivity toward formate (up to 92.7%) and oxalate (up to 45.3%) in glycerol electroreforming[98]. The selectivity exhibited an approximately linear correlation with Hammett parameters, underscoring a strong electronic effect. In situ infrared reflection absorption spectroscopy revealed a continuous increase in formate-associated peaks for both catalysts, with the 1,351 cm-1 peak intensity lower for Ni-phen-NO2 consistent with its reduced faradaic efficiency for formate. Conversely, the 1,307 cm-1 peak, indicative of oxalate, was significantly stronger for Ni-phen-NO2. Similar results were observed when glycerol was substituted with glycolaldehyde, corroborating the proposed intermediate pathway. In situ Raman and ultraviolet photoelectron spectroscopy (UPS) further elucidated the interplay between catalyst structure and electronic effects. UPS showed that although all Ni-phen catalysts exhibited similar work functions, their valence band positions differed; electron-withdrawing substituents lowered the valence band, thereby modulating oxidation capacity and altering reaction pathways. By integrating intermediate analysis with spectroscopic techniques, the study demonstrated that tuning the valence band through key intermediates (e.g., glycolaldehyde) provides an effective means of balancing C-C bond cleavage and oxidation.
4.3.5 Support engineering
The choice of support materials, such as carbon-based materials or metal oxides, critically influences the electronic structure of catalysts by facilitating charge transfer and dispersing active site[99,100]. Shi et al. synthesized an Ag/Co3O4 catalyst via spontaneous redox between a ZIF-67 precursor and AgNO3, exploiting strong metal-support interactions (SMSI) to generate electron-deficient Agδ+ interfacial sites[101]. This system achieved outstanding FOR activity, delivering current densities of 10 mA·cm-2 at 0.32 V vs. RHE and 100 mA·cm-2 at 0.65 V vs. RHE. When integrated into an Ag/Co3O4||20% Pt/C electrolyzer, it demonstrated hydrogen efficiency above 195% and formic acid selectivity exceeding 98%, sustaining stability for over 60 hours. DFT and in situ infrared spectroscopy confirmed that Agδ+ sites lower the C-H bond cleavage barrier of the H2C(OH)O intermediate, thereby boosting reaction efficiency. In alkaline AORs, rapid OH- consumption at the anode often outpaces its diffusion, leading to local acidification (surface pH decline) that suppresses kinetics and shifts product distributions. For instance, during GOR, current densities as low as 10 mA·cm-2 can decrease the surface pH from 13 to 12, corresponding to a tenfold drop in OH- concentration. To address this, Shi et al. introduced an anode-electrolyte acidity regulation strategy using Lewis acidic CeO2-x supports decorated with noble metal nanoparticles (Au, Pt, Pd)[102]. Oxygen vacancies and Ce3+ sites in CeO2-x strongly adsorb OH-, yielding a diffusion coefficient 20.4 times higher than that of pure Au and maintaining a higher interfacial pH. This promotes alcohol deprotonation to alkoxide intermediates, accelerating the conversion of glyceraldehyde to dihydroxyacetone and further to lactate via a base-catalyzed Cannizzaro rearrangement, achieving 81% selectivity. Kinetic enhancement was also notable, with current densities reaching 693 mA·cm-2. Mechanistic studies confirmed that enhanced OH- adsorption and transport facilitate both deprotonation and intermediate transformation, offering a generalizable strategy for improving diverse AORs.
4.3.6 Oxidation state regulation
The oxidation state of metal catalysts plays a decisive role in governing electrocatalytic activity and product selectivity in reactions such as FOR and glucose oxidation[103-105]. Cu-based catalysts, for example, are highly effective for FOR at ultra-low potentials, but their stability under high current densities remains a major challenge due to limited mechanistic understanding. To address this, Wang et al. designed a partially reduced CuO@Cu foam (CuxO@CF) catalyst that exploits the synergistic interplay between Cu0 and Cu+ sites, achieving efficient FOR at 0.35 V vs. RHE with high current density and simultaneous hydrogen production at both electrodes[106]. In situ studies and DFT calculations revealed that Cu⁰ lowers the barrier (0.07 eV) for HOCH₂O + OH reactions, facilitating intermediate transformation, while Cu+ promotes C-H bond cleavage (0.23 eV barrier), accelerating hydrogen atom accumulation. Neither oxidation state alone provides optimal activity, but the Cuo-Cu+ interface markedly reduces energy barriers relative to single-state surfaces. Notably, all evolved hydrogen originated from HCHO, and the catalyst maintained > 98% conversion, yield, and selectivity over 17 cycles, underscoring its stability. Similarly, Bitter et al. developed a combined high-performance liquid chromatography (HPLC) and high-pressure anion exchange chromatography (HPAEC) strategy to probe how Pt oxidation states modulate glucose oxidation pathways[107]. They found that Pt0 exhibits sevenfold higher activity for primary alcohol dehydrogenation than for oxidation, favoring glucodialdehyde formation. In contrast, Pt0 displays comparable activity for both processes, promoting gluconate production. Furthermore, Pt0 selectively catalyzes dehydrogenation at the primary alcohol group of gluconate, while Pt0 also promotes secondary alcohol dehydrogenation, leading to more complex product distributions.
5. Reactor Innovation
Electrocatalytic AORs provides a sustainable alternative to the OER by operating at lower anodic potentials while simultaneously generating high-value chemicals and hydrogen. Nevertheless, conventional liquid-phase electrolyzers suffer from mass transfer limitations, competing side reactions, and challenges in product separation, which collectively diminish FE, selectivity, and scalability[108]. Recent advances in reactor engineering have begun to overcome these obstacles through improved mass transport, suppression of parasitic reactions, and enhanced operational stability. This section summarizes recent breakthroughs in reactor innovations for AOR, with a focus on novel architectures and strategies that surpass the performance of conventional configurations such as H-cell, GDE/flow cells, and MEAs equipped with AEM, PEM or BPM. While these established systems provide the baseline for AOR research, the emphasis here is on emerging designs that effectively address existing limitations and drive future development.
Two-electrode electrolyzers enable the simultaneous production of high-value chemicals and hydrogen, thereby enhancing both the sustainability and economic feasibility of electrochemical processes. A representative example is provided by Gao et al., who designed a Co9S8@Ni3S2/NF heterostructure catalyst for an anion exchange membrane (AEM) electrolyzer, coupling the selective oxidation of HMF to FDCA at the anode with the HER at the cathode[109]. This heterojunction optimizes H* adsorption for HER while improving the adsorption/desorption dynamics of organic substrates for HMFOR, leading to markedly enhanced reaction kinetics. At a current density of 50 mA cm−2, the electrolyzer continuously produced H2 and FDCA at a low cell voltage of 1.61 V, achieving production rates of 45.6 L·h-1·m-2 for H2 and 7.25 mg·h-1·cm-2 for FDCA, with FE of approximately 100% and 93.0%, respectively. In situ Raman spectroscopy and DFT calculations revealed that electron transfer from Ni3S2 to Co9S8 at the heterojunction interface facilitates the formation of Ni/CoOOH, the active species for HMFOR. This electronic modulation lowers the energy barrier for HMF oxidation, thereby improving both selectivity and efficiency, and underscores the promise of heterostructure-based electrolyzers for sustainable biomass valorization.
Selective oxidation of alcohols and biomass offers a viable alternative to the OER in conventional membrane electrode assembly (MEA) electrolyzers, lowering cathodic overpotential (e.g., 0.14 V for benzyl alcohol to benzaldehyde) and enabling the continuous generation of high-value aldehydes instead of undesired oxygen gas. Traditional MEA systems employ aqueous electrolyte-alcohol mixtures to initiate oxidation via water-derived active oxygen species, which also maintain ionic conductivity. However, this approach entails additional material and purification costs due to the solubility of target products in the electrolyte. Competing reactions, including water oxidation and overoxidation by active oxygen species, restrict the operational voltage window for selective aldehyde production, resulting in reduced selectivity, increased energy consumption, higher waste generation, and complicated product separation. Moreover, anode corrosion driven by water oxidation remains a major limitation to the durability of MEA electrolyzers in both hydrogen production and electrochemical organic synthesis. To overcome these challenges, the development of electrolyte-free electrocatalytic oxidation of pure organic liquids is highly desirable, although it has long been considered impractical due to the low charge mobility inherent in neat alcohols. Li et al. addressed this challenge by developing an innovative all-solid proton generator-transfer electrolyzer for the direct electrocatalytic splitting of pure alcohols into aldehydes and hydrogen gas[110]. In this system, Pt nanoparticles act as the proton generator, efficiently activating and cleaving O-H and C-H bonds to supply sufficient ions. A 3D conducting network composed of ionomers and carbon spheres facilitates proton diffusion, enabling smooth alcohol oxidation at the anode using pure alcohol as the sole reactant. By eliminating water oxidation and overoxidation reactions, this electrolyzer achieves exceptional selectivity for aldehydes (> 99%) with high FEs (> 95%) at a low cell voltage of 1.06 V and a current density of 10 mA·cm-2. Importantly, the absence of oxygen-containing active species, ensured by the solid proton generator and transfer pathway, significantly enhances electrode durability. The system maintains stability over 10 cycles (> 10 days) at a cell voltage of 1.2 V, retaining high FEs (80-93%) for electrocatalytic alcohol splitting.
Ethanol is an ideal feedstock for substituting fossil fuels and producing high-value chemicals, as its dehydrogenation reaction simultaneously generates hydrogen (a clean energy carrier) and acetaldehyde (an industrial intermediate with annual production exceeding 106 tons). Conventional electrocatalytic pathways, however, require high voltages (~0.8 V) to overcome kinetic barriers, resulting in substantial energy consumption. Thermal catalytic routes are limited by thermodynamic equilibrium, exhibit weak ethanol C-H bond dissociation, and are prone to side reactions and acetaldehyde decomposition at moderate temperatures, leading to low conversion and selectivity. To address these challenges, Wang et al. developed a coupled thermo-electrocatalytic strategy using a bifunctional Ru/C catalyst to achieve efficient ethanol dehydrogenation at a low temperature of 120 °C and an ultra-low voltage of 0.06 V, producing hydrogen and acetaldehyde with high efficiency[111]. The integration of an electrochemical hydrogen pump (EHP) rapidly removes anodic hydrogen, shifting the reaction equilibrium forward and achieving hydrogen and acetaldehyde yields of 1,020 mmol g-1·h-1 and 1,185 mmol g-1·h-1, respectively, threefold higher than standalone thermal dehydrogenation, with an acetaldehyde selectivity of 97.9%. This approach also substantially reduces energy consumption compared to traditional electrolytic systems. DFT calculations confirmed that the C-H bond cleavage barrier on the Ru(101) surface (0.83 eV) is lower than the O-H bond cleavage barrier on Pt(111) (0.92 eV). Furthermore, elevating the temperature to 473 K (200 °C) reduces the rate-determining step barrier from 0.63 eV to 0.40 eV, markedly enhancing reaction kinetics.
Biomass molecules (e.g., glucose, HMF) containing reactive functional groups such as aldehydes and hydroxyls are prone to spontaneous decomposition (e.g., glucose to lactate, glyceraldehyde to lactate) or polymerization (e.g., HMF to dark humic substances) in alkaline electrolytes. These side reactions result in significant carbon loss, hindering high-concentration, large-scale production. To address this, Duan et al. developed a single-pass continuous flow reactor (SPCFR) system featuring a high electrode-area-to-electrolyte-volume ratio, short substrate residence time, and separate feeding of substrate and alkaline solution, effectively suppressing non-Faradaic degradation[112]. Figure 11a illustrates a conventional flow reactor, where premixed substrate and alkaline solution undergo cyclic feeding, promoting non-Faradaic degradation. Figure 11b,c highlight the core design of the SPCFR, which mitigates degradation through three key features: a high electrode-area-to-electrolyte-volume ratio (≈ 2.5 cm2/mL), separate substrate and alkaline solution feeds, and a short residence time. Figure 11d shows a physical image of a single-module SPCFR, revealing its simple structure (30 cm2 CoOOH/NF anode, 30 cm2 nickel foam cathode, no ion-exchange membrane). Figure 11e compares the performance of the SPCFR with batch reactors for polyol oxidation (e.g., ethylene glycol, glycerol) to formate using a bar chart, demonstrating that the SPCFR achieves formate yields above 80% with selectivities of 90%-97%, significantly outperforming batch reactors. Figure 11f compares product distributions for HMF oxidation to FDCA, showing that SPCFR increases FDCA selectivity from 37.8% in batch reactors to 91.3%, while substantially reducing humic substance formation. The stability curve indicates that SPCFR maintains FDCA selectivity of ≈ 95% and single-pass conversion of ≈ 94% over 100 hours of continuous operation at a constant current of 5 A, without performance decay, clearly demonstrating its ability to suppress non-Faradaic degradation and achieve efficient, stable conversion. Furthermore, a nine-module stacked SPCFR system (electrode area of 270 cm2) was developed, enabling efficient conversion of glucose to formate (single-pass conversion 81.8%, selectivity 76.5%, concentration 562.8 mM) and HMF to FDCA (single-pass conversion 95.8%, selectivity 96.9%, concentration 556.9 mM). Kilogram-scale validation was successfully achieved (0.7 kg of potassium formate from lignocellulose/soybean oil and 1.17 kg of FDCA from HMF). Mechanistic studies further confirmed that the true intermediate in glucose electrooxidation to formate is an aldehyde, rather than the traditionally assumed aldonic acid, providing a new strategy for scalable biomass electrosynthesis.
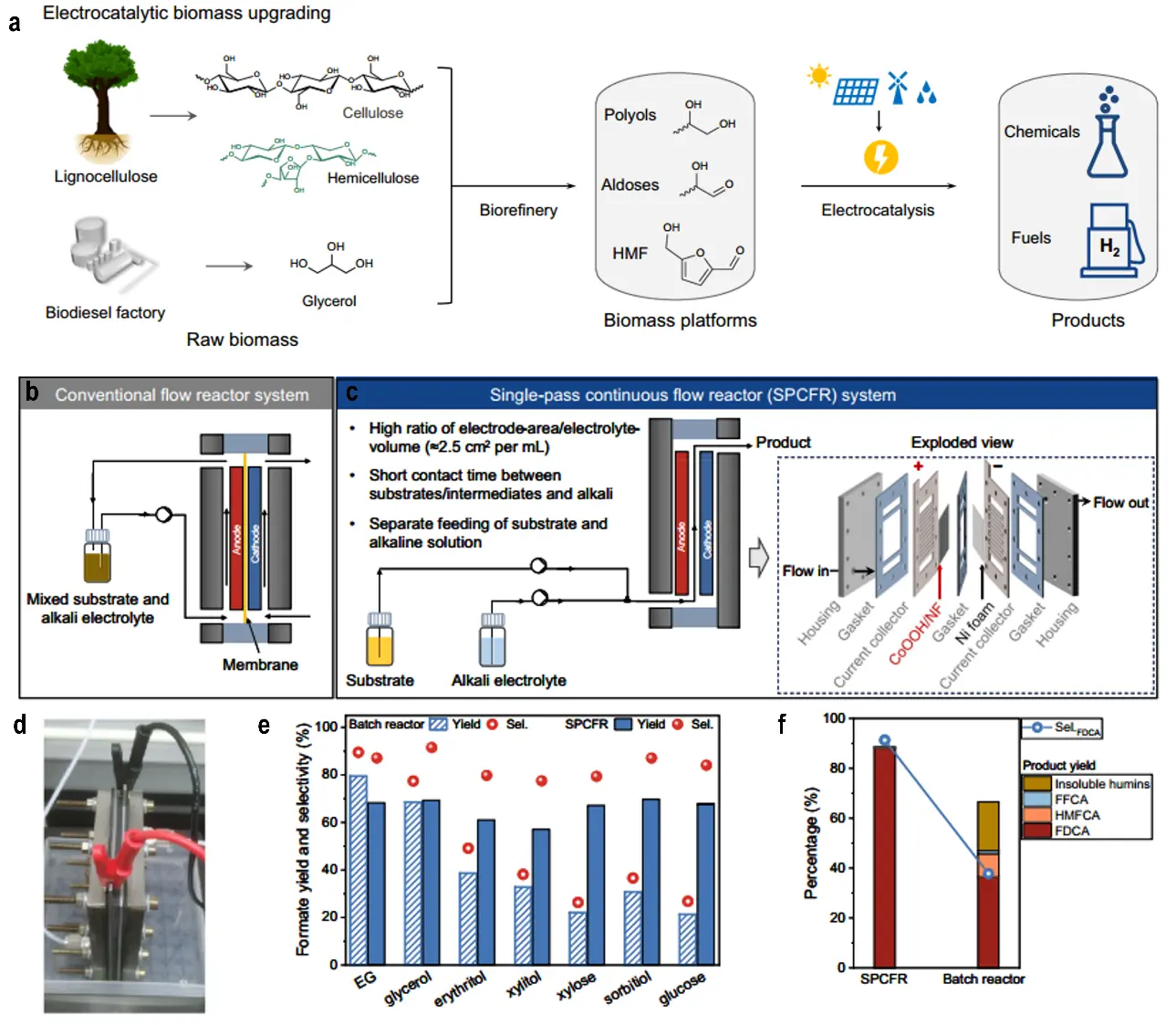
Figure 11. (a) Electrocatalytic biomass platforms upgrading toward chemicals and fuels by integrating with upstream biorefinery processes; (b) Traditional flow reactor system; (c) Designed SPCFR system; (d) Photograph of single-module SPCFR. Catalytic performances of SPCFR system and batch reactor for electrooxidation of polyhydroxy compounds-to-formate (e) and HMF-to-FDCA (f)[112]. SPCFR: single-pass continuous flow reactor; HMF: 5-hydroxymethylfurfural; FDCA: 2,5-furandicarboxylic acid.
The scale-up of electrocatalytic processes, such as HMF oxidation to FDCA, faces significant challenges, including mass transfer limitations that reduce FE for target products, non-Faradaic side reactions leading to carbon loss, and low mass transfer efficiency in liquid-phase electrolysis (with molecular diffusion coefficients in liquids are ~1/100 of those in gases). These limitations have hindered kilowatt-scale demonstrations. Efficient large-scale HMF-to-FDCA conversion requires high current densities, moderate cell voltages (to minimize energy consumption), high selectivity and FE, and elevated product concentrations (to reduce separation costs). Duan et al. addressed these challenges by developing an engineered solid polymer electrolyte (SPE) reactor capable of regulating both Faradaic and non-Faradaic reactions to achieve selective HMF electrooxidation to FDCA[113]. The reactor’s core innovation lies in the synergistic integration of critical components and optimized operating conditions to overcome these bottlenecks. Specifically, optimized porous transport layers (PTLs) and serpentine flow fields generate chaotic flow, enhancing radial convective mass transfer and suppressing Faradaic side reactions such as the OER. A cooling chamber maintains the reaction temperature at ~29 °C, inhibiting non-Faradaic HMF condensation. Furthermore, the catalyst-coated membrane design, combined with a liquid-free cathode electrolyte, minimizes molecular cross-diffusion and water permeation, thereby increasing FDCA concentration. A static mixer reduces HMF residence time in alkaline media, further mitigating side reactions. At a current density of 1.5 A·cm-2, the reactor achieves a power output of 654 W, with 97.0% selectivity, 88.2% FE, and an FDCA concentration of ~1.24 M. Continuous operation at 0.5 A·cm-2 for 140 hours showed minimal voltage degradation (1.25 mV·h-1), with stable performance metrics, limited primarily by commercial AEM degradation rather than catalyst or PTL limitations. By parallelizing eight SPE reactors, a total power of 4.3 kW (1,600 A, 2.71 V) was achieved at 1.0 A·cm-2, yielding an FDCA production rate of 1.38 kg/h (33 kg/day), representing a 1-2 orders of magnitude increase over previous electrochemical reports. Economically, the electrochemical production cost of FDCA is estimated at $2,176/ton, reducible to $1301.2/ton by valorizing byproducts such as K2SO4 and H2, making it competitive with thermocatalytic FDCA ($1,490/ton) and petroleum-based terephthalic acid (TPA, $1445/ton). FDCA serves as a key precursor for bioplastics, including polyethylene furanoate for 3D-printable thermoplastics and epoxy-based thermosets for molding applications. This scalable, efficient, and cost-competitive approach highlights the potential of electrocatalytic HMF oxidation for sustainable bioplastic production.
6. Conclusion and Perspective
Electrocatalytic AORs have emerged as pivotal technologies for sustainable chemical synthesis, offering a low-carbon, energy-efficient alternative to conventional thermochemical processes. Recent advancements in catalyst design, mechanistic elucidation, and process integration have significantly advanced the field, enabling the selective transformation of alcohols and aldehydes into high-value chemicals. Despite these advancements, enduring challenges in catalyst stability, selectivity, and process scalability continue to hinder the widespread industrial adoption of AORs. This section synthesizes these challenges, delineates strategic research directions, and underscores the transformative potential of AORs in advancing sustainable chemistry and decarbonizing chemical manufacturing (Figure 12).

Figure 12. Schematic illustration of the future perspectives of AORs. AOR: alcohol oxidation reactions.
Despite considerable progress, several critical barriers impede the practical deployment of AORs:
Catalyst stability: To achieve long-term stability for non-noble metal catalysts, such as Ni, Cu, and Co, which are critical for cost-effective electrochemical applications, remains a major challenge due to their susceptibility to degradation under harsh operational conditions. These catalysts often suffer from surface restructuring, metal leaching, or poisoning by reaction intermediates and byproducts, leading to reduced performance and limited lifespans. For instance, Ni-based catalysts in alkaline electrolytes may undergo phase transformations, such as converting from Ni to Ni(OH)2, or complete dissolution, compromising their catalytic efficiency. In contrast, noble metal catalysts, such as Pt and Au, demonstrate superior durability but are hindered by prohibitively high costs and limited global availability, rendering them impractical for large-scale industrial applications. To overcome these challenges, a multifaceted approach is required. First, developing hybrid catalysts by integrating non-noble metals with robust, high-surface-area supports, such as carbon nanotubes, graphene, or metal-organic frameworks (MOFs), can enhance structural integrity and resistance to degradation while maintaining catalytic activity. Second, advanced in-situ characterization techniques, such as X-ray absorption spectroscopy (XAS), scanning electrochemical microscopy (SECM), and operando Raman spectroscopy, enable real-time monitoring of catalyst behavior under working conditions, providing critical insights into degradation mechanisms and informing the design of more resilient materials. Third, protective strategies, such as applying nanoscale coatings (e.g., metal oxides or polymers) or alloying non-noble metals with stable elements like Mo, W, or Cr, can significantly mitigate leaching and poisoning while improving overall stability and catalytic performance. Additionally, computational modeling and machine learning could guide the rational design of these catalysts by predicting stable compositions and optimizing their electrochemical properties. By combining these strategies, researchers can develop cost-effective, durable, and high-performing non-noble metal catalysts, paving the way for sustainable and scalable solutions in energy conversion, storage, and other electrochemical technologies.
Selectivity control: Controlling selectivity in AORs for polyhydric alcohols like glycerol and HMF is challenging due to the formation of diverse intermediates, such as glyceraldehyde, dihydroxyacetone, and formic acid, driven by complex reaction networks and competing adsorption on catalyst surfaces, which often leads to suboptimal product distributions. To design catalysts that preferentially yield desired products while suppressing side reactions requires a deep understanding of reaction pathways, intermediate dynamics, and surface interactions. Strategies to achieve this include the development of tailored catalysts, such as bimetallic systems (e.g., Pt-Au or Pd-Ni), to modulate adsorption energies, and the use of surface modifications, for instance, nitrogen-doped carbon supports, to minimize undesired reaction pathways. Real-time monitoring using in-situ techniques, such as infrared spectroscopy or X-ray absorption spectroscopy, provides critical insights into intermediate evolution and catalyst behavior. Additionally, computational tools like DFT and machine learning can predict selectivity trends, while optimizing reaction conditions, such as pH or temperature, can further enhance product specificity. These integrated strategies enable precise control over AOR selectivity, promoting efficient and sustainable production of high-value chemicals from biomass-derived feedstocks.
Scalability and system integration: Scaling AORs from laboratory batch systems to industrial processes necessitates significant advancements to overcome limitations in reactor design, catalyst performance, and system integration. Existing electrochemical setups, such as those for HMF oxidation, suffer from low single-pass conversion efficiencies and are limited by mass transport constraints, which reduce overall productivity. Although flow-based reactors offer the potential for continuous operation, challenges including non-uniform current distribution, catalyst degradation over extended operation, and the high cost of noble metal catalysts (e.g., Pt or Au) hinder scalability. To address these barriers, advanced flow reactors featuring optimized electrode geometries, such as microchannel or 3D-printed designs, can enhance mass transport and efficiency. The development of durable, cost-effective non-noble metal catalysts (e.g., Ni or Co-based systems) supported on robust materials like carbon nanotubes or metal-organic frameworks can mitigate degradation while reducing costs. In-situ monitoring techniques, including electrochemical impedance spectroscopy and X-ray absorption spectroscopy, enable real-time process control, and integrating AOR systems with renewable energy sources, such as solar or wind, further improves energy efficiency. Comprehensive process optimization, coupled with techno-economic analysis, is essential to ensure scalability and commercial viability, paving the way for sustainable, large-scale production of high-value chemicals from biomass-derived feedstocks.
To overcome these challenges and unlock the full potential of AORs, future research should prioritize the following interdisciplinary strategies:
Advanced catalyst design: Developing robust and cost-effective catalysts is paramount for industrial AOR applications. Strategies such as heteroatom doping (e.g., P, S, N) and vacancy engineering can finely tune the electronic structure of non-noble metal catalysts, enhancing both stability and activity. For instance, phosphorus-doped Ni catalysts have shown improved resistance to surface passivation in alkaline media. Multi-element alloying and the application of protective coatings (e.g., carbon or metal oxide layers) offer additional avenues to mitigate catalyst leaching and degradation. Moreover, high-throughput screening and combinatorial synthesis approaches can accelerate the discovery of novel catalyst compositions tailored for specific AORs.
In Situ and operando characterization: Gaining real-time insights into catalyst dynamics and reaction mechanisms are critical for optimizing AOR performance. Advanced in situ and operando techniques, such as Raman spectroscopy, FTIR, XANES, and EXAFS, allow monitoring of catalyst surface evolution and intermediate adsorption under operational conditions. Expanding these techniques to study complex substrates like glycerol or HMF will deepen understanding of reaction pathways, enabling the rational design of catalysts with enhanced selectivity and durability.
Paired electrolysis systems: Coupling AORs with cathodic HER offers a dual-benefit approach by replacing the energy-intensive OER with value-added anodic oxidation. This strategy not only reduces overall energy consumption but also generates green hydrogen as a co-product, enhancing economic viability. Future efforts should focus on developing bifunctional catalysts that efficiently drive both AOR and HER, as well as designing scalable flow reactors with integrated anode-cathode configurations to maximize energy efficiency and product yields.
Innovative reactor design: To address scalability challenges, priority should be given to novel reactor designs such as flow-based and membrane-less electrolyzers. Such systems enhance mass transport, improve current distribution, and reduce ohmic losses. For example, 3D-printed electrodes with tailored geometries or microfluidic reactors with optimized flow patterns can minimize transport limitations and enhance reaction efficiencies. Integrating AOR reactors with renewable energy sources, such as solar or wind, further aligns these systems with global decarbonization objectives, ensuring sustainable operation at scale.
Computational and data-driven approaches: Computational modeling, including DFT and machine learning (ML), offers powerful tools for accelerating catalyst discovery and process optimization. DFT simulations can elucidate the electronic and structural properties of catalysts, predicting optimal adsorption energies for key intermediates. ML algorithms can analyze large datasets to identify stable, high-performance catalyst compositions and guide doping or alloying strategies. Extending these models to simulate complex reaction networks, such as those in glycerol or HMF oxidation, will enable the rational design of catalysts with tailored selectivity and activity.
Authors contribution
Chen L: Data analysis, investigation, resources, validation, writing-original draft, writing-review & editing.
Yuan ZY: Conceptualization, project administration, supervision, validation, writing-review & editing.
All authors have given approval to the final version of the manuscript.
Conflicts of interest
Zhong-Yong Yuan is the Editor-in-Chief of Smart Materials and Devices. Another author has no conflicts of interest to declare.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
All data are available in the main text.
Funding
This work was supported by the National Natural Science Foundation of China (22179065).
Copyright
© The Author(s) 2025.
References
-
1. Ahmad M, Hussain MB, Mushtaq MA, Raza W, Mehmood A, Afzal S, et al. Advances in electrocatalytic hydrogen evolution coupled with alcohol and aldehyde oxidation: mechanistic insights and economic feasibility. Adv Mater. 2025;37(33):2502966.[DOI]
-
2. Zhao Y, Björk EM, Yan Y, Schaaf P, Wang D. Recent progress in transition metal based catalysts and mechanism analysis for alcohol electrooxidation reactions. Green Chem. 2024;26(9):4987-5003.[DOI]
-
3. Wu W, Zhai H, Holubnyak E. Technological evolution of large-scale blue hydrogen production toward the US Hydrogen Energy Earthshot. Nat Commun. 2024;15(1):5684.[DOI]
-
4. Bojarska Z, Karnas MJ, Godula-Jopek A, Mandrek S, Makowski Ł. Simultaneous hydrogen generation and organic oxidation in low-temperature electrolyzers. Appl Energy. 2025;389:125697.[DOI]
-
5. Kottaichamy AR, Marichelvam T, Tzadikov J, Vaza RC, Volokh M, Barzilai S, et al. Energy-efficient hydrogen generation via peroxide-mediated electrocatalytic pathways. Angew Chem Int Ed. 2025;64(28):e202502735.[DOI]
-
6. Ren JT, Chen L, Wang HY, Tian WW, Yuan ZY. Water electrolysis for hydrogen production: from hybrid systems to self-powered/catalyzed devices. Energy Environ Sci. 2024;17(1):49-113.[DOI]
-
7. Vitulano F, Solida A, Sorti L, Morelli CF, Minguzzi A, Vertova A. Alcohol and carbonyl redox reactions in electrochemical organic synthesis. ChemistryEurope. 2025;3(4):e202500013.[DOI]
-
8. Liu C, Chen F, Zhao BH, Wu Y, Zhang B. Electrochemical hydrogenation and oxidation of organic species involving water. Nat Rev Chem. 2024;8(4):277-293.[DOI]
-
9. Hu F, Wang P, Lu Z, Chen K, Ding Y, Wang L, et al. Electrocatalytic aldehyde oxidation: an emerging anodic reaction for efficient electrolytic systems. Sustain Energy Fuels. 2025;9(4):904-920.[DOI]
-
10. Gao W, Wang C, Wen W, Wang S, Zhang X, Yan D, et al. Electrochemical hydrogen production coupling with the upgrading of organic and inorganic chemicals. Adv Mater. 2025;37(32):2503198.[DOI]
-
11. Zhang XY, Hu SM, Xu HG, Yang HG, Liu PF. Integrated electrolytic hydrogen production for boosting energy utilization. ChemCatChem. 2024;16(16):e202400345.[DOI]
-
12. Li J, Xie W, Zhou H, Li Z, Shao M. Techno-economic analysis of electrochemical hydrogen production coupled with alternative oxidation. Chem Eng Sci. 2024;298:120322.[DOI]
-
13. Wang HY, Sun ML, Ren JT, Yuan ZY. Circumventing challenges: design of anodic electrocatalysts for hybrid water electrolysis systems. Adv Energy Mater. 2023;13(4):2203568.[DOI]
-
14. Li J, Duan H. Recent progress in energy-saving hydrogen production by coupling with value-added anodic reactions. Chem. 2024;10(10):3008-3039.[DOI]
-
15. Kumar R, Singh R, Dutta S. Review and outlook of hydrogen production through catalytic processes. Energy Fuels. 2024;38(4):2601-2629.[DOI]
-
16. Ren JT, Yao Y, Yuan ZY. Fabrication strategies of porous precious-metal-free bifunctional electrocatalysts for overall water splitting: Recent advances. Green Energy Environ. 2021;6(5):620-643.[DOI]
-
17. Qiu J, Forbes T, Lin T. Tailoring the oxidation of benzyl alcohol and its derivatives with (photo)electrocatalysis. Chem Commun. 2025;61:3421-3435.[DOI]
-
18. Gao T, An Q, Tang X, Yue Q, Zhang Y, Li B, et al. Recent progress in energy-saving electrocatalytic hydrogen production via regulating the anodic oxidation reaction. Phys Chem Chem Phys. 2024;26(29):19606-19624.[DOI]
-
19. Lee J, Lee SA, Lee TH, Jang HW. Unlocking the potential of chemical-assisted water electrolysis for green hydrogen production. Ind Chem Mater. 2025;3:277-310.[DOI]
-
20. Liu Y, Yang Z, Zou Y, Wang S, He J. Interfacial micro-environment of electrocatalysis and its applications for organic electro-oxidation reaction. Small. 2024;20(4):2306488.[DOI]
-
21. Wang P, Zheng J, Xu X, Zhang YQ, Shi QF, Wan Y, et al. Unlocking efficient hydrogen production: nucleophilic oxidation reactions coupled with water splitting. Adv Mater. 2024;36(35):2404806.[DOI]
-
22. Gao G, Sun Z, Chen X, Zhu G, Sun B, Huang XL, et al. Recent advances in hydrogen production coupled with alternative oxidation reactions. Coord Chem Rev. 2024;509:215777.[DOI]
-
23. van der Ham MP, Creus J, Bitter JH, Koper MT, Pescarmona PP. Electrochemical and non-electrochemical pathways in the electrocatalytic oxidation of monosaccharides and related sugar alcohols into valuable products. Chem Rev. 2024;124(21):11915-11961.[DOI]
-
24. Shi J, Ma J, Ma E, Li J, Hu Y, Fan L, et al. Electrochemical alcohol oxidation reaction on Precious-Metal-Free catalysts: Mechanism, activity, and selectivity. Carbon Neutral. 2024;3(2):285-312.[DOI]
-
25. Wang T, Tao L, Zhu X, Chen C, Chen W, Du S, et al. Combined anodic and cathodic hydrogen production from aldehyde oxidation and hydrogen evolution reaction. Nat Catal. 2022;5(1):66-73.[DOI]
-
26. Mao Q, Deng K, Yu H, Xu Y, Wang Z, Li X, et al. In situ reconstruction of partially hydroxylated porous Rh metallene for ethylene glycol-assisted seawater splitting Adv Funct Mater. 2022;32(31):2201081.[DOI]
-
27. Li Z, Yan Y, Xu SM, Zhou H, Xu M, Ma L, et al. Alcohols electrooxidation coupled with H2 production at high current densities promoted by a cooperative catalyst. Nat Commun. 2022;13(1):147.[DOI]
-
28. Li R, Kuang P, Wang L, Tang H, Yu J. Engineering 2D NiO/Ni3S2 heterointerface electrocatalyst for highly efficient hydrogen production coupled with benzyl alcohol oxidation. Chem Eng J. 2022;431:134137.[DOI]
-
29. Guo Y, Yang X, Liu X, Tong X, Yang N. Coupling methanol oxidation with hydrogen evolution on bifunctional Co-doped Rh electrocatalyst for efficient hydrogen generation. Adv Funct Mater. 2022;33(2):2209134.[DOI]
-
30. Ramos NC, Holewinski A. Recent advances in anodic hydrogen production: electrochemical oxidative dehydrogenation of aldehydes to carboxylates. Curr Opin Electrochem. 2024;45:101484.[DOI]
-
31. Fu H, Chen Z, Chen X, Jing F, Yu H, Chen D, et al. Modification strategies for development of 2D material-based electrocatalysts for alcohol oxidation reaction. Adv Sci. 2024;11(37):2306132.[DOI]
-
32. Qian Q, Zhu Y, Ahmad N, Feng Y, Zhang H, Cheng M, et al. Recent advancements in electrochemical hydrogen production via hybrid water splitting. Adv Mater. 2024;36(4):2306108.[DOI]
-
33. Fan L, Wang D, Ma K, Zhou CA, Yue H. Recent advances in hydrogen production from hybrid water electrolysis through alternative oxidation reactions. ChemCatChem 2024;16(5):e202301332.[DOI]
-
34. Mathur A, Diesendruck CE. Advanced device architecture strategies for decoupled water splitting: a review. ACS Mater Lett. 2024;6(7):2725-2737.[DOI]
-
35. Tang D, Lu G, Shen Z, Hu Y, Yao L, Li B, et al. A review on photo-, electro- and photoelectro- catalytic strategies for selective oxidation of alcohols. J Energy Chem. 2023;77:80-118.[DOI]
-
36. Chen L. Electrocatalytic seawater splitting from direct electrolysis to hybrid electrolysis: challenges and opportunities. Mater Today. 2025;86:282-316.[DOI]
-
37. Cheng J, Xiang Y, Huang X, Wei Z. Reducing energy costs during hydrogen production from water electrolysis by coupling small molecule oxidation: from molecular catalysis to industrial exploration. Precis Chem. 2024;2(9):447-470.[DOI]
-
38. Ahmad M, Hussain MB, Chen J, Yang Y, Wu X, Chen H, et al. Direct methanol fuel cell with porous carbon-supported PtRu single-atom catalysts for coproduction of electricity and value-added formate. ACS Catal. 2024;14(12):9134-9143.[DOI]
-
39. Yang C, Zhang L, Lu Y, Zou Y, Wang S. Designing efficient catalysts for electrocatalytic organic synthesis: from electronic structure to adsorption behavior. Matter. 2024;7(2):456-474.[DOI]
-
40. Kolbe H. Untersuchungen über die Elektrolyse organischer Verbindungen. Justus Liebigs Ann Chem. 1849;69(3):257-294.[DOI]
-
41. Shono T. Electroorganic chemistry in organic synthesis. Tetrahedron. 1984;40(5):811-850.[DOI]
-
42. Baizer MM. Electrolytic reductive coupling: I. acrylonitrile. J Electrochem Soc. 1964;111(2):215.[DOI]
-
43. Yamamoto K, Kuriyama M, Onomura O. Anodic oxidation for the stereoselective synthesis of heterocycles. Acc Chem Res. 2020;53(1):105-120.[DOI]
-
44. Siu JC, Fu N, Lin S. Catalyzing electrosynthesis: a homogeneous electrocatalytic approach to reaction discovery. Acc Chem Res. 2020;53(3):547-560.[DOI]
-
45. Novaes LF, Liu J, Shen Y, Lu L, Meinhardt JM, Lin S. Electrocatalysis as an enabling technology for organic synthesis. Chem Soc Rev. 2021;50(14):7941-8002.[DOI]
-
46. Meyer TH, Choi I, Tian C, Ackermann L. Powering the future: how can electrochemistry make a difference in organic synthesis? Chem. 2020;6(10):2484-2496.[DOI]
-
47. Ouyang D, Gao D, Hong J, Jiang Z, Zhao X. Promoting and controlling electron transfer of furfural oxidation efficiently harvest electricity, furoic acid, hydrogen gas and hydrogen peroxide. J Energy Chem. 2023;79:135-147.[DOI]
-
48. Liu L, He Y, Ma DD, Wu XT, Zhu QL. Directional editing of self-supported nanoarray electrode for adaptive paired-electrolysis. J Colloid Interface Sci. 2023;640:423-433.[DOI]
-
49. Li Z, Zhang Y, Yang Q, Wu J, Ren Z, Si F, et al. Hydrogen co-production via nickel-gold electrocatalysis of water and formaldehyde. iScience. 2023;26(10):107994.[DOI]
-
50. Li F, Liu C, Lin H, Sun Y, Yu H, Xue S, et al. High activity of bifunctional Ni2P electrocatalyst for benzyl alcohol oxidation coupled with hydrogen evolution. J Colloid Interface Sci. 2023;640:329-337.[DOI]
-
51. Li D, Tu J, Lu Y, Zhang B. Recent advances in hybrid water electrolysis for energy-saving hydrogen production. Green Chem Eng. 2023;4(1):17-29.[DOI]
-
52. Kishore MA, Lee S, Yoo JS. Fundamental limitation in electrochemical methane oxidation to alcohol: a review and theoretical perspective on overcoming it. Adv Sci. 2023;10(31):2301912.[DOI]
-
53. Zhang Y, Wang H, Lee JM, Zhang Q. Energy-saving electrochemical hydrogen production via co-generative strategies in hybrid water electrolysis: recent advances and perspectives. Chinese J Catal. 2023;55:44-115.[DOI]
-
54. Hao Y, Yu D, Zhu S, Kuo CH, Chang YM, Wang L, et al. Methanol upgrading coupled with hydrogen product at large current density promoted by strong interfacial interactions. Energy Environ Sci. 2023;16(3):1100-1110.[DOI]
-
55. Guo L, Zhang X, Gan L, Pan L, Shi C, Huang ZF, et al. Advances in selective electrochemical oxidation of 5-hydroxymethylfurfural to produce high-value chemicals. Adv Sci. 2023;10(4):2205540.[DOI]
-
56. Chen Y, Fu Y, Peng W, Wang S. Minireview of coupled electrochemical hydrogen production and organic-oxidation for low energy consumption. Energy Fuels. 2023;37(23):17915-17931.[DOI]
-
57. Zhao M, Guo C, Liu C, Gao L, Ren X, Yang H, et al. An amorphous Ni-Fe catalyst for electrocatalytic dehydrogenation of alcohols to value-added chemicals. Nanoscale. 2023;15(38):15600-15607.[DOI]
-
58. Meng F, Wu Q, Elouarzaki K, Luo S, Sun Y, Dai C, et al. Essential role of lattice oxygen in methanol electrochemical refinery toward formate. Sci Adv. 2023;9(34):eadh9487.[DOI]
-
59. Qiu Y, Zhang J, Jin J, Sun J, Tang H, Chen Q, et al. Construction of Pd-Zn dual sites to enhance the performance for ethanol electro-oxidation reaction. Nat Commun. 2021;12(1):5273.[DOI]
-
60. Wu G, Dong X, Mao J, Li G, Zhu C, Li S, et al. Anodic glycerol oxidation to formate facilitating cathodic hydrogen evolution with earth-abundant metal oxide catalysts. Chem Eng J. 2023;468:143640.[DOI]
-
61. Zhu B, Dong B, Wang F, Yang Q, He Y, Zhang C, et al. Unraveling a bifunctional mechanism for methanol-to-formate electro-oxidation on nickel-based hydroxides. Nat Commun. 2023;14(1):1686.[DOI]
-
62. Zhou P, Hai G, Zhao G, Li R, Huang X, Lu Y, et al. CeO2 as an “electron pump” to boost the performance of Co4N in electrocatalytic hydrogen evolution, oxygen evolution and biomass oxidation valorization. Appl Catal B Environ. 2023;325:122364.[DOI]
-
63. Yang Y, Wu X, Ahmad M, Si F, Chen S, Liu C, et al. A direct formaldehyde fuel cell for CO2-emission free co-generation of electrical energy and valuable chemical/hydrogen. Angew Chem Int Ed. 2023;62(21):e202302950.[DOI]
-
64. Yao ZC, Chai J, Tang T, Ding L, Jiang Z, Fu J, et al. Manipulating hydrogenation pathways enables economically viable electrocatalytic aldehyde-to-alcohol valorization. Proc Natl Acad Sci U.S.A. 2025;122(8):e2423542122.[DOI]
-
65. Yang G, Jiao Y, Yan H, Xie Y, Wu A, Dong X, et al. Interfacial engineering of MoO2-FeP heterojunction for highly efficient hydrogen evolution coupled with biomass electrooxidation., Adv Mater. 2020;32:2000455.[DOI]
-
66. Zhu P, Shen Y, Zhang ZM, Wang D, Xi B, An X, et al. Modulating alcohol adsorption modes for boosting electrooxidation-assisted hydrogen production. ACS Catal. 2024;14(10):7674-7683.[DOI]
-
67. Du R, Chen Z, Wang S, Zeng S, Jia R, Zhang K, et al. Manipulating the interfacial hydrophobic microenvironment via electrolyte engineering promotes electrocatalytic fatty alcohol oxidation coupled with hydrogen production. JACS Au. 2025;5(4):1974-1982.[DOI]
-
68. Xu L, Huang Z, Yang M, Wu J, Chen W, Wu Y, et al. Salting-Out aldehyde from the electrooxidation of alcohols with 100% selectivity. Angew Chem Int Ed. 2022;61(45):e202210123.[DOI]
-
69. Xu L, Chen W, Wang C, Wu W, Yao Y, Huang Z, et al. Combining anodic alcohol oxidative coupling for C-C bond formation with cathodic ammonia production. Natl Sci Rev. 2024;11(5):nwae134.[DOI]
-
70. Zhao S, Wang S, Qiu C, Xu Z, Ren L, Wang Y, et al. Enhanced electrocatalytic alcohol oxidation mediated with ultra-low concentration of aminoxyl radicals via an Intermediate-Rich local microenvironment. Chem Eng Sci. 2024;286:119653.[DOI]
-
71. Badreldin A, Youssef E, Djire A, Abdala A, Abdel-Wahab A. A critical look at alternative oxidation reactions for hydrogen production from water electrolysis. Cell Rep Phys Sci. 2023;4(6):101427.[DOI]
-
72. Li YN, Hong QL, Miao BQ, Wang TJ, Ding Y, Chen Y. Platinum-tellurium alloy metallene toward formic acid oxidation reaction. Renewables. 2023;1(1):90-99.[DOI]
-
73. You X, Han J, Del Colle V, Xu Y, Chang Y, Sun X, et al. Relationship between oxide identity and electrocatalytic activity of platinum for ethanol electrooxidation in perchlorate acidic solution. Commun Chem. 2023;6(1):101.[DOI]
-
74. Yan Y, Cai X, Yang J, Fu Y, Shi Q, Hao P, et al. Tandem electrocatalytic benzylic alcohol oxidation and aldol condensation for efficient valuable α,β-unsaturated ketone production. Green Chem. 2025;27(21):6016-6026.[DOI]
-
75. Shi Q, Zhu YQ, Liu X, Yuan BJ, Tang W, Wang X, et al. Electrocatalytic ethylene glycol to long-chain C3+ alpha-hydroxycarboxylic acids via cross-coupling with primary alcohols. J Am Chem Soc. 2025;147(16):14004-14014.[DOI]
-
76. Yan Y, Zhou H, Xu SM, Yang J, Hao P, Cai X, et al. Electrocatalytic upcycling of biomass and plastic wastes to biodegradable polymer monomers and hydrogen fuel at high current densities. J Am Chem Soc. 2023;145(11):6144-6155.[DOI]
-
77. Lu Q, Huang J, Han C, Sun L, Yang X. Facile synthesis of composition-tunable PtRh nanosponges for methanol oxidation reaction. Electrochim Acta. 2018;266:305-311.[DOI]
-
78. Li G, Han G, Wang L, Cui X, Moehring NK, Kidambi PR, et al. Dual hydrogen production from electrocatalytic water reduction coupled with formaldehyde oxidation via a copper-silver electrocatalyst. Nat Commun. 2023;14(1):525.[DOI]
-
79. Zhao L, Lv Z, Shi Y, Zhou S, Liu Y, Han J, et al. Simultaneous generation of furfuryl alcohol, formate, and H2 by co-electrolysis of furfuryl and HCHO over bifunctional CuAg bimetallic electrocatalysts at ultra-low voltage. Energy Environ Sci. 2024;17(2):770-779.[DOI]
-
80. Sun H, Li L, Chen Y, Kim H, Xu X, Guan D, et al. Boosting ethanol oxidation by NiOOH-CuO nano-heterostructure for energy-saving hydrogen production and biomass upgrading. Appl Catal B Environ. 2023;325:22388.[DOI]
-
81. Hausmann JN, Menezes PV, Vijaykumar G, Laun K, Diemant T, Zebger I, et al. In-liquid plasma modified nickel foam: NiOOH/NiFeOOH active site multiplication for electrocatalytic alcohol, aldehyde, and water oxidation. Adv Energy Mater. 2022;12(38):2202098.[DOI]
-
82. Goetz MK, Bender MT, Choi KS. Predictive control of selective secondary alcohol oxidation of glycerol on NiOOH. Nat Commun. 2022;13(1):5848.[DOI]
-
83. Yu H, Zhang L, Jiang S, Liu W, Deng K, Wang Z, et al. Dual hydrogen production system: synergistic effect of Ru and Ce over Cu2O nanotubes drives hydrogen evolution and formaldehyde oxidation. Small. 2024;20(47):2406107.
-
84. Yang M, Jiang Y, Dong CL, Xu L, Huang Y, Leng S, et al. A self-reactivated PdCu catalyst for aldehyde electro-oxidation with anodic hydrogen production. Nat Commun. 2024;15(1):9852.[DOI]
-
85. Bu J, Chang S, Li J, Yang S, Ma W, Liu Z, et al. Highly selective electrocatalytic alkynol semi-hydrogenation for continuous production of alkenols. Nat Commun. 2023;14(1):1533.[DOI]
-
86. Zhang Q, Zhang G, Guan S, Wang J, Li K, Wang C, et al. N-CoFeP/NF electrocatalyst for coupling hydrogen production and oxidation reaction of various alcohols. J Colloid Interface Sci. 2024;662:686-694.[DOI]
-
87. Si F, Liu J, Zhang Y, Zhao B, Liang Y, Wu X, et al. Surface spin enhanced high stable NiCo2S4 for energy-saving production of H2 from water/methanol coelectrolysis at high current density. Small. 2023;19(2):2205257.[DOI]
-
88. Pang Q, Feng C, Fan X, Sun K, Xiang K, Dong L, et al. The identification of dual active routes for highly efficient furfural oxidation reaction coupling with hydrogen production over Re modulated Co(OH)2/CC. Chem Eng J. 2023;469:143995.[DOI]
-
89. Wang H, Niu C, Liu W, Tao S. d-Electron tuned CoMoP for enhance 5-hydroxymethylfurfural oxidation and HER. Appl Catal B Environ. 2024;340:123249.[DOI]
-
90. Shi J, He H, Guo Y, Ji F, Li J, Zhang Y, et al. Enabling high-efficiency ethanol oxidation on NiFe-LDH via deprotonation promotion and absorption inhibition. J Energy Chem. 2023;85:76-82.[DOI]
-
91. Chen W, Shi J, Wu Y, Jiang Y, Huang YC, Zhou W, et al. Vacancy-induced catalytic mechanism for alcohol electrooxidation on nickel-based electrocatalyst. Angew Chem Int Ed. 2024;63(4):e202316449.[DOI]
-
92. Tian C, Li XY, Nelson VE, Ou P, Zhou D, Chen Y, et al. Paired electrosynthesis of H2 and acetic acid at A/cm2 current densities. ACS Energy Lett. 2023;8(10):4096-4103.[DOI]
-
93. Wu Y, Wang T, Liu C, Qin G, Li S. Building synergy between Cu and Ag for efficient hydrogen production from formaldehyde reforming. Appl Surf Sci. 2023;639:158201.[DOI]
-
94. Li T, Wang Q, Wu J, Sui Y, Tang P, Liu H, et al. Strain and shell thickness engineering in Pd3Pb@ Pt bifunctional electrocatalyst for ethanol upgrading coupled with hydrogen production. Small. 2024;20(7):2306178.
-
95. Qian Q, He X, Li Z, Chen Y, Feng Y, Cheng M, et al. Electrochemical biomass upgrading coupled with hydrogen production under industrial-level current density. Adv Mater. 2023;35(25):2300935.[DOI]
-
96. Chen L, Ren JT, Yuan ZY. Enabling internal electric fields to enhance energy and environmental catalysis. Adv Energy Mater. 2023;13(11):2203720.[DOI]
-
97. Ren JT, Chen L, Wang HY, Yuan ZY. High-entropy alloys in electrocatalysis: from fundamentals to applications. Chem Soc Rev. 2023;52(23):8319-8373.[DOI]
-
98. Wu J, Liu X, Hao Y, Wang S, Wang R, Du W, et al. Ligand hybridization for electro-reforming waste glycerol into isolable oxalate and hydrogen. Angew Chem Int Ed. 2023;62(9):e202216083.[DOI]
-
99. Yu H, Wang W, Mao Q, Deng K, Wang Z, Xu Y, et al. Pt single atom captured by oxygen vacancy-rich NiCo layered double hydroxides for coupling hydrogen evolution with selective oxidation of glycerol to formate. Appl Cata B Environ. 2023;330:122617.[DOI]
-
100. Zhang L, Wei H, Yuan R, Li P, Ren JT. Interface engineering of transition-metal-based electrocatalysts for alkaline water splitting. Coord Chem Rev. 2025;545:217009.[DOI]
-
101. Mao P, Chen B, Huang R, Jing Y, Xiao L, Zhang B, et al. Modulating silver performance in electrocatalytic oxidation of HCHO via SMSI between Ag-Co3O4 interfaces. Small. 2024;20(48):2405358.[DOI]
-
102. Huang B, Yan J, Li Z, Chen L, Shi J. Anode‐electrolyte interfacial acidity regulation enhances electrocatalytic performances of alcohol oxidations. Angew Chem Int Ed. 2024;63(40):e202409419.[DOI]
-
103. Chen L, Ren JT, Yuan ZY. Design strategies of phosphorus-containing catalysts for photocatalytic, photoelectrochemical and electrocatalytic water splitting. Green Chem. 2022;24(2):713-747.[DOI]
-
104. Ren JT, Chen L, Wang HY, Feng Y, Yuan ZY. Hydrogen oxidation electrocatalysts for anion-exchange membrane fuel cells: activity descriptors, stability regulation, and perspectives. Energy Environ Sci. 2024;17(12):3960-4009.[DOI]
-
105. Chen W, Shi J, Xie C, Zhou W, Xu L, Li Y, et al. Unraveling the electrophilic oxygen-mediated mechanism for alcohol electrooxidation on NiO. Natl Sci Rev. 2023;10(5):nwad099.[DOI]
-
106. Pan Y, Li Y, Dong CL, Huang YC, Wu J, Shi J, et al. Unveiling the synergistic effect of multi-valence Cu species to promote formaldehyde oxidation for anodic hydrogen production. Chem. 2023;9(4):963-977.[DOI]
-
107. van Der Ham MP, van Keulen E, Koper MT, Tashvigh AA, Bitter JH. Steering the selectivity of electrocatalytic glucose oxidation by the Pt oxidation state. Angew Chem Int Ed. 2023;62(33):e202306701.[DOI]
-
108. Wang D, Wang P, Wang S, Chen YH, Zhang H, Lei A. Direct electrochemical oxidation of alcohols with hydrogen evolution in continuous-flow reactor. Nat Commun. 2019;10(1):2796.[DOI]
-
109. Zhang R, Gao F, Yang C, Bian Y, Wang G, Xue K, et al. Boosting hydrogen evolution via anodic oxidation of 5-hydroxymethylfurfural in anion exchange membrane electrolyzer over a metallic heterostructure. Mater Today Nano. 2023;23:100373.[DOI]
-
110. Zhang Z, Leng BL, Zhang SN, Xu D, Li QY, Lin X, et al. Electrocatalytic aromatic alcohols splitting to aldehydes and H2 gas. J Am Chem Soc. 2024;146(39):27179-27185.[DOI]
-
111. Wu Y, Zhu X, Du S, Huang G, Zhou B, Lu Y, et al. Promoted hydrogen and acetaldehyde production from alcohol dehydrogenation enabled by electrochemical hydrogen pumps. Proc Natl Acad Sci U.S.A. 2023;120(27):e2300625120.[DOI]
-
112. Zhou H, Ren Y, Yao B, Li Z, Xu M, Ma L, et al. Scalable electrosynthesis of commodity chemicals from biomass by suppressing non-Faradaic transformations. Nat Commun. 2023;14(1):5621.[DOI]
-
113. Ren Y, Kong W, Li Y, Zhan W, Zhang C, Miao Y, et al. Selective electrooxidation of 5-hydroxymethylfurfural at pilot scale by engineering a solid polymer electrolyte reactor. Nat Catal. 2025;8:771-783.[DOI]
Copyright
© The Author(s) 2025. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Publisher’s Note
Science Exploration remains a neutral stance on jurisdictional claims in published
maps
and institutional affiliations. The views expressed in this article are solely those
of
the author(s) and do not reflect the opinions of the Editors or the publisher.
Share And Cite



Science Exploration Style
Chen L, Yuan ZY. Electrocatalytic alcohol and aldehyde oxidation: advances in catalysts and reaction mechanisms for sustainable chemical synthesis. Smart Mater Devices. 2026;2:202531. https://doi.org/10.70401/smd.2025.0017
Tips
Copy completed.
Submit a Manuscript
Author Instructions
Cite this Article
Article Metrics
0
View
0
Download
Article Updates

- Abstract
- Keywords
- 1. Introduction
- 2. History and Research Status of AOR
- 3. Reaction Mechanisms
- 4. Catalyst Design for AOR
- 5. Reactor Innovation
- 6. Conclusion and Perspective
- Authors contribution
- Conflicts of interest
- Ethical approval
- Consent to participate
- Consent for publication
- Availability of data and materials
- Funding
- Copyright
Science Exploration Style
Chen L, Yuan ZY. Electrocatalytic alcohol and aldehyde oxidation: advances in catalysts and reaction mechanisms for sustainable chemical synthesis. Smart Mater Devices. 2026;2:202531. https://doi.org/10.70401/smd.2025.0017
copy