The application of attention mechanisms in biological sequence analysis
*Correspondence to:
Wenzheng Bao, School of Information Engineering (School of Big Data), Xuzhou University of Technology, Xuzhou 221018, Jiangsu, China.
E-mail: baowz55555@126.com
Comput Biomed. 2026;1:202602. 10.70401/cbm.2026.0014
Received: January 19, 2026Accepted: May 13, 2026Published: May 15, 2026
Abstract
In recent years, attention mechanisms have gained widespread application and significant advancements in the field of biological sequence analysis. This paper systematically summarizes the fundamental principles of attention mechanisms and their latest research progress in biological sequence analysis. First, the development history of attention mechanisms is introduced, with a focus on classic mechanisms such as self-attention, cross-attention, and multi-head attention, along with their improved variants. Next, a brief overview of the classification and characteristics of bioinformatics databases is provided. Subsequently, the application of attention mechanisms in the analysis of DNA, RNA, and protein sequences is highlighted. In the realm of DNA sequence analysis, attention mechanisms have been applied to tasks such as epigenetic analysis and regulatory element identification; in RNA sequence analysis, they play a crucial role in single-cell RNA sequencing, RNA function prediction, and structure prediction; in protein sequence analysis, attention mechanisms are widely used in protein classification, function prediction, structure prediction, site prediction, and interaction prediction. Furthermore, this paper summarizes the applications of attention mechanisms in other biological sequence analysis tasks, such as multi-omics analysis and enzyme analysis. The attention mechanisms can significantly improve the accuracy, interpretability, and computational efficiency of biological sequence analysis, providing powerful computational tools for bioinformatics research.
Keywords
Attention mechanism, biological sequence analysis, DNA sequence, RNA sequence, protein sequence, bioinformatics
1. Introduction
The attention mechanism originally stems from research on the human visual cognitive system. Vaswani et al. introduced the Transformer architecture in their paper “Attention Is All You Need”, bringing the attention mechanism to new heights[1]. Traditional attention mechanisms mainly include two forms: self-attention and cross-attention. The self-attention mechanism captures the internal dependencies of a sequence by calculating the relevance between elements within the sequence. The core idea is to map the input sequence into three matrices: Query, Key, and Value. The weight of the Value is determined by the similarity between the Query and Key.
To further enhance the model’s expressive power, the Transformer introduces the multi-head attention mechanism. Multi-head attention projects the input into multiple subspaces, computing attention independently in each subspace and finally concatenating the outputs from the various heads. To address the issues of high computational complexity and large memory usage in traditional attention mechanisms, researchers have proposed various improved attention variants. Numerous experiments have demonstrated that the self-attention mechanism can capture long-distance dependencies, aiding in the modeling of global features in input information[2]. Cross-attention mechanisms establish relationships between different feature representations, facilitating the fusion of features across levels and types. For instance, cross-attention can connect features between encoders and decoders, addressing semantic gaps and other challenges[3]. In contrast to single-scale attention, which can only process and analyze input data at a single feature scale, multi-scale attention (MSA), which can construct and utilize features at multiple different scales, is better suited for handling complex tasks[4]. However, such attention mechanisms do not typically exist in isolation. Such methods are often constructed together with other attention mechanisms.
The global attention mechanism has seen significant development and innovation in recent years. Researchers have proposed various improved global attention mechanisms tailored for different application scenarios, effectively addressing the limitations of traditional attention mechanisms in terms of computational efficiency and performance. For instance, the multi-scale linear attention mechanism proposed by Cai et al. aims to tackle the computational cost and hardware efficiency issues in high-resolution dense prediction tasks[5]. In the field of remote sensing image analysis, Yang et al. developed AMMUNet, which introduces an innovative attention mechanism. Its core components include the granular multi-head self-attention (GMSA) module and the attention map merging mechanism (AMMM)[6]. GMSA efficiently captures global information through dimension correspondence strategies, while AMMM merges MSA maps using fixed mask templates. The super token attention (STA) mechanism proposed by Huang et al. improves visual transformers from another perspective[7]. This mechanism has achieved excellent results in multiple computer vision tasks, particularly reaching an 86.4% top-1 accuracy rate in the ImageNet-1K image classification task. The Table 1 demonstrated the comparison of Basic Attention Mechanisms and Their Main Variants.
Table 1. Comparison of basic attention mechanisms and their main variants.
| Mechanism Type | Core Components | Attention Scope |
| Self-Attention | Three weight matrices: Q, K, V | All positions in the sequence |
| Multi-Head Attention | Parallel computation of multiple self-attention layers (“heads”), each focusing on a different subspace | All positions in the sequence |
| Global Attention | Similar to self-attention, but emphasizes global information capture | Computes correlation strength between all positions, considering global context |
| Local Attention | Limits attention computation scope, typically through window partitioning | Specific parts of the sequence |
Q: query; K: key; V: value.
The multi-scale dilated attention proposed by Jiao et al. represents a breakthrough in local attention mechanisms[8]. In the audio domain, the Asynchronous Hybrid Attention Module and Multi-Scale Window Attention Module developed by Li et al. provide new solutions for audio question answering tasks[9]. The varying window attention mechanism proposed by Yan et al. has made important contributions in the field of semantic segmentation[10].
The combination of local and global attention is an important research direction for enhancing model performance, and recent studies have made significant progress in this area. The HiLo attention mechanism proposed by Pan et al. provides an efficient solution for the practical application of visual Transformers[11]. Wang et al. proposed innovative solutions for single image super-resolution tasks, including multi-scale large kernel attention and gated spatial attention units[12].
The development of sparse attention mechanisms aims to address the issues of heavy computational burden and high memory usage associated with traditional attention mechanisms. Recent research has proposed two representative improvement schemes: bidirectional routing attention (BRA) and single-head self-attention (SHSA) mechanisms. Among them, the BRA mechanism proposed by Zhu et al. employs a dual-layer structural design, optimizing computational efficiency through a “coarse-to-fine” cascading filtering strategy[13]. The SHSA mechanism proposed by Yun and Ro focuses on addressing the computational redundancy in multi-head attention mechanisms[14].
When it comes to the bioinformatics with attention methods, several typical methods can be listed in this field. For instance, the g-Inspector model applies the attention mechanism to investigate the significance of each region[15]. The IMAGO model employs the Transformer pre-training process, integrates multi-head attention mechanisms and regularization techniques, and optimizes the loss function to effectively reduce overfitting and noise issues during training[16]. Zhang et al. proposed the Topological-aware Residue-Gene Ontology Attention Network for protein function prediction[17]. The EGA-Ploc model employed a linear attention mechanism optimized for high-resolution IHC images[18]. The Hierarchical and Dynamic Graph Attention Network has been proposed to predict drug-disease associations[19]. Yella and Jegga proposed a multi-view graph attention network for indication discovery (MGATRx)[20]. Xu et al. proposed a Multistructure graph classification method with Attention mechanism and convolutional neural network (CNN), called MAC[21]. Sun et al. proposed SADLN: A self-attention based deep learning network of integrating multi-omics data for cancer subtype recognition[22]. Zhou et al. proposed a graph neural network framework combining graph convolutional network, Transformer with cross-attention, and multi-layer perceptron classifier, called GTCM, to improve the accuracy of identifying cancer driver genes[23]. Wu et al. proposed a framework that unites specificity-aware GATs and cross-modal attention to integrate different omics data (MOSGAT)[24]. Liu et al. proposed Collaborative Attention Contrast Learning framework, which integrates a genetic attention module to capture key intra-omics features and an omics attention module to enhance inter-omics relationships and optimizes the collaborative attention models through the strategic utilization of a contrastive loss function[25]. Moon and Lee proposed a novel multi-task attention learning algorithm for multi-omics data, termed MOMA, which captures important biological processes for high diagnostic performance and interpretability[26]. Wang et al. proposed a novel approach termed ML-FGAT. The approach begins by extracting node information of proteins from sequence data, physical-chemical properties, evolutionary insights, and structural details[27]. Yan et al. proposed the AttentionSplice model, a hybrid construction combined with multi-head self-attention, CNN, bidirectional long short-term memory network[28]. The HybProm model uses DNA2Vec to transform DNA sequences into low-dimensional vectors, followed by a CNN-BiLSTM-Attention architecture to extract features and predict promoters across species, including E. coli, humans, mice, and plants[29]. Aleb defined a new mutual attention-based model for DTBA prediction. We represent both compounds and targets by sequences[30]. Han et al. proposed an effective sequence-based approach, called RattnetFold, to identify protein fold types. The basic concept of RattnetFold is to employ a stack CNN with the attention mechanism that acts as a feature extractor to extract fold-specific features from protein residue-residue contact maps[31]. Zheng et al. presented a new graph neural network framework, line graph attention networks (LGAT)[32]. Fan et al. developed a novel architecture for extracting an effective graph representation by introducing structured multi-head self-attention in which the attention mechanism consists of three different forms, i.e., node-focused, layer-focused and graph-focused[33]. Zhang et al. proposed AMter, the first end-to-end model designed for predicting transcriptional terminators, leveraging attention mechanisms[34]. Cai et al. proposed a neural attention mechanism that dynamically assigns differentiated weights to the subbands and the channels of EEG signals to derive discriminative representations for AAD[35]. The Attention-based Adaptive Graph ProbSparse Hybrid Network was proposed[36]. Brain network structures were dynamically optimised using variational inference and the information bottleneck principle, refining the adjacency matrix for improved epilepsy classification. Wang et al. proposed a new model AMDECDA for predicting circRNA-disease association by combining attention mechanism and data ensemble strategy[37]. Yang et al. proposed attention-aware multi-scale fusion network. Our network is with dense convolutions to perceive microscopic capillaries[38].
In the field of biological sequence analysis, several attention methods have been utilized. This work focuses on the application of Attention Mechanisms in Biological Sequence Analysis at three levels, including DNA, RNA, and protein sequences. Figure 1 demonstrated the attention utilization in biological sequences.
2. Biological Databases
In this work, bioinformatics databases are classified into core and specialized databases based on multidimensional metrics, including data volume, update frequency, usage prevalence, and impact factor. This classification framework not only encapsulates the holistic attributes of bioinformatics databases but also fully elucidates the professional value of each database within specific research domains.
Core databases are characterized by large data volumes, high usage frequency, and significant academic influence, with representative examples summarized in Table 2. Such databases typically exhibit the following salient features: (1) an extensive data scale, as exemplified by UniProt, which curates more than 250 million protein sequences, and PubChem, which archives over 119 million chemical compounds; (2) a high-frequency update mechanism, with the majority of core databases implementing weekly or monthly iteration protocols; (3) a broad user community, registering annual access volumes that generally surpass one million visits; (4) a well-integrated toolchain encompassing data submission, curation, and analytical workflows; and (5) sustained institutional support from leading research organizations such as the National Institutes of Health and the European Molecular Biology Laboratory. Collectively, core databases furnish fundamental data infrastructure for interdisciplinary bioinformatics research.
Table 2. Core biological databases.
| Category | Database Name | Main Data Scale/Characteristics |
| Protein Database | UniProt[154] | 572,619 reviewed sequences and more than 250 million unreviewed sequences |
| PDB/RCSB PDB[155] | 229,380 experimental structures and over 1.06 million computational structure models | |
| AlphaFold[156] | Over 200 million protein structure predictions | |
| Genome Database | UCSC Genome Browser[157] | Genome data covering more than 100 species |
| ENCODE[158] | Covers all functional elements of the human genome | |
| Sequence Database | SRA[159] | The world’s largest sequencing database |
| GEO[160] | Over 7.55 million samples and over 240,000 series data | |
| Chemical Database | PubChem[161] | Over 119 million compounds and over 327 million substances |
| Pathway Database | KEGG[162] | Covers all known metabolic and regulatory pathways |
| Biomedical Database | NLM[163] | The world’s largest biomedical library |
| Interaction Database | STRING[164] | Over 500,000 chemical substances and over 9.6 million proteins |
PDB: protein data bank; RCSB: Research Collaboratory for Structural Bioinformatics; UCSC: University of California Santa Cruz; SRA: sequence read archive; GEO: gene expression omnibus; NLM: US National Library of Medicine; STRING: search tool for the retrieval of interacting genes.
In contrast, specialized databases offer in-depth, domain-specific data resources for focused research areas, as shown in Table 3. While these databases may not rival core databases in terms of raw data volume, they possess irreplaceable professional significance within their respective research niches. Their distinctive attributes are as follows: (1) refined and domain-specialized data annotation schemas; (2) well-defined research themes and scenario-specific application scopes; (3) an intimate integration with specialized experimental techniques and research methodologies; and (4) the provision of customized algorithmic support and analytical tools tailored to the demands of domain-specific research endeavors.
Table 3. Specialized biological databases.
| Category | Database Name | Main Features |
| RNA Specialized Databases | Rfam[165] | 4,178 RNA families |
| NONCODE[166] | Focus on non-coding RNA | |
| CircInteractome[167] | Circular RNA interactions | |
| CSCD[168] | 2.9 million+ circular RNAs | |
| miRNA Databases | HMDD[169] | Human microRNA disease data |
| miRcode[169] | MicroRNA target prediction | |
| Epigenetics Databases | MethSMRT[170] | DNA methylation data |
| DNAmod[171] | DNA modified nucleotides | |
| Functional Databases | GO[172] | 40,000+ GO terms, 8 million+ annotations |
| BRENDA[173] | Enzyme information database | |
| AAindex[174] | Amino acid property indicators | |
| Disease Databases | TCGA[175] | 20,000+ cancer sample data |
| Circ2Disease[175] | Circular RNA disease associations | |
| Interaction Specialized Databases | HVIDB[176] | 48,000+ human-virus interactions |
| PiSITE[177] | Protein interaction sites | |
| NPInter[178] | Non-coding RNA interactions | |
| Structural Specialized Databases | SAbDab[179] | Antibody structure data |
| PDBbind[180] | Molecular binding data | |
| DUD-E[181] | Molecular docking benchmark data | |
| Biochemical Reaction Databases | Rhea[182] | More than 17,000 reaction data |
| SABIO-RK[183] | Biochemical reaction kinetics | |
| RxnFinder[184] | Biochemical reaction search engine | |
| Other Specialized Databases | SEdb 2.0[185] | More than 1.71 million super enhancers |
| Phospho.ELM[186] | More than 42,000 phosphorylation sites | |
| Greengenes2[187] | Microbial classification data | |
| BioSNAP[188] | Biomedical network data | |
| MetaCyc[189] | 3,153 metabolic pathways | |
| SilencerDB[190] | More than 33,000 silencer sequences |
CSCD: cancer-specific circRNA database; HMDD: human microRNA disease database; GO: gene ontology; TCGA: The Cancer Genome Atlas; HVIDB: human–virus interaction dataBase; PDB: protein data bank; DUD-E: directory of useful decoys: enhanced.
3. Application of Attention Mechanism in Biological Sequence Analysis
In recent years, the attention mechanism has made significant progress in the field of biological sequence analysis. This chapter will detail the main applications of the Attention mechanism in the analysis of DNA, RNA, and protein sequences.
3.1 DNA sequence analysis
3.1.1 DNA epigenetics analysis
DNA methylation, as a key epigenetic modification process, includes various forms such as 4-methylcytosine (4mC), 5-methylcytosine, and 6-methyladenine (6mA), and plays an important role in biological processes such as gene expression regulation, cell differentiation, and environmental adaptation. In recent years, with the development of deep learning technologies, researchers have developed various innovative computational models to enhance the accuracy and efficiency of DNA methylation analysis.
These studies mainly focus on the following aspects: first, in the field of nanopore sequencing data analysis, the PoreFormer model developed by Dai et al. utilizes the attention mechanism of the Transformer framework, capable of directly processing raw current signals and extracting hierarchical features, shown in Figure 2, demonstrating excellent performance in the recognition of multiple types of methylation. Secondly, in DNA methylation prediction, the DeepPGD model proposed by Teragawa et al. integrates temporal convolutional networks, BiLSTM, and the attention mechanism, achieving significant results on datasets from multiple species[39].
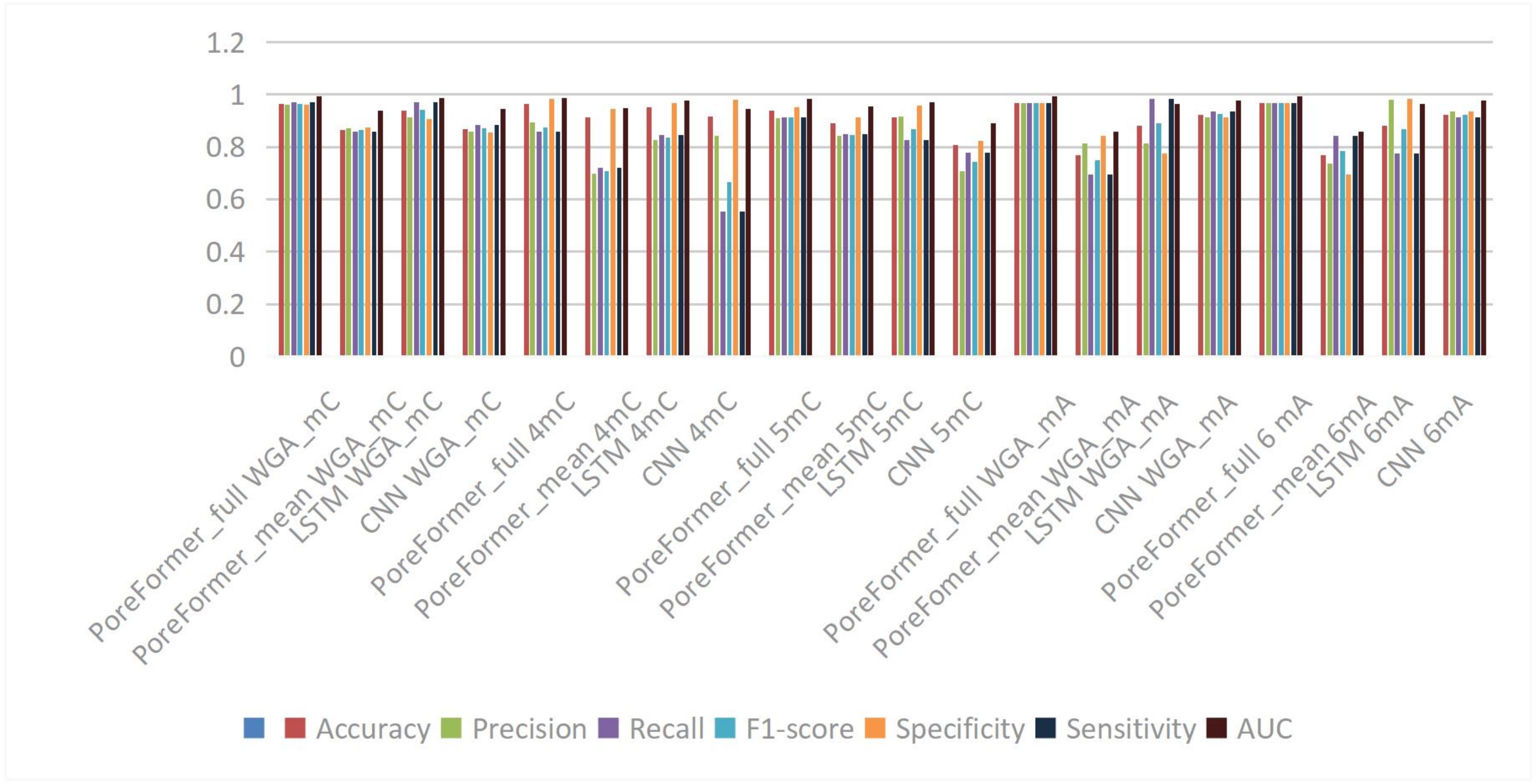
In the identification of specific methylation sites, the LA6mA and AL6mA models developed by Zhang et al. address the prediction problem of 6mA sites by combining Bi-LSTM and self-attention mechanisms, achieving high prediction accuracy[40]. In the field of single-cell methylome analysis, the CpG Transformer model proposed by De Waele et al. innovatively combines axial attention and sliding window self-attention mechanisms, effectively solving the data missing problem in single-cell DNA methylation sequencing[41].
Additionally, in the prediction research of extrachromosomal circular DNA, Chang et al. adopted an improved BERT model (DNABERT), achieving high prediction accuracy through self-attention mechanisms[42]. In the prediction of 4mC sites, Yu et al. integrated the attention mechanism into a CNN-RNN hybrid model, achieving significant improvements in prediction tasks across multiple species[43].
These research efforts fully demonstrate the broad application value of the attention mechanism in DNA methylation analysis, providing powerful computational tools and methodological support for DNA epigenetics research. The development of these models not only enhances the accuracy of methylation analysis but also offers new research ideas for understanding gene regulation mechanisms and disease occurrence mechanisms.
3.1.2 DNA regulatory element recognition
Transcription is the initial step of gene expression, and transcription factors regulate the transcription process by binding to specific DNA sequences. Accurately predicting transcription factor binding sites (TFBSs) is crucial for identifying genomic features, understanding sequence expression programs, and addressing issues in molecular biology and medicine. In recent years, with the development of deep learning technologies, attention mechanisms have shown significant advantages in the field of DNA regulatory element recognition.
In the prediction of TFBSs, researchers have proposed various innovative attention mechanisms. The species attention mechanism proposed by Lim et al. effectively integrates sequence data with evolutionary information by assigning attention weights to different species[44]. The DeepGRN tool developed by Chen et al. employs single attention and paired attention modules, enhancing the ability of deep neural networks to capture long-range dependencies[45]. The funnel-structured attention mechanism proposed by Han et al. improves prediction performance by adjusting feature weights through bottom-up and top-down approaches[46]. Wang et al. combined self-attention mechanisms with Fourier transform-enhanced multi-head attention in the TBCA framework, significantly improving the prediction accuracy of the model[46]. The Figure 3 shown in the performances of different methods.
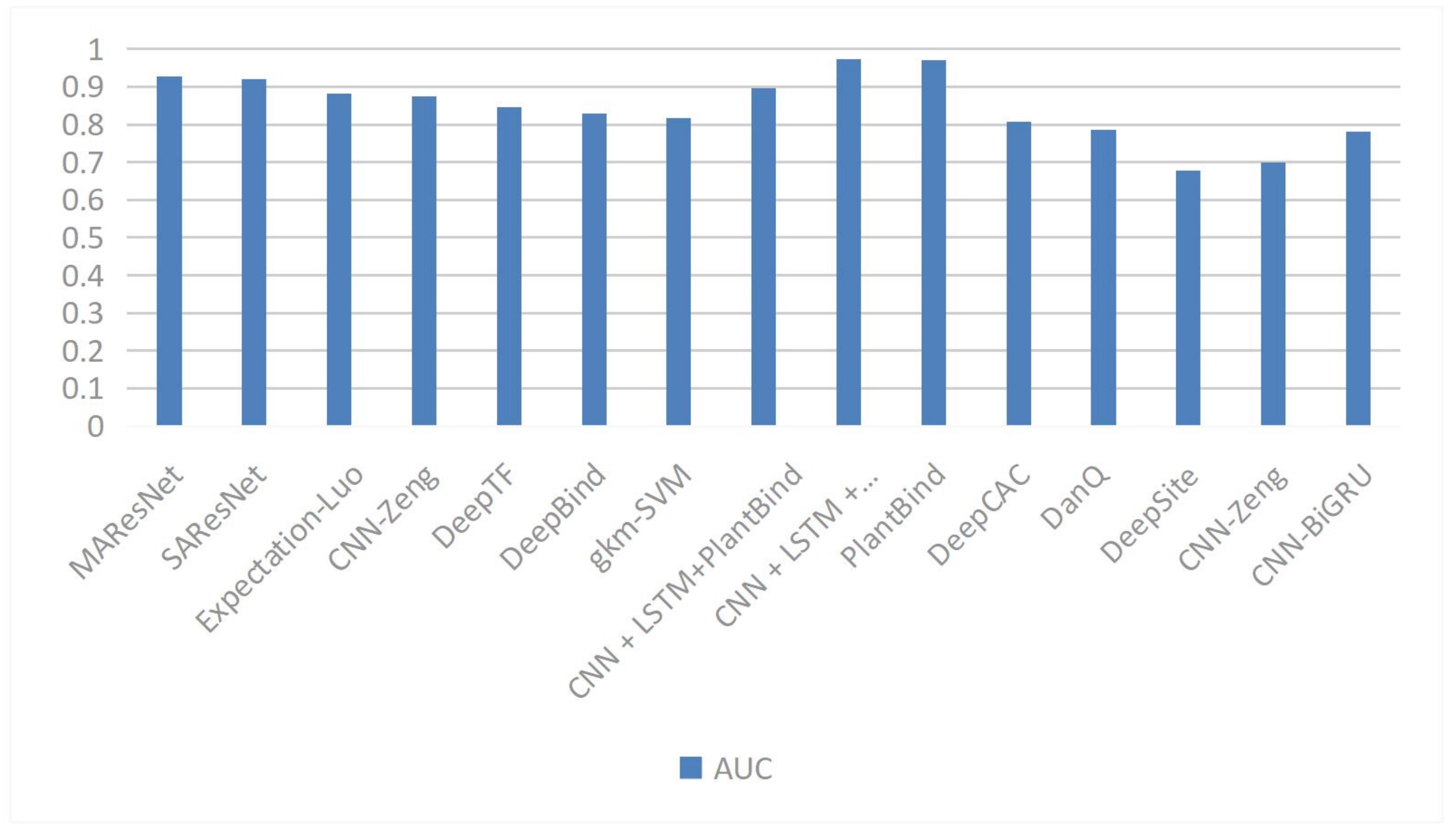
Figure 3. The AUC of different methods in TFBSs. AUC: area under the curve; TFBSs: transcription factor binding sites.
The PlantBind model developed by Yan et al. utilizes attention mechanisms to allow the model to focus on key regions of DNA sequences while predicting multiple TFBSs and identifying their binding motifs[47]. The MHSA mechanism proposed by Zhang et al. can simultaneously capture both local and long-distance dependency features, enhancing prediction performance while reducing model parameters[48]. The convolutional block attention module (CBAM) designed by Wang et al. enhances feature extraction capabilities through channel and spatial attention operations[49]. The STA mechanism proposed by Zhang et al. decomposes global attention into a multiplication operation of sparse association graphs and low-dimensional attention, effectively capturing spatial dependencies in DNA shape data[50]. Yu et al. applied self-attention mechanisms in the DSAC dual-branch network to achieve weighted modeling of features at different positions within sequences, significantly improving prediction accuracy[51].
In terms of model architecture innovation, multi-head attention mechanisms have been widely applied in various models. The PCLAtt model by Bhukya et al. and the DeepSTF model by Ding et al. both demonstrate the advantages of multi-head attention mechanisms in enhancing prediction performance[52,53]. The channel attention mechanism proposed by Zhang et al. focuses on refining high-order features within sequence and shape characteristics, improving the representational power and interpretability of the model[54].
Attention mechanisms also show promising application prospects in other DNA regulatory element recognition tasks. In the field of promoter recognition, Zhu et al. combined attention mechanisms with Siamese neural networks, improving the prediction accuracy of bacterial promoters[55]. In gene regulation modeling, the GraphReg method proposed by Karbalayghareh et al. successfully integrates remote regulatory element information using GAT[56]. In enhancer and silencer recognition, the ADH-Enhancer framework by Mehmood et al. and the DeepICSH framework by Zhang et al. respectively apply attention mechanisms to enhance prediction performance[57-59].
3.1.3 Other applications of attention mechanisms in DNA sequence analysis
Significant progress has been made in the application of attention mechanisms in areas such as DNA functional annotation, sequence embedding and edit distance, DNA storage, and sequence reconstruction.
In the field of DNA functional annotation, researchers have developed several innovative models. Yang et al. proposed the MIXBend model, which predicts DNA bending ability through attention layers in both sequence encoders and physicochemical encoders, effectively identifying important motifs in the genome[58]. Li et al. introduced a category-attention neural network, which improves the accuracy of predicting non-coding regulatory functions of DNA and performs exceptionally well in identifying the functional effects of DNA sequences[59]. In the area of DNA sequence embedding and edit distance, the Needleman-Wunsch Attention framework proposed by Lee and No significantly enhances the ability to capture edit distances between sequences by combining the NW matrix with attention maps[60]. The Flow Attention mechanism included in DeepFormer developed by Yao et al. not only reduces computational complexity from O(n2) to O(n) based on the conservation laws of flow networks but also effectively captures long-range interactions among chromatin features.
In terms of DNA storage and sequence reconstruction, researchers have proposed targeted solutions: Qin et al. utilized attention mechanisms to score the quality of DNA sequences, effectively addressing contamination issues in multi-sequence reconstruction; the DNA storage coding system proposed by Cao et al. combines graph convolutional networks (GCN) with self-attention mechanisms to improve the efficiency and speed of DNA storage coding while enhancing storage density without compromising code quality[61,62].
3.2 RNA sequence analysis
3.2.1 Single-cell RNA sequencing (scRNA-seq)
scRNA-seq is a key technological tool for studying cellular heterogeneity and developmental processes. Compared to traditional bulk RNA sequencing, it offers higher resolution and sensitivity, allowing for precise descriptions of cellular states within tissues and organs, thereby transforming the landscape of transcriptomics research. However, scRNA-seq data generally exhibit characteristics such as high noise and sparsity, with a significant number of zero values present in gene expression profiles, posing substantial challenges for data analysis. The main steps of scRNA-seq are demonstrated in Figure 4.
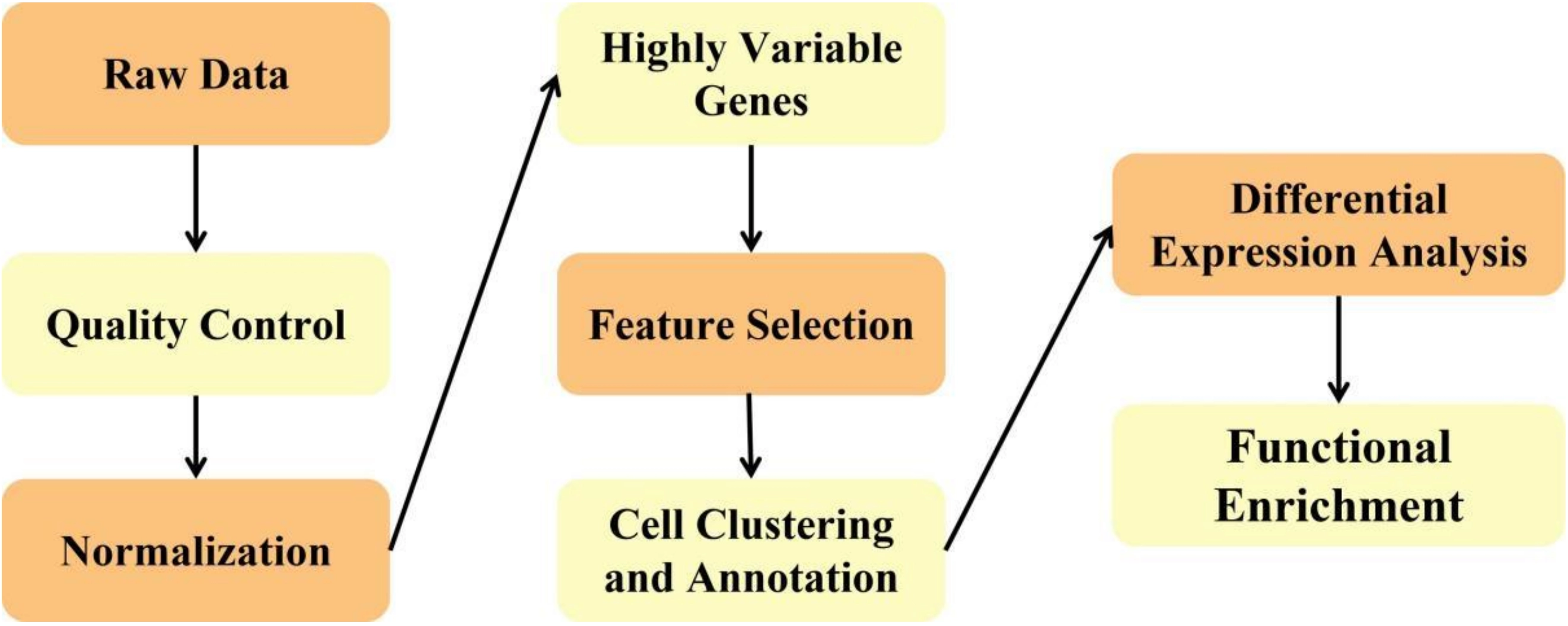
In recent years, researchers have proposed various innovative methods based on attention mechanisms to address these challenges. In the field of cell clustering analysis, the MSA mechanism proposed by Tan et al. effectively enhances model performance by integrating feature information from different layers of a multi-scale autoencoder[63]. The AGAC model developed by Zhou et al. employs a mixed attention mechanism to achieve flexible feature aggregation, demonstrating excellent performance across multiple real datasets[64]. scVGATAE (Single-cell Variational Graph Attention Autoencoder) method proposed by Liu et al. significantly improves clustering accuracy through a variational graph attention autoencoder[65]. Dayu Hu’s scDFC model enhances the accuracy of structural information in cell clustering using a graph attention network[66]. Xiang Feng’s scDGAE model, based on directed graph neural networks, shows exceptional generalization capability in addressing gene imputation and cell clustering issues[67].
In the area of gene regulatory network (GRN) inference, the view-level attention mechanism introduced by Yuan et al. dynamically adjusts the relative importance of node embedding representations, thereby improving the accuracy of GRN inference[68]. The GATCL model developed by Liu et al. captures the relationships between transcription factors and genes more accurately using graph attention networks, achieving high AUROC values across multiple datasets, shown in Figure 5[69]. The method proposed by Lin et al. further enhances network inference performance through multi-view contrastive learning[70].
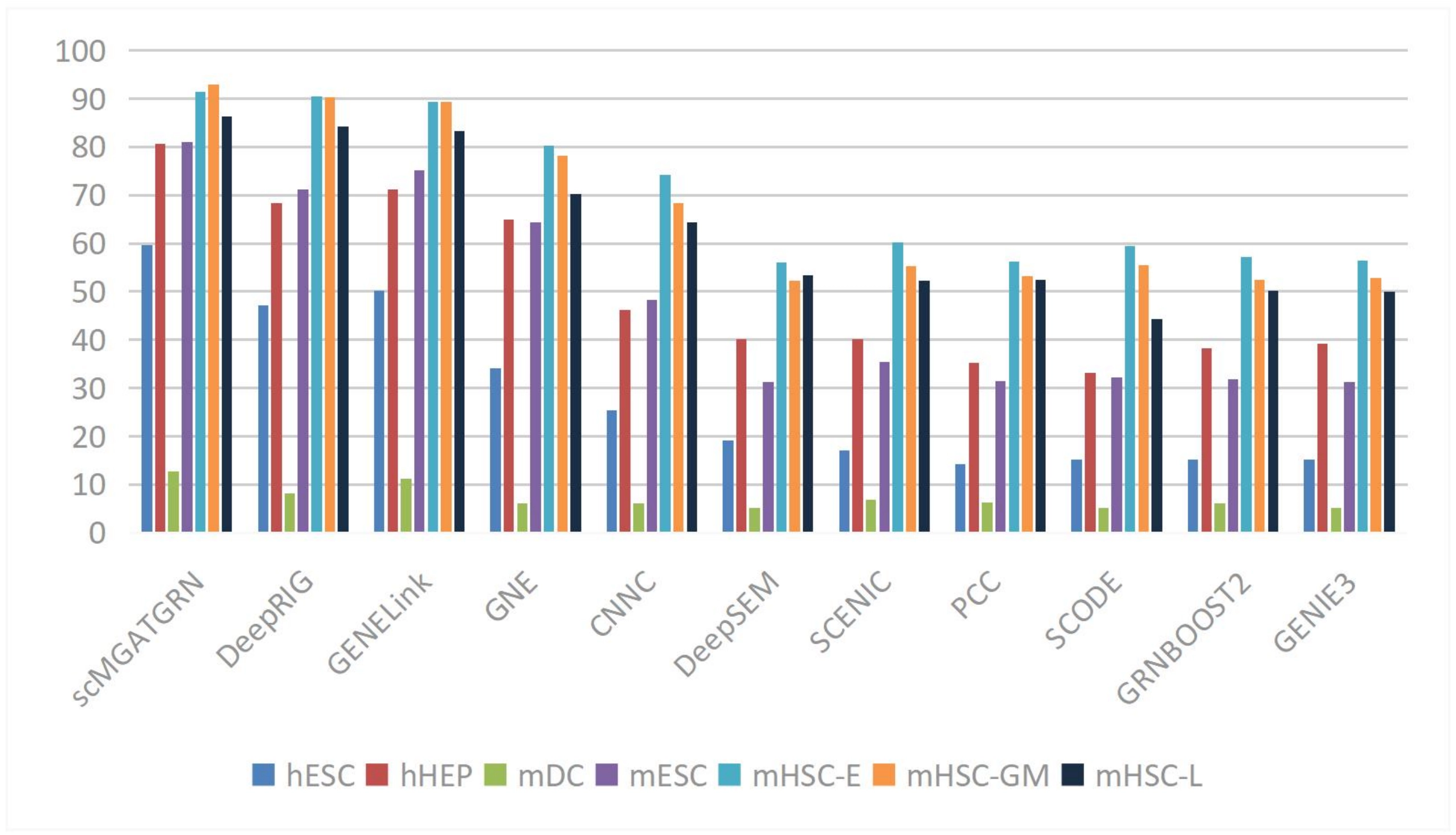
Figure 5. Average AUPRC of the benchmark scRNA-seq datasets with cell-type-specific networks. Reproduced from[70]. CC BY 4.0. AUPRC: area under the precision-recall curve; scRNA-seq: single-cell RNA sequencing.
In the field of spatial transcriptomics analysis, the STGAT method proposed by Li et al. significantly improves the deconvolution accuracy of spatial transcriptomic data using graph attention networks[71]. The GTAD method developed by Zhang et al. based on GAT, demonstrates outstanding performance in inferring the spatial composition of cells, accurately revealing information about cell types, hierarchical structures, and spatial distributions[72].
Additionally, in data integration and dimensionality reduction, the multi-view graph convolutional network model proposed by Sun et al. enhances the accuracy and stability of cell type identification through a multi-view graph convolutional network[73]. The scCompressSA method developed by Zhang et al. based on SA mechanisms, employs a dual-channel self-attention mechanism to improve feature extraction while maintaining computational efficiency[74].
3.2.2 RNA sequence classification
Teragawa and Wang proposed the cross multi-head attention mechanism[75]. This mechanism is used to address the issue of predicting non-coding RNA (ncRNA) families. Its role is to facilitate cross-interaction between two different feature sets, allowing each feature to consider information from other feature sets, thereby better capturing the interactions and correlations between features. The advantage lies in its ability to better capture long-distance dependencies and local patterns at different positions within sequences when handling tasks like ncRNA family classification, enhancing the model’s expressive and modeling capabilities, allowing a more comprehensive understanding of sequence features to improve classification accuracy, and maintaining consistent performance when dealing with ncRNA sequences of varying lengths. Through experimental comparisons, the ConF model using this mechanism outperformed other benchmark algorithms in accuracy, sensitivity, precision, and F1-score and other evaluation metrics, shown in Figure 6.
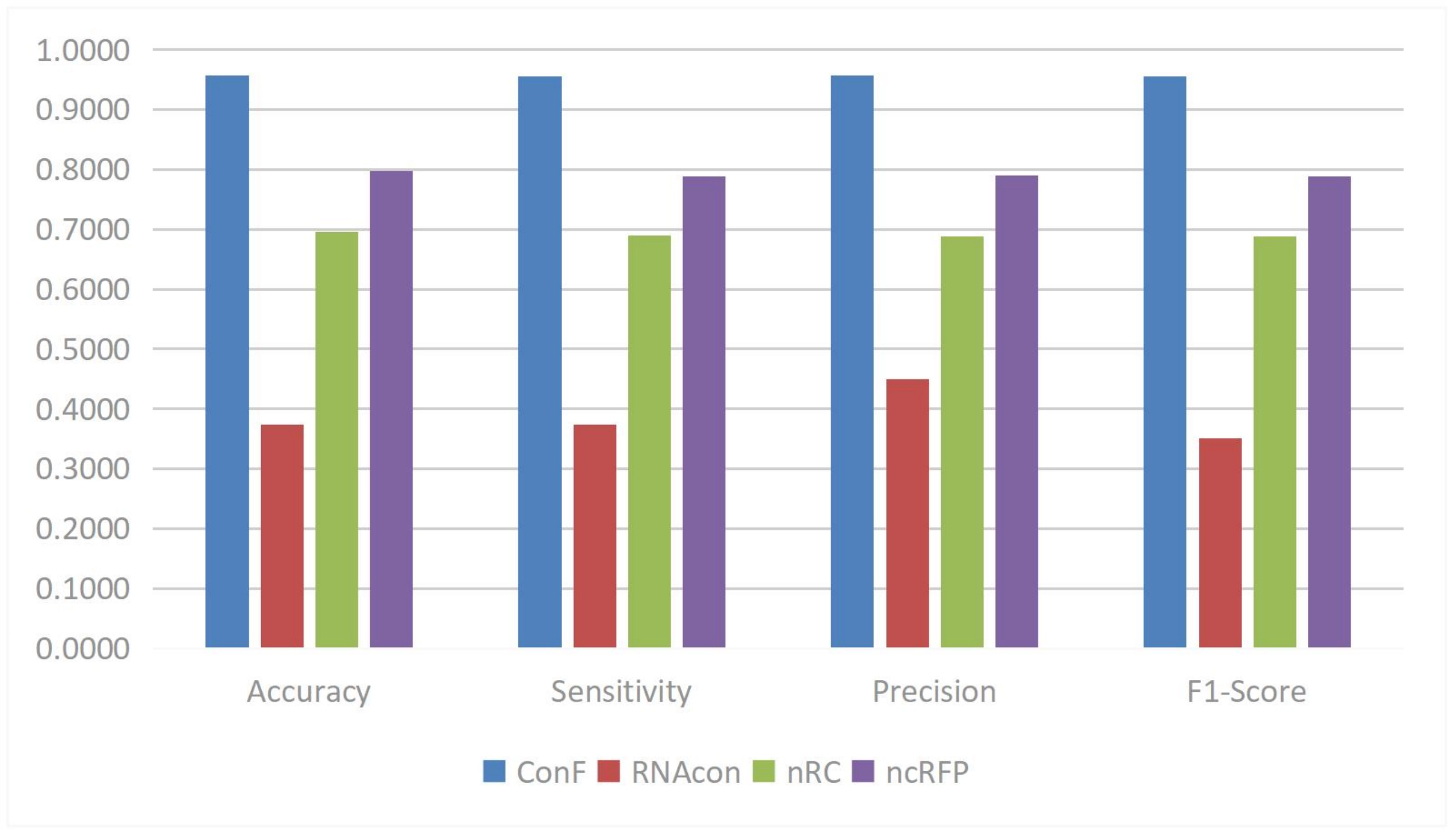
Figure 6. The performances of SOTA RNA Sequence Classification model. Reproduced from[76]. CC BY 4.0. SOTA: state-of-the-art.
3.2.3 RNA epigenetics analysis
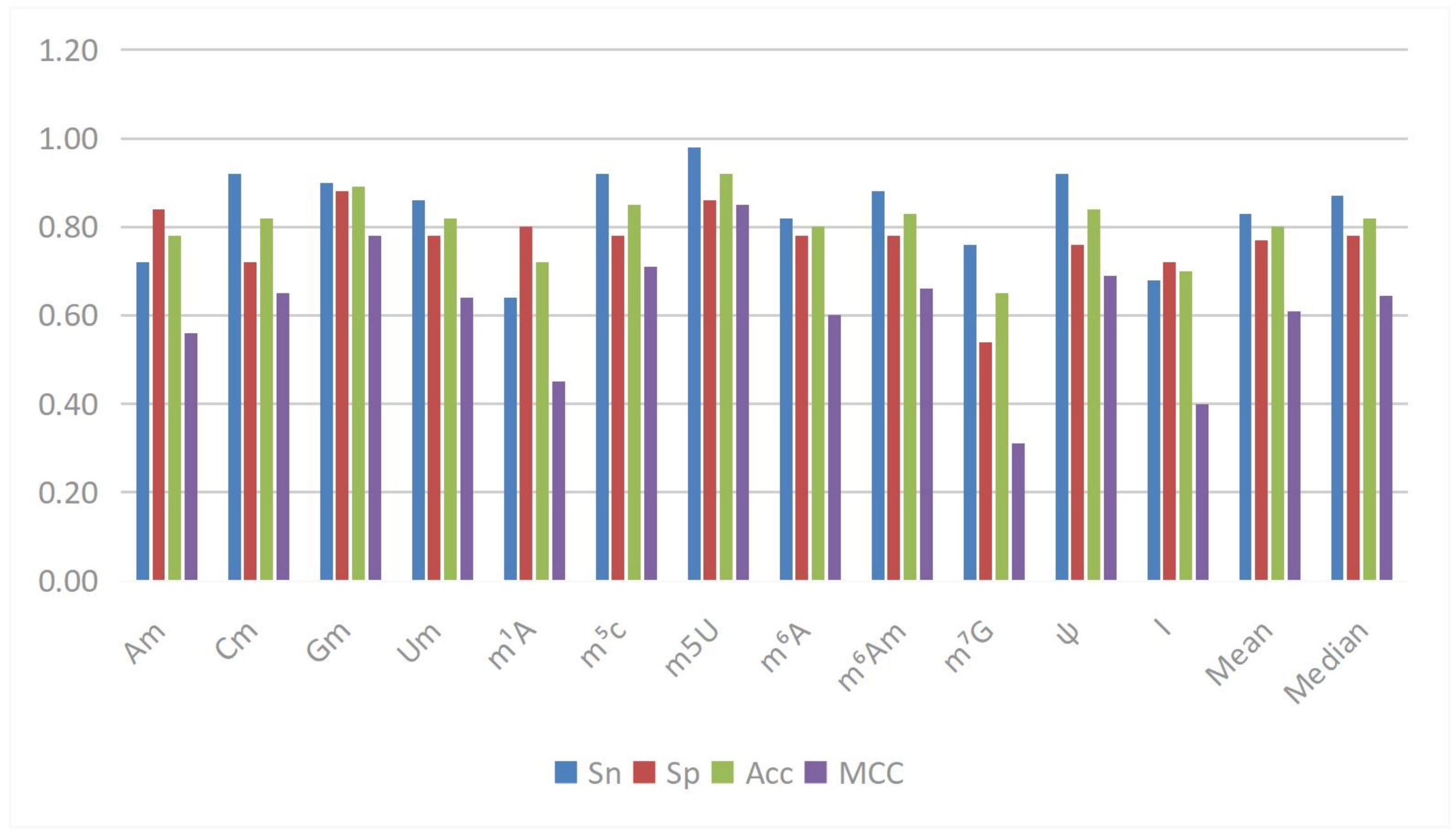
In RNA epigenetics research, Yuan et al. proposed the DPNN-N4-acetylcytidine (ac4C) model[76], which is based on the MHSA mechanism and is used to identify ac4C sites in mRNA. This mechanism employs multiple parallel “heads” to focus on different subspaces of the input sequence, independently computing attention weights and integrating the results, thereby simultaneously capturing various patterns and contextual information in the sequence data. Its advantage lies in its ability to globally process sequence data, directly connecting any two elements in the sequence while retaining local features and enhancing global contextual information, ultimately improving model performance. Experimental results indicate that this mechanism effectively increases the model’s accuracy, specificity, Matthews correlation coefficient (MCC), and area under the curve (AUC) when identifying ac4C sites. Wang et al. proposed the MSCAN model[77], which employs multi-scale self-attention and cross-attention mechanisms to address the prediction problem of RNA methylation sites. Traditional methods such as BiLSTM have limitations in parallel computation and training time, CNNs struggle to effectively learn long-distance sequence dependencies, and Transformers lack the ability to interact across different scale sequence information. MSCAN effectively extracts deep features of sequences with 41 base pairs at different scales and captures the relationships between sequences of different scales. This model outperformed existing predictors in the prediction of 12 modified sites, demonstrating strong generalization capability and revealing significant associations between different types of RNA modifications within the relevant sequence context through cross-modification validation experiments. Additionally, Liang et al. proposed the Rm-LR model[77], based on the Bilinear Attention Network, to improve the accuracy and interpretability of RNA modification predictions. This model computes a paired interaction matrix through a bilinear interaction layer to capture interactions between different patterns in the input sequence, and calculates context vectors based on task importance, allowing the model to focus on key parts of the input sentences. In the prediction of various types of RNA modifications, Rm-LR significantly outperformed traditional classifiers, achieving the highest average accuracy, which can reach 0.9113, and demonstrated good local and global sequence information capture abilities in ablation experiments, enhancing the model’s interpretability in conjunction with visualized attention weights. Song et al. proposed the MultiRM model based on attention mechanisms, aimed at addressing the prediction and interpretation of RNA modification sites[78]. This model employs the Bahdanau attention mechanism, enabling the model to focus on relevant RNA sequence regions according to different modification types, thereby achieving simultaneous predictions for multiple RNA modifications (such as m6A, m1A, and twelve others) and returning key sequence content that contributes most to positive predictions. MultiRM significantly improved prediction performance when processing 51-bp sequence input, aiding in the understanding of potential associations between different RNA modifications and identifying strong association regions that may play key roles in gene regulation, shown in Figure 7.
Cao et al. proposed a Pre-norm based attention mechanism for the circRNA-RBP recognition task[79]. Its purpose is to avoid the risk of gradient vanishing or explosion brought by deep networks, allowing for more stable gradient updates during network training. The effects and advantages include: after adopting this mechanism, the CircSSNN model demonstrated good parallelism, stability, and transferability in multiple experiments across 37 circRNA datasets and 31 linRNA datasets. It effectively captures sequential semantics and global context to generate discriminative features, exhibiting overall advantages in predictive performance, stability, and parallelism, while providing good recognition performance, scalability, and a wide application range without the need for parameter fine-tuning for different tasks.
3.2.4 RNA structure prediction
In the field of RNA secondary structure prediction, an innovative approach was introduced utilizing a Transformer Encoder structure with self-attention mechanisms, which is inspired by successful applications in natural language processing (e.g., BERT model). Unlike traditional RNA prediction algorithms that rely on specific bioinformatics models or simple neural network structures, this cross-domain model fusion provides a novel perspective for RNA structure prediction[80]. By leveraging the self-attention mechanism of the Transformer Encoder, it effectively captures long-distance dependencies in RNA sequences, a characteristic that is particularly crucial in RNA structure prediction. Traditional models often face limitations when handling long-range interactions, whereas the introduction of the Transformer Encoder significantly enhances the modeling capability of these long-distance relationships. Additionally, Zhao et al. proposed the attention mechanism-based deep learning model Read2Pheno, which combines CNNs and recurrent neural networks to effectively predict classification and sample-related attributes[81]. This model automatically identifies key nucleotide regions, avoiding the cumbersome preprocessing and manual interpretation required by traditional microbiome sequence classification methods, thereby achieving read-level phenotypic prediction. Experimental results show that Read2Pheno performs comparably to traditional methods across various datasets, while its results offer greater interpretability, providing deeper biological insights. The KnotFold method proposed by Gong et al. demonstrated superiority in predicting RNA secondary structures[82]. This method considers long-range interactions in RNA sequences when predicting base pairing probabilities through attention mechanisms, rather than solely focusing on pairs of bases. This innovation better captures long-distance interactions and non-nested base pairs, significantly enhancing the ability to identify pseudoknot structures. Evaluation results on the PKTest dataset indicate that KnotFold’s average F1 score surpasses other existing methods, demonstrating its potential in RNA secondary structure prediction.
3.2.5 RNA-RNA interactions
In the study of RNA-RNA interactions, various innovative models have emerged in recent years, utilizing advanced attention mechanisms to enhance the predictive capability of interactions between long non-coding RNAs (lncRNAs) and microRNAs (miRNAs). The MGCAT (Multi-view Graph Neural Network with Cascaded Attention) model proposed by Li et al. employs a cascaded attention mechanism that encompasses view-level, node-level, and layer-level attention[83]. This mechanism aims to address the complexity in the prediction of lncRNA-miRNA interactions (LMI). View-level attention dynamically integrates multi-view features to effectively capture the inherent properties of nodes; node-level attention enhances the accuracy of node features by iteratively aggregating neighborhood information; and layer-level attention adaptively integrates features to make node representations more effective. Extensive experiments across four benchmark datasets demonstrate that MGCAT excels in LMI prediction, showing potential for discovering novel interactions.
The CFHAN model proposed by He et al. combines node-level attention and semantic-level attention mechanisms to tackle the issues of low semantics and noise in predicting interactions between plant lncRNAs and miRNAs[84]. Node-level attention updates feature representations by calculating attention scores between the target node and its neighboring nodes, thereby enhancing the understanding of connection importance; meanwhile, semantic-level attention aggregates different meta-path embeddings of similar nodes by learning the semantic relationships between meta-paths and the target node. These mechanisms not only improve the model’s robustness against graph noise but also enable CFHAN to perform exceptionally well in predicting plant LMI, achieving an average AUC of 0.9953 and an average ACC of 0.9733, demonstrating strong cross-species predictive capability.
Additionally, the model based on multi-head attention mechanisms proposed by Yang et al. focuses on identifying piRNA-targeted mRNA sites in Caenorhabditis elegans[85]. This model mimics the target recognition process of biological piRNAs to extract hidden binding rules. It achieved an AUC of 93.3% on an independent test set, improving by 6.7% compared to a pure CNN model. This achievement not only demonstrates outstanding recognition performance but also provides in-depth insights into the preferences of intracellular piRNA binding, further advancing the study of piRNA functional mechanisms.
3.2.6 RNA-protein interactions
In the field of RNA-protein interactions, the application of attention mechanisms has led to a breakthrough improvement in the performance of predictive models. In recent years, researchers have proposed various innovative attention mechanism models to address critical issues such as insufficient prediction accuracy, difficulty in capturing long-distance dependencies, and data sparsity.
For example, Wang et al. introduced the CBAM, effectively addressing challenges related to insufficient sample data and model instability[86]. This method enhances accuracy by 1.84% through adaptive learning to output key feature information from different channels and local regions, significantly boosting the model’s robustness and predictive ability.
In predicting circRNA-RBP binding sites, the CRIECNN model proposed by Lasantha et al. introduced a self-attention mechanism that autonomously captures key local and global contextual information from attribute descriptors[87]. By adaptively prioritizing important information while disregarding less significant parts, it significantly improves prediction accuracy. Relatedly, the iCRBP-LKHA model developed by Lin Yuan and others employs a hybrid channel-space attention mechanism (CBAM-1D) to effectively address the issue of traditional CNNs or LSTMs being unable to capture long-distance dependencies in processing long circRNA nucleotide sequence data[88].
In feature extraction and fusion, the MAN proposed by Du et al. innovatively applies attention mechanisms to fuse features from different scale branches, demonstrating excellent performance in motif prediction tasks by automatically highlighting useful features while suppressing unhelpful ones[89]. Wei Lan and others applied graph attention mechanisms to circRNA-disease association prediction, effectively mitigating data sparsity issues by distinguishing the importance of entity neighbor information, enabling the model to capture high-order neighbor information[90], shown in Figure 8.
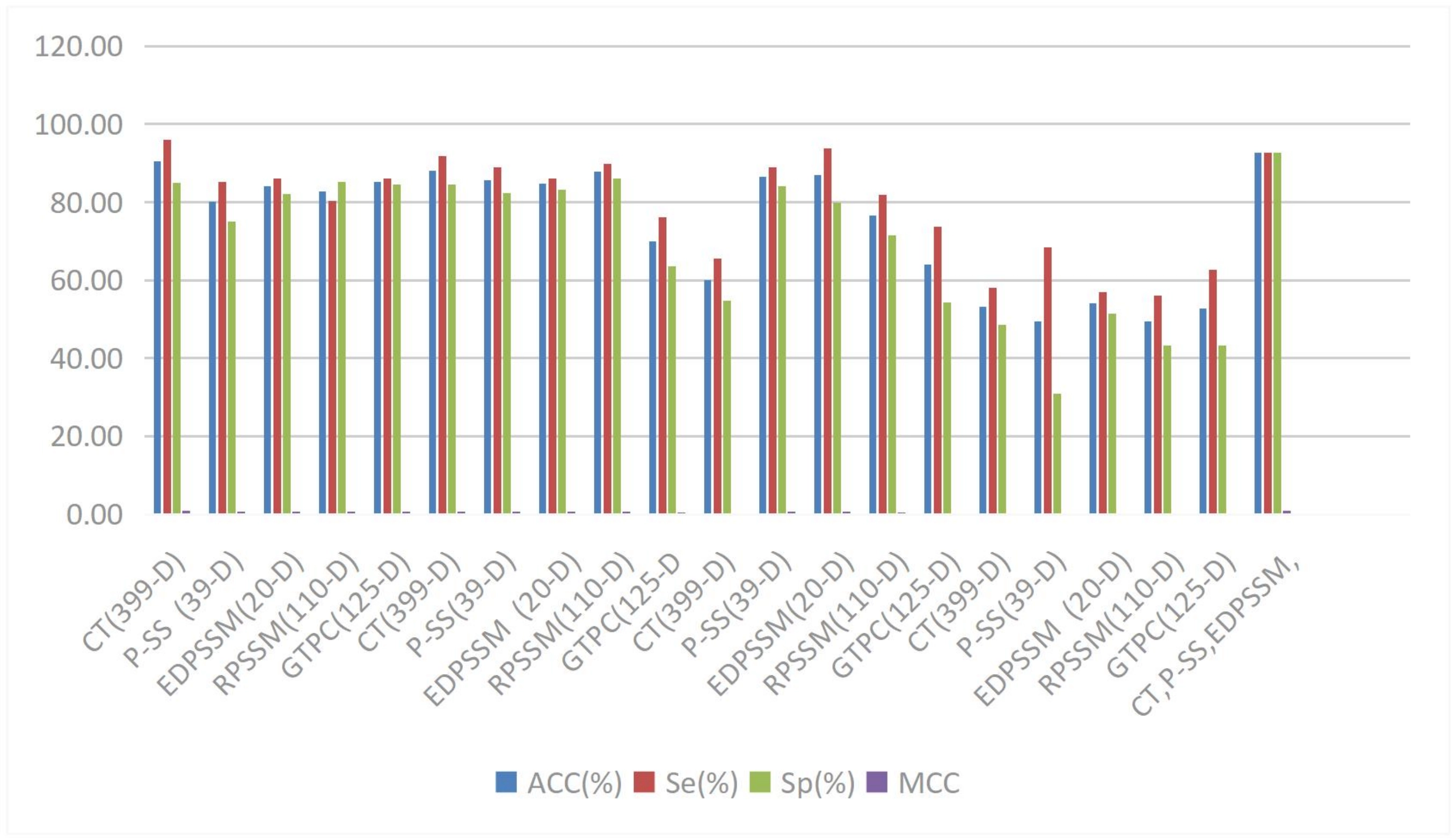
Figure 8. The comparison of prediction results with different feature extraction algorithms on dataset Rpi488. Reproduced with permission from reference[86]. Copyright © 2023 Elsevier.
In terms of model interpretability, the ncRPI-LGAT model developed by Yong Han and Shao-Wu Zhang employs a multi-head graph attention network (GATv2), which not only improves the prediction accuracy of ncRNA-protein interactions but also provides interpretability of model decisions through analysis of attention weight distributions[91]. The DeCban model proposed by Liangliang Yuan and Yang Yang introduces a self-attention mechanism that can automatically integrate semantic information across different branches and abstraction levels, demonstrating excellent performance and computational efficiency when processing full-length circRNA sequence predictions[92]. In the plant field, the GPLPI model developed by Jael Sanyanda Wekesa and others utilizes graph attention mechanisms to improve the accuracy of predicting plant lncRNA-protein interactions by optimizing the objective function[93].
3.2.7 Other research associated with RNA sequence analysis
In other studies related to RNA sequence analysis, the attention mechanism has demonstrated extensive application value and significant performance advantages. These studies cover several important fields, each with its unique innovations and breakthrough progress.
Firstly, important breakthroughs have been achieved in the field of disease association prediction. The GraphCDA framework developed by Dai et al. efficiently learns feature representations for circRNAs and diseases by innovatively employing GAT[94]. This framework adopts a MHSA mechanism, assigning differentiated weights to different nodes in the neighborhood, achieving remarkable results across multiple public databases, with AUROC and AUPR values reaching 0.9882 and 0.9895, respectively. At the same time, Zhao et al. innovatively combined LSTM with heterogeneous context graph attention networks in their research on lncRNA-disease association prediction, significantly enhancing prediction accuracy by effectively capturing the sequential structural information in meta-path instances[95]. Significant progress has also been made in spatial transcriptomics research. The PAST framework proposed by Li et al. utilizes a self-attention mechanism to effectively capture local spatial patterns, allowing for adaptive learning of attention weights and aggregating information from adjacent points in the same domain, which significantly improves clustering effects and annotation accuracy[96]. Yang et al. successfully addressed the inference of intercellular communication at single-cell resolution by applying graph attention networks, achieving precise analysis of cellular behavior in complex biological processes[97].
In the field of RNA solvent accessibility prediction, the gated local attention mechanism proposed by Huang et al. enhances prediction performance through feature rearrangement[98]. Moreover, the attention mechanism also plays an important role in other RNA-related research: the DeepLDA model developed by Meihong Gao and Xuequn Shang predicts lncRNA-drug resistance associations using graph attention mechanisms[99]. The HATZFS framework by Hong et al. effectively identifies pancreatic cancer driver biomarkers through GAT[100]. Asim et al. apply attention mechanisms to enhance model interpretability in RNA subcellular localization prediction[101]. Jiang et al. optimize prediction outcomes in circRNA prediction by combining various attention mechanisms[102].
3.3 Protein sequence analysis
3.3.1 Protein classification
In the field of protein classification research, several research teams have employed various types of attention mechanisms to enhance model performance. Zhang et al. used a Transformer encoder in SiameseCPP to learn peptide sequence information[103], effectively capturing local and global features of sequences through MHSA blocks and feedforward networks. Wang et al. proposed the ESMSec framework[104], which employs a multi-head attention mechanism to predict secretory proteins in human body fluids, achieving an average accuracy of 0.8390 across tests in plasma, cerebrospinal fluid, and semen, demonstrating excellent predictive performance. Zhao et al. introduced a self-attention mechanism in DeepTP to address the problem of predicting thermally stable proteins[105], extracting key information through weight allocation, and optimizing feature extraction capabilities by combining CNN and BiLSTM modules.
The CFAGO model developed by Wu et al. employs a multi-head attention mechanism, achieving superior results across multiple evaluation metrics on human and mouse datasets by cross-fusing protein-protein interaction (PPI) networks and protein biological attribute information[106]. Lin et al. proposed a dual-scale attention mechanism that implements attention distribution on spatial and positional scales in the DAttProt model[107], showcasing strong competitiveness and interpretability in enzyme protein classification prediction tasks, shown in Figure 9.
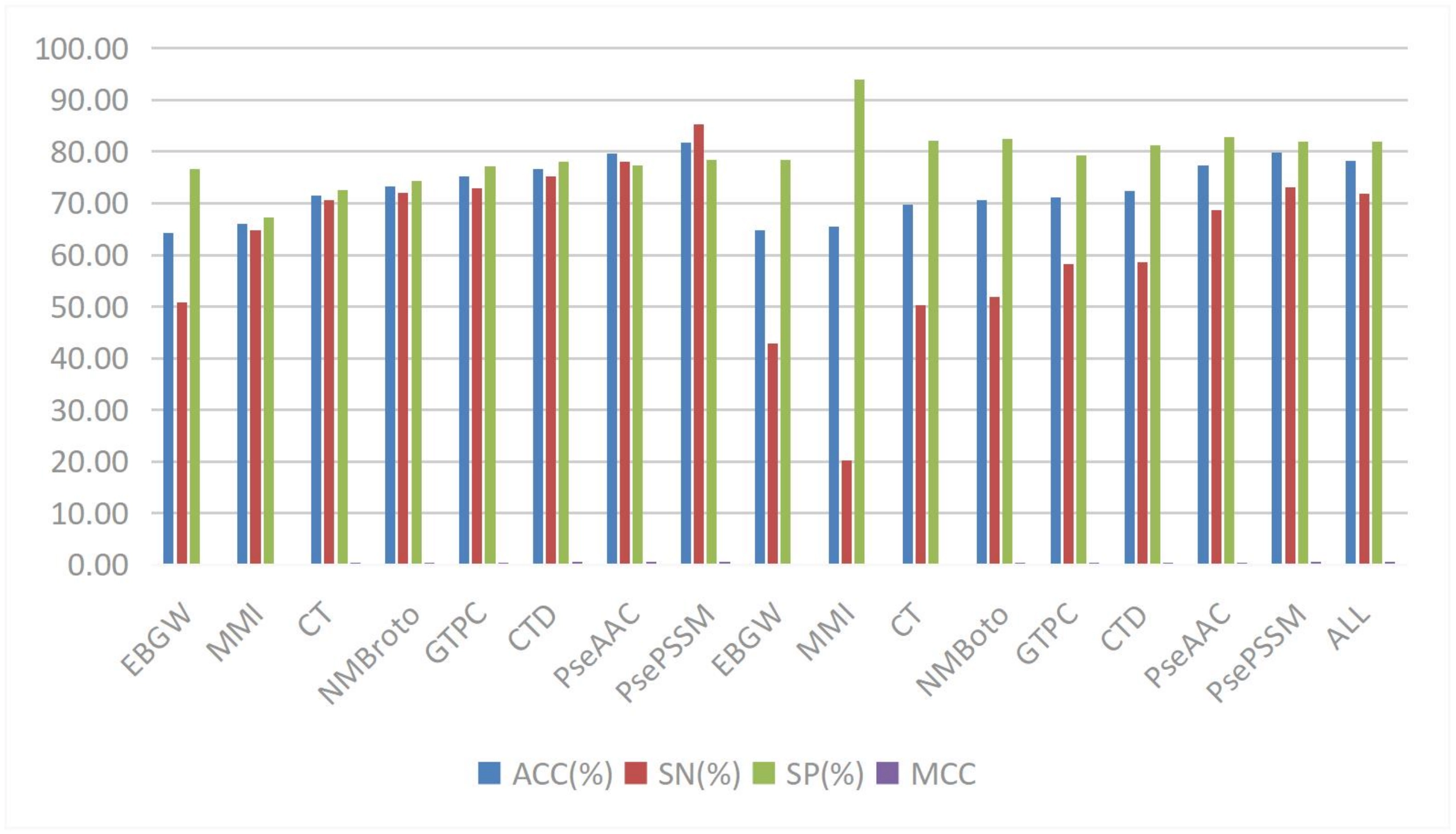
Figure 9. The different methods for RBPs on training dataset. Reproduced from[147]. CC BY 4.0. RBPs: RNA-binding proteins.
In the field of DNA and RNA binding protein prediction, the DRBPPred-GAT model proposed by Zhang et al. integrates a graph multi-head attention network, optimizing the feature extraction of inter-node dependencies by assigning differentiated weights to different neighboring nodes[108], and demonstrating excellent predictive accuracy and stability in 10-fold cross-validation. These research findings indicate that attention mechanisms play an important role in protein classification tasks, effectively enhancing the model’s feature extraction capabilities and predictive accuracy, providing new technical directions and solutions for protein classification research.
3.3.2 Protein function prediction
In recent years, significant progress has been made in protein function prediction methods based on attention mechanisms.
In the area of peptide toxicity prediction, the ATSE attention mechanism proposed by Yu et al. can learn distinguishing features from graph structures and evolutionary information, achieving superior predictive results through the integration of the Toxicity-vib model[109]. In the field of protein function prediction, the attention mechanism-based graph neural network HNetGO model developed by Zhang et al. utilizes attention mechanisms to learn embedding vectors of nodes in heterogeneous networks, achieving leading levels in cellular components and molecular function branches on human datasets[110]. For specific function prediction, the category attention mechanism applied in the RecGOBD model by Xia et al. is particularly suitable for predicting functions of brain development-related proteins, outperforming baseline models in multiple metrics including AUROC, AUPR, and Fmax[111].
In terms of protein function annotation, the PFresGO model developed by Pan et al. employs a dual multi-head attention mechanism that not only captures the correlations between gene ontology (GO) terms but also identifies key functional regions, performing excellently in predictions across all GO categories[112]. Additionally, the GAT-based method proposed by Boqiao Lai and Jinbo Xu integrates protein sequence representations and predicted residue contact maps[112], achieving accurate function predictions even when the similarity between test proteins and training proteins is low.
3.3.3 Protein structure prediction
In the field of protein structure prediction, the application of attention mechanisms has made significant progress. Firstly, the multi-head attention module in the Transformer network developed by Google’s DeepMind demonstrated excellent performance in the CASP14 experiment, effectively capturing the correlations between amino acid (AA) residues across different dimensions[113]. The DCMA model developed by Zhang et al. utilizes the multi-head attention mechanism to predict the dihedral angles of protein backbone torsion[114]. This model not only captures long-range dependencies but also offers advantages such as lightweight design and short training times.
In the area of secondary structure prediction, the improved channel attention (ICA) mechanism proposed by Jin et al. enhances the number of parameters through increased convolution operations, better expressing the importance of each channel in PSSP[115]. The attention mechanism employed in the TLCrys model by Jin et al. simultaneously focuses on global and local features, significantly improving the accuracy of protein crystallization predictions[116]. This model uses dilated convolution to expand the receptive field, with the computation parameters increasing only linearly with sequence length.
In the domain boundary prediction field, the method developed by Mahmud et al. based on the multi-head attention mechanism integrates long-range information from 1D and 2D inputs, resulting in a 29.56% improvement in F1 score for multi-domain target prediction in CASP14, and a 13.28% increase in classification accuracy for distinguishing single-domain from multi-domain proteins[117]. Additionally, the GLTM model proposed by Ma et al. is applied to single-channel membrane protein dimer motif localization, achieving an accuracy of 97.5% on benchmark datasets by reallocating weights of local subsequences[118].
In summary, attention mechanisms demonstrate unique advantages in the field of protein structure prediction: they effectively handle sequence data, capture long-range dependencies, integrate multi-scale features, and exhibit high computational efficiency and good generalization performance. These advances provide new ideas and methods for protein structure prediction research.
3.3.4 Protein site prediction
In recent years, attention mechanisms have been widely applied in the field of protein site prediction, with researchers proposing various innovative prediction models. In the prediction of carbonylation sites, the Carsite_AGan model proposed by Chen et al. employs an attention-based dual rebalancing strategy using generative adversarial networks, effectively addressing the issue of class imbalance in the data[119]. In the field of phosphorylation site prediction, the Attenphos method developed by Song et al. utilizes a self-attention mechanism to successfully tackle long-distance dependency issues, significantly enhancing prediction accuracy[120].
In the prediction of protein-peptide binding sites, the GAPS (Geometric Attention Network for Peptide Binding Site Recognition) model proposed by Zhu et al. integrates geometric features and multiple attention mechanisms[121], showcasing excellent robustness and versatility through a transfer learning strategy. The ULDNA model developed by Zhu et al. applies an LSTM-attention network to predict DNA binding sites[122], improving prediction accuracy by reinforcing the association between evolutionary diversity features and residue-level DNA binding patterns. Zheng et al. improved the gated multi-head attention mechanism in the EGPDI model[123], effectively addressing the issue of information redundancy in multi-view graph embedding fusion.
In the prediction of protein-protein binding sites, the DeepBindPPI model proposed by Sunny et al. utilizes an attention-based GCN[124], enhancing prediction precision through multi-head attention layers and cross-attention layers. Abdin et al. introduced a MHSA mechanism in the PepNN model[125], effectively addressing the flexibility issues of peptides. The PITHIA model developed by SeyedMohsen Hosseini and Lucian Ilie combines multiple sequence alignments with the Transformer architecture[125], significantly outperforming existing models across multiple evaluation metrics.
3.3.5 PPI
In the field of PPI prediction research, deep learning methods based on attention mechanisms have made significant progress in recent years. Researchers have developed various innovative attention mechanism models to improve the accuracy and efficiency of PPI prediction. The TUnA (Transformer-based Uncertainty Aware Model for PPI prediction) model proposed by Ko et al. employs a MHSA mechanism that effectively captures long-range dependencies in protein sequences and identifies distant residues that interact in three-dimensional structures[126], demonstrating excellent performance on cross-species datasets while significantly reducing computational costs.
In the realm of cross-attention mechanisms, the Pair-EGRET model developed by Alam et al. quantifies the mutual influences between ligand and receptor residues through multi-head cross-attention layers, achieving outstanding results on datasets such as DBD5[127]. Similarly, the cross-attention PHV proposed by Sho Tsukiyama and Hiroyuki Kurata focuses on PPI prediction between humans and viruses, outperforming existing models on multiple evaluation metrics[128]. These cross-attention-based methods provide new research insights for analyzing complex protein interaction networks.
Graph attention mechanisms have also played an important role in PPI prediction. The Graph Attention Network proposed by Baranwal et al., named Struct2Graph[129], can identify residues contributing to protein complex formation in an unsupervised manner. The SN-GGAT developed by Zhijie Xiang’s team innovatively optimizes node feature updates by employing the principle of “low-order high attention[130], high-order low attention”. Meanwhile, GraphPBSP proposed by Sun et al. combines pre-training models, demonstrating excellent performance and good generalization capabilities across multiple test sets[131].
Additionally, the DEAttentionDTA model developed by Chen et al. adopts a multi-head attention mechanism that better captures relationships between different parts of sequences by segmenting feature dimensions and applying attention mechanisms separately[132], achieving significant results in predicting protein-ligand binding affinity. This model has shown superiority across various evaluation metrics when compared with multiple existing tools.
Although these methods based on different attention mechanisms each have their unique characteristics, they have significantly enhanced the accuracy, interpretability, and computational efficiency of PPI predictions. They have not only demonstrated excellent performance on standard datasets but also shown good adaptability and generalization in specific application scenarios (such as virus-host interactions and binding site predictions). These research advancements provide powerful computational tools for a deeper understanding of protein interaction networks and have accelerated rapid development in this field. Overall, attention mechanisms have shown tremendous application potential in the PPI prediction domain, paving new directions for future research.
3.3.6 Drug-target interaction prediction
In recent years, attention mechanisms have made significant progress in the field of drug-target interaction prediction. Various scholars have proposed different types of attention mechanisms to address this issue. The hierarchical multimodal self-attention mechanism proposed by Bian et al. effectively captures the complex interactions between proteins and drugs[133], demonstrating significant advantages across multiple benchmark datasets. Luo et al. introduced a pocket-guided cooperative attention mechanism and a paired multimodal attention mechanism within the DrugLAMP framework[134]. The former uses protein pocket information to guide the attention distribution of drug features, while the latter focuses on maintaining the uniqueness of features, performing excellently in both standard and real-world scenarios.
In terms of drug-target affinity prediction, the dynamic graph attention network (DGDTA) proposed by Zhai et al. overcomes the limitations of traditional graph attention networks by considering the importance of different atoms and edges within the drug graph[135], achieving significant advancements on the Davis and KIBA datasets. Ghimire et al. developed a convolutional model with a self-attention mechanism called CSatDTA[136], which successfully addressed the issue of long-range dependencies by combining convolutional models with self-attention mechanisms, outperforming traditional models on various evaluation metrics.
Additionally, the interpretable cross-attention network proposed by Hiroyuki Kurata and Sho Tsukiyama not only enhances prediction performance but also reveals, for the first time, the statistical significance of the association between experimental binding sites and attention weights[137]. The GraphATT-DTA model designed by Haelee Bae and Hojung Nam constructs new features reflecting the relationship between compound substructures and protein subsequences through attention mechanisms[138], achieving advancements in both performance prediction and interpretability, while providing unique biological insights.
Overall, these research efforts demonstrate the important role of attention mechanisms in drug-target interaction prediction, not only improving prediction accuracy but also enhancing the interpretability of models, thus providing powerful computational tools for drug development.
3.3.7 Proteins and other ligands
In the study of interactions between proteins and other ligands, the application of attention mechanisms has made significant progress. Researchers have proposed various innovative attention mechanism models to address different types of molecular interaction prediction problems.
In the field of drug-target binding prediction, the ArkDTA model proposed by Gim et al. introduces an NCI-aware attention regularization mechanism, which guides the attention distribution through non-covalent interactions, enhancing the interpretability of the model[138]. The distance-aware attention-based affinity prediction method developed by Rahman et al. combines distance features with attention mechanisms, significantly improving prediction accuracy[139]. The adaptive graph attention mechanism (CurvAGN) proposed by Wu et al. effectively addresses the heterogeneity and geometric structure issues in protein-ligand complexes[140]. The hybrid attention CNN designed by Kyro et al. achieves high-accuracy predictions while maintaining low computational costs[141].
In the area of molecular interaction feature recognition, the attention mechanism (BAPA) developed by Seo et al. effectively identifies key descriptors[142]. The cross-attention mechanism (CAPLA) proposed by Jin et al. captures the interaction features between protein binding pockets and ligands[143]. The self-attention mechanism (SAResNet) developed by Shen et al. shows excellent performance in DNA-protein binding prediction[144]. The convolutional attention module adopted by Ning Wang enhances the recognition of DNA and RNA binding residues[145].
In other molecular interaction prediction tasks, the local affinity attention method (FOTF-CPI) proposed by Yin et al. improves the accuracy of compound-protein interaction predictions[146]. Campana et al. combined self-attention and cross-attention[147], achieving good results in metabolite-protein interaction predictions. The cross-TCR-interpreter model developed by Koyama et al. demonstrates outstanding performance and interpretability in T cell receptor-peptide binding predictions[148].
These studies indicate that attention mechanisms play an important role in molecular interaction prediction, not only improving prediction accuracy but also enhancing the interpretability of models, providing powerful tools for drug development and molecular recognition research.
3.3.8 Protein generation
In the field of protein generation, Bo Ni and others proposed an Attention-based Diffusion Model[149], which aims to generate new protein sequences based on secondary structure constraints. This method effectively predicts AA sequences and their three-dimensional protein structures by minimizing the L2 difference between actual and predicted noise levels through a stepwise denoising strategy. The main advantage of this model lies in its ability to generate proteins that conform to expected secondary structures and its robustness towards imperfect or unrealistic design goals. Additionally, this method has the potential to discover new protein sequences that have not been explored in nature and can be extended to generate new proteins with other characteristics. This innovative research provides new ideas and tools for protein design and is expected to promote the development of related fields.
3.3.9 Antibody design and optimization
In the field of antibody research, the application of attention mechanisms has provided an effective solution for important input elements in prediction tasks[150]. This mechanism successfully overcomes the information compression bottleneck faced by traditional models when handling long input sequences by calculating a context vector, which is the weighted average of all intermediate encoder outputs. This approach not only significantly enhances performance, especially in sequence-to-sequence tasks, but also reveals the contextual information of the entire sequence that influences the prediction results. For example, in antibody-related predictions, the attention mechanism enables the model to make effective predictions without being limited to features such as invariant disulfide bonds in antibodies, thereby broadening the application potential of antibody design and optimization.
3.4 Other types of biological sequence analysis
3.4.1 Multi-omics sequence analysis
In current bioinformatics research, multi-omics sequence analysis is gradually becoming an important tool for addressing complex biological problems. Below is a summary of two related studies.
Firstly, the moBRCA-net model proposed by Joung Min Choi and Heejoon Chae aims to address the challenges in breast cancer subtype classification, particularly in the integration of multi-omics data[151]. This model employs an omics-level attention mechanism, which effectively identifies the relative importance of various features in the input data, thus focusing on key areas and enhancing the prediction accuracy of breast cancer subtypes. Validation shows that moBRCA-net significantly outperforms traditional machine learning classifiers across multiple metrics, including average accuracy, F1 score, and MCC. Furthermore, by analyzing attention scores, the model reveals significant differences in feature importance among different subtypes, further enhancing its interpretability and practical value.
Secondly, the STGAT (Spatial Transcriptomics Graph Attention Network) developed by Sudipto Baul and others applies the attention mechanism to the integrated analysis of spatial transcriptomics and large-scale RNA sequencing[151]. The SEG module in this model prioritizes information from neighboring points, enhancing the learning capacity of meaningful connections in the data through neighbor attention coefficients and self-attention coefficients. STGAT demonstrates outstanding performance in gene expression prediction, particularly showing higher correlation and lower mean squared error in tests involving the “breast cancer dataset” and “HER2+ dataset”. Additionally, STGAT exhibits good application effects in distinguishing tumor from non-tumor tissue points, cell type classification, and various downstream analyses of TCGA data.
In summary, these two studies demonstrate the significant applications of attention mechanisms in multi-omics sequence analysis, providing new ideas and tools for precision medicine and personalized treatment.
3.4.2 Analysis of enzymes
In recent years, research on enzyme screening and prediction has provided us with new solutions. Xing et al. proposed a Multi-head Attention Mechanism based on the Transformer architecture, aiming to address the prediction problem of the complex relationship between enzymes and substrate-product pairs. By assigning different weights to each element in the input protein sequence, this mechanism can meticulously extract features of the enzyme and effectively handle the interactions between substrate-product pairs and enzymes. The model SPEPP performed particularly well on the test data, achieving ROC-AUC and PRC-AUC values of 0.993 and 0.994, respectively. Furthermore, this model was also able to accurately identify the majority of instances in zero-shot validation, demonstrating higher accuracy compared to the existing model ESP.
In another study, Wang et al. proposed the DeepEnzyme model[152], which enhances the prediction capability of enzyme turnover number (kcat) by employing a Neural Attention Mechanism. This mechanism captures the attention weights of various residue sites within the enzyme, thereby integrating features extracted from enzymes and substrates more effectively for kcat prediction. With the combination of Transformer and GCN components, DeepEnzyme excelled in predicting kcat values, achieving a coefficient of determination (R2) of approximately 0.6 on the test dataset. Compared to other models such as TurNuP, DLKcat, and DLTKcat, DeepEnzyme significantly improved prediction accuracy and demonstrated strong robustness when handling enzymes with low sequence similarity to those in the training dataset. Additionally, this model has the capability to assess the impact of point mutations on enzyme catalytic activity.
4. Conclusions
In recent years, significant advancements have been made in the field of biological sequence analysis through the use of attention mechanisms. This article systematically summarizes the fundamental principles of attention mechanisms and their applications in the analysis of DNA, RNA, and protein sequences, highlighting the effectiveness of this mechanism and its importance in bioinformatics research.
The attention mechanism originates from visual cognition, and the introduction of classical mechanisms such as self-attention and cross-attention has greatly propelled the development of deep learning models. The introduction of multi-head attention mechanisms enhances the model’s ability to process complex information. In terms of bioinformatics databases, they are categorized into core databases and specialized databases; the former provides extensive data resources, while the latter offers in-depth data support for specific fields.
In DNA sequence analysis, attention mechanisms are widely applied in epigenetic analysis and regulatory element identification, optimizing model performance and prediction accuracy. Moreover, the prediction capability of TFBSs has been improved through various attention mechanisms. In RNA sequence analysis, particularly in the field of scRNA-seq, attention mechanisms have significantly enhanced the processing capabilities of scRNA-seq data, showing exceptional performance in cell clustering and GRN inference. Additionally, various innovative models have demonstrated the advantages of attention mechanisms in recognizing RNA modification sites and structural prediction.
In the analysis of protein sequences, attention mechanisms have substantially improved the accuracy of protein classification and functional prediction, especially in recognizing DNA and RNA binding proteins. Furthermore, attention mechanisms effectively address the complexities of site prediction and protein-protein interactions, enhancing the robustness and interpretability of the models. In drug-target interaction prediction, attention mechanisms exhibit good performance, optimizing prediction accuracy, and model interpretability.
Overall, attention mechanisms provide powerful computational tools for bioinformatics research by enhancing the accuracy, interpretability, and computational efficiency of biological sequence analysis. The widespread application of this mechanism not only advances research progress in the bioinformatics field but also opens up new possibilities for applications in medicine and biotechnology. In the future, as technology continues to develop, attention mechanisms will play an even greater role in solving various biological problems.
Acknowledgements
Grammarly was employed during manuscript preparation, solely for language polishing to improve the fluency of the text. The authors take full responsibility for the integrity, originality, and accuracy of the work.
Authors contribution
Tang Y: Formal analysis, resources, writing-original draft.
Bao W: Writing-review & editing.
Conflicts of interest
The authors declare no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
This work was supported by the National Natural Science Foundation of China (Grant No. 61902337), Xuzhou Science and Technology Plan Project (Grant No. KC21047), Jiangsu Provincial Natural Science Foundation (Grant No. SBK2019040953), Natural Science Fund for Colleges and Universities in Jiangsu Province (Grant No. 19KJB520016).
Copyright
© The Author(s) 2026.
References
-
1. Vaswani A, Shazeer N, Parmar N, Uszkoreit J, Jones L, Gomez AN, et al. Attention is all you need. Adv Neural Inf Process Syst. 2017. Available from: https://proceedings.neurips.cc/paper/2017/hash/3f5ee243547dee91fbd053c1c4a845aa-Abstract.html
-
2. Zhao C, Cai W, Dong C, Hu C. Wavelet-based fourier information interaction with frequency diffusion adjustment for underwater image restoration. In: 2024 IEEE/CVF Conference on Computer Vision and Pattern Recognition (CVPR); 2024 Jun 16-22; Seattle, USA. Piscataway: IEEE; 2024. p. 8281-8291.[DOI]
-
3. Ates GC, Mohan P, Celik E. Dual Cross-Attention for medical image segmentation. Eng Appl Artif Intell. 2023;126:107139.[DOI]
-
4. Shu M, Xue L, Yu N, Martín-Martín R, Xiong C, Goldstein T, et al. Hierarchical point attention for indoor 3D object detection. In: 2024 IEEE International Conference on Robotics and Automation (ICRA); 2024 May 13-17; Yokohama, Japan. Piscataway: IEEE; 2024. p. 4245-4251.[DOI]
-
5. Cai H, Li J, Hu M, Gan C, Han S. EfficientViT: Multi-scale linear attention for high-resolution dense prediction. arXiv:2205.14756 [Preprint]. 2022.[DOI]
-
6. Yang Y, Zheng S, Wang X, Ao W, Liu Z. AMMUNet: Multiscale attention map merging for remote sensing image segmentation. IEEE Geosci Remote Sens Lett. 2025;22:6000705.[DOI]
-
7. Huang H, Zhou X, Cao J, He R, Tan T. Vision transformer with super token sampling. arXiv:221111167 [Preprint]. 2022.[DOI]
-
8. Jiao J, Tang YM, Lin KY, Gao Y, Ma AJ, Wang Y, et al. DilateFormer: Multi-scale dilated transformer for visual recognition. IEEE Trans Multimed. 2023;25:8906-8919.[DOI]
-
9. Li G, Xu Y, Hu D. Multi-scale attention for audio question answering. arXiv:2305.17993 [Preprint]. 2023.[DOI]
-
10. Yan H, Wu M, Zhang C. Multi-scale representations by varying window attention for semantic segmentation. arXiv:2404.16573 [Preprint]. 2024.[DOI]
-
11. Pan Z, Cai J, Zhuang B. Fast vision transformers with hilo attention. Adv Neural Inf Process Syst. 2022;35:14541-14554.[DOI]
-
12. Wang Y, Li Y, Wang G, Liu X. Multi-scale attention network for single image super-resolution. In: 2024 IEEE/CVF Conference on Computer Vision and Pattern Recognition (CVPR); 2024 Jun 16-22; Seattle, USA. Piscataway: IEEE; 2024. p. 5950-5960.[DOI]
-
13. Zhu L, Wang X, Ke Z, Zhang W, Lau R. BiFormer: Vision transformer with bi-level routing attention. In: 2023 IEEE/CVF Conference on Computer Vision and Pattern Recognition (CVPR); 2023 Jun 17-24; Vancouver, Canada. Piscataway: IEEE; 2023. p. 10323-10333.[DOI]
-
14. Yun S, Ro Y. SHViT: Single-head vision transformer with memory efficient macro design. In: 2024 IEEE/CVF Conference on Computer Vision and Pattern Recognition (CVPR); 2024 Jun 16-22; Seattle, USA. Piscataway: IEEE; 2024. p. 5756-5767.[DOI]
-
15. Luo Z, Cui Y, Zhao S, Yin J. g-inspector: Recurrent attention model on graph. IEEE Trans Knowl Data Eng. 2020;34(2):680-690.[DOI]
-
16. Liu M, Liang L, Wang Q, Zhang Y, Shi L, Zhang T, et al. IMAGO: An improved model based on attention mechanism for enhanced protein function prediction. Biomolecules. 2025;15(12):1667.[DOI]
-
17. Zhang Q, Yang F, Huang W, Feng J, Liu J. TRGOA: Topological-aware residue-gene ontology attention network for protein function prediction. IEEE Trans Comput Biol Bioinform. 2025;22(2):603-613.[DOI]
-
18. Wan B, Huang X, Qiao Y, Peng J, Yang F. EGA-Ploc: An efficient global-local attention model for multi-label protein subcellular localization prediction on the immunohistochemistry images. IEEE J Biomed Health Inform. 2025;29(12):9299-9311.[DOI]
-
19. Huang S, Wang M, Zheng X, Chen J, Tang C. Hierarchical and dynamic graph attention network for drug-disease association prediction. IEEE J Biomed Health Inform. 2024;28(4):2416-2427.[DOI]
-
20. Yella JK, Jegga AG. MGATRx: Discovering drug repositioning candidates using multi-view graph attention. IEEE/ACM Trans Comput Biol Bioinform. 2021;19(5):2596-2604.[DOI]
-
21. Xu Y, Wang J, Guang M, Yan C, Jiang C. Multistructure graph classification method with attention-based pooling. IEEE Trans Comput Soc Syst. 2022;10(2):602-613.[DOI]
-
22. Sun Q, Cheng L, Meng A, Ge S, Chen J, Zhang L, et al. SADLN: Self-attention based deep learning network of integrating multi-omics data for cancer subtype recognition. Front Genet. 2023;13:1032768.[DOI]
-
23. Zhou R, Li G, Mao Y, Liu T, Chen Y, Wu J, et al. Identifying cancer driver genes using a neural network framework with cross-attention mechanism. IEEE Trans Comput Biol Bioinform. 2025;22(5):2206-2215.[DOI]
-
24. Wu W, Wang S, Zhang Y, Yin W, Zhao Y, Pang S. MOSGAT: Uniting specificity-aware GATs and cross modal-attention to integrate multi-omics data for disease diagnosis. IEEE J Biomed Health Inform. 2024;28(9):5624-5637.[DOI]
-
25. Liu C, Chen Y, Wu X. Collaborative attention contrast learning for cancer subtype identification based on multi-omics data. IEEE Trans Comput Biol Bioinform. 2025;22(5):2105-2116.[DOI]
-
26. Moon S, Lee H. MOMA: A multi-task attention learning algorithm for multi-omics data interpretation and classification. Bioinformatics. 2022;38(8):2287-2296.[DOI]
-
27. Wang C, Wang Y, Ding P, Li S, Yu X, Yu B. ML-FGAT: Identification of multi-label protein subcellular localization by interpretable graph attention networks and feature-generative adversarial networks. Comput Biol Med. 2024;170:107944.[DOI]
-
28. Yan W, Zhang B, Zuo M, Zhang Q, Wang H, Mao D. AttentionSplice: An interpretable multi-head self-attention based hybrid deep learning model in splice site prediction. Chin J Electronics. 2022;31(5):870-887.[DOI]
-
29. Luo R, Liu J, Guan L, Li M. HybProm: An attention-assisted hybrid CNN-BiLSTM model for the interpretable prediction of DNA promoter. Methods. 2025;235:71-80.[DOI]
-
30. Aleb N. A mutual attention model for drug target binding affinity prediction. IEEE/ACM Trans Comput Biol Bioinform. 2022;19(6):3224-3232.[DOI]
-
31. Han K, Liu Y, Xu J, Song J, Yu DJ. Performing protein fold recognition by exploiting a stack convolutional neural network with the attention mechanism. Anal Biochem. 2022;651:114695.[DOI]
-
32. Zheng K, Zhang XL, Wang L, You ZH, Zhan ZH, Li HY. Line graph attention networks for predicting disease-associated Piwi-interacting RNAs. Brief Bioinform. 2022;23(6):bbac393.[DOI]
-
33. Fan X, Gong M, Xie Y, Jiang F, Li H. Structured self-attention architecture for graph-level representation learning. Pattern Recognit. 2020;100:107084.[DOI]
-
34. Zhang H, Li J, Hu F, Lin H, Ma J. AMter: An end-to-end model for transcriptional terminators prediction by extracting semantic feature automatically based on attention mechanism. Concurr Comput Pract Exp. 2024;36(13):e8056.[DOI]
-
35. Cai S, Su E, Xie L, Li H. EEG-based auditory attention detection via frequency and channel neural attention. IEEE Trans Human Mach Syst. 2022;52(2):256-266.[DOI]
-
36. Dong C, Liu Y, Sun D. Multi-view seizure classification based on attention-based adaptive graph ProbSparse hybrid network. CAAI Trans Intell Technol. 2025;10(6):1783-1798.[DOI]
-
37. Wang L, Wong L, You ZH, Huang DS. AMDECDA: Attention mechanism combined with data ensemble strategy for predicting CircRNA-disease association. IEEE Trans Big Data. 2024;10(4):320-329.[DOI]
-
38. Yang Q, Ma B, Cui H, Ma J. AMF-NET: Attention-aware multi-scale fusion network for retinal vessel segmentation. In: 2021 43rd Annual International Conference of the IEEE Engineering in Medicine & Biology Society (EMBC); 2021 Nov 01-05; Mexico. Piscataway: IEEE; 2021. p. 3277-3280.[DOI]
-
39. Teragawa S, Wang L, Liu Y. DeepPGD: A deep learning model for DNA methylation prediction using temporal convolution, BiLSTM, and attention mechanism. Int J Mol Sci. 2024;25(15):8146.[DOI]
-
40. Zhang Y, Liu Y, Xu J, Wang X, Peng X, Song J, et al. Leveraging the attention mechanism to improve the identification of DNA N6-methyladenine sites. Brief Bioinform. 2021;22(6):bbab351.[DOI]
-
41. De Waele G, Clauwaert J, Menschaert G, Waegeman W. CpG Transformer for imputation of single-cell methylomes. Bioinformatics. 2022;38(3):597-603.[DOI]
-
42. Chang KL, Chen JH, Lin TC, Leu JY, Kao CF, Wong JY, et al. Short human eccDNAs are predictable from sequences. Brief Bioinform. 2023;24(3):bbad147.[DOI]
-
43. Yu L, Zhang Y, Xue L, Liu F, Chen Q, Luo J, et al. Systematic analysis and accurate identification of DNA N4-methylcytosine sites by deep learning. Front Microbiol. 2022;13:843425.[DOI]
-
44. Lim D, Baek C, Blanchette M. Graphylo: A deep learning approach for predicting regulatory DNA and RNA sites from whole-genome multiple alignments. iScience. 2024;27(2):109002.[DOI]
-
45. Chen C, Hou J, Shi X, Yang H, Birchler JA, Cheng J. DeepGRN: Prediction of transcription factor binding site across cell-types using attention-based deep neural networks. BMC Bioinform. 2021;22:38.[DOI]
-
46. Han K, Shen LC, Zhu YH, Xu J, Song J, Yu DJ. MAResNet: Predicting transcription factor binding sites by combining multi-scale bottom-up and top-down attention and residual network. Brief Bioinform. 2022;23:bbab445.[DOI]
-
47. Yan W, Li Z, Pian C, Wu Y. PlantBind: An attention-based multi-label neural network for predicting plant transcription factor binding sites. Brief Bioinform. 2022;23(6):bbac425.[DOI]
-
48. Zhang J, Liu B, Wu J, Wang Z, Li J. DeepCAC: A deep learning approach on DNA transcription factors classification based on multi-head self-attention and concatenate convolutional neural network. BMC Bioinform. 2023;24:345.[DOI]
-
49. Wang K, Zeng X, Zhou J, Liu F, Luan X, Wang X. BERT-TFBS: A novel BERT-based model for predicting transcription factor binding sites by transfer learning. Brief Bioinform. 2024;25(3):bbae195.[DOI]
-
50. Zhang Y, Wang Z, Ge F, Wang X, Zhang Y, Li S, et al. MLSNet: A deep learning model for predicting transcription factor binding sites. Brief Bioinform. 2024;25(6):bbae489.[DOI]
-
51. Yu Y, Ding P, Gao H, Liu G, Zhang F, Yu B. Cooperation of local features and global representations by a dual-branch network for transcription factor binding sites prediction. Brief Bioinform. 2023;24(2):bbad036.[DOI]
-
52. Bhukya R, Kumari A, Dasari CM, Amilpur S. An attention-based hybrid deep neural networks for accurate identification of transcription factor binding sites. Neural Comput Appl. 2022;34(21):19051-19060.[DOI]
-
53. Ding P, Wang Y, Zhang X, Gao X, Liu G, Yu B. DeepSTF: Predicting transcription factor binding sites by interpretable deep neural networks combining sequence and shape. Brief Bioinform. 2023;24(4):bbad231.[DOI]
-
54. Zhang Y, Wang Z, Zeng Y, Liu Y, Xiong S, Wang M, et al. A novel convolution attention model for predicting transcription factor binding sites by combination of sequence and shape. Brief Bioinform. 2022;23:bbab525.[DOI]
-
55. Zhu Y, Li F, Guo X, Wang X, Coin LJM, Webb GI, et al. TIMER is a Siamese neural network-based framework for identifying both general and species-specific bacterial promoters. Brief Bioinform. 2023;24(4):bbad209.[DOI]
-
56. Karbalayghareh A, Sahin M, Leslie CS. Chromatin interaction–aware gene regulatory modeling with graph attention networks. Genome Res. 2022;32(5):930-944.[DOI]
-
57. Mehmood F, Arshad S, Shoaib M. ADH-Enhancer: An attention-based deep hybrid framework for enhancer identification and strength prediction. Brief Bioinform. 2024;25(2):bbae030.[DOI]
-
58. Yang M, Zhang S, Zheng Z, Zhang P, Liang Y, Tang S. Employing bimodal representations to predict DNA bendability within a self-supervised pre-trained framework. Nucleic Acids Res. 2024;52(6):e33.[DOI]
-
59. Li J, Pu Y, Tang J, Zou Q, Guo F. DeepATT: A hybrid category attention neural network for identifying functional effects of DNA sequences. Brief Bioinform. 2021;22(3):bbaa159.[DOI]
-
60. Lee K, No A. Needleman-Wunsch attention: A framework for enhancing DNA sequence embedding. IEEE Access. 2024;12:69087-69096.[DOI]
-
61. Qin Y, Zhu F, Xi B, Song L. Robust multi-read reconstruction from noisy clusters using deep neural network for DNA storage. Comput Struct Biotechnol J. 2024;23:1076-1087.[DOI]
-
62. Cao B, Wang B, Zhang Q. GCNSA: DNA storage encoding with a graph convolutional network and self-attention. iScience. 2023;26(3):106231.[DOI]
-
63. Tan D, Yang C, Wang J, Su Y, Zheng C. scAMAC: Self-supervised clustering of scRNA-seq data based on adaptive multi-scale autoencoder. Brief Bioinform. 2024;25(2):bbae068.[DOI]
-
64. Zhou W, Song W, Zhang Z, Zhang F, Teng Z, Tian Z. Hierarchical feature aggregation with mixed attention mechanism for single-cell RNA-seq analysis. Expert Syst Appl. 2025;260:125340.[DOI]
-
65. Liu L, Wu X, Yu J, Zhang Y, Niu K, Yu A. scVGATAE: A variational graph attentional autoencoder model for clustering single-cell RNA-seq data. Biology. 2024;13(9):713.[DOI]
-
66. Hu D, Liang K, Zhou S, Tu W, Liu M, Liu X. scDFC: A deep fusion clustering method for single-cell RNA-seq data. Brief Bioinform. 2023;24(4):bbad216.[DOI]
-
67. Feng X, Zhang H, Lin H, Long H. Single-cell RNA-seq data analysis based on directed graph neural network. Methods. 2023;211:48-60.[DOI]
-
68. Yuan L, Zhao L, Jiang Y, Shen Z, Zhang Q, Zhang M, et al. scMGATGRN: A multiview graph attention network–based method for inferring gene regulatory networks from single-cell transcriptomic data. Brief Bioinform. 2024;25(6):bbae526.[DOI]
-
69. Liu J, Zhou S, Ma J, Zang M, Liu C, Liu T, et al. Graph attention network with convolutional layer for predicting gene regulations from single-cell ribonucleic acid sequence data. Eng Appl Artif Intell. 2024;136:108938.[DOI]
-
70. Lin Z, Le OY. Inferring gene regulatory networks from single-cell gene expression data via deep multi-view contrastive learning. Brief Bioinform. 2023;24:bbac586.[DOI]
-
71. Li W, Zhang H, Wang L, Wang P, Yu K. STGAT: Graph attention networks for deconvolving spatial transcriptomics data. Comput Methods Programs Biomed. 2024;257:108431.[DOI]
-
72. Zhang T, Zhang Z, Li L, Dong B, Wang G, Zhang D. GTAD: A graph-based approach for cell spatial composition inference from integrated scRNA-seq and ST-seq data. Brief Bioinform. 2023;25:bbad469.[DOI]
-
73. Sun H, Qu H, Duan K, Du W. scMGCN: A multi-view graph convolutional network for cell type identification in scRNA-seq data. Int J Mol Sci. 2024;25(4):2234.[DOI]
-
74. Zhang W, Yu R, Xu Z, Li J, Gao W, Jiang M, et al. scCompressSA: Dual-channel self-attention based deep autoencoder model for single-cell clustering by compressing gene–gene interactions. BMC Genom. 2024;25:423.[DOI]
-
75. Teragawa S, Wang L. ConF: A deep learning model based on BiLSTM, CNN, and cross multi-head attention mechanism for noncoding RNA family prediction. Biomolecules. 2023;13(11):1643.[DOI]
-
76. Yuan J, Wang Z, Pan Z, Li A, Zhang Z, Cui F. DPNN-ac4C: A dual-path neural network with self-attention mechanism for identification of N4-acetylcytidine (ac4C) in mRNA. Bioinformatics. 2024;40(11):btae625.[DOI]
-
77. Wang H, Huang T, Wang D, Zeng W, Sun Y, Zhang L. MSCAN: Multi-scale self- and cross-attention network for RNA methylation site prediction. BMC Bioinform. 2024;25:32.[DOI]
-
78. Song Z, Huang D, Song B, Chen K, Song Y, Liu G, et al. Attention-based multi-label neural networks for integrated prediction and interpretation of twelve widely occurring RNA modifications. Nat Commun. 2021;12:4011.[DOI]
-
79. Cao C, Yang S, Li M, Li C. CircSSNN: CircRNA-binding site prediction via sequence self-attention neural networks with pre-normalization. BMC Bioinform. 2023;24:220.[DOI]
-
80. Justyna M, Antczak M, Szachniuk M. Machine learning for RNA 2D structure prediction benchmarked on experimental data. Brief Bioinform. 2023;24(3):bbad153.[DOI]
-
81. Zhao Z, Woloszynek S, Agbavor F, Mell JC, Sokhansanj BA, Rosen GL. Learning, visualizing and exploring 16S rRNA structure using an attention-based deep neural network. PLoS Comput Biol. 2021;17(9):e1009345.[DOI]
-
82. Gong T, Ju F, Bu D. Accurate prediction of RNA secondary structure including pseudoknots through solving minimum-cost flow with learned potentials. Commun Biol. 2024;7:297.[DOI]
-
83. Li H, Wu B, Sun M, Ye Y, Zhu Z, Chen K. Multi-view graph neural network with cascaded attention for lncRNA-miRNA interaction prediction. Knowl Based Syst. 2023;268:110492.[DOI]
-
84. He Y, Ning Z, Zhu X, Zhang Y, Liu C, Jiang S, et al. Plant lncRNA-miRNA interaction prediction based on counterfactual heterogeneous graph attention network. Interdiscip Sci Comput Life Sci. 2025;17(2):244-256.[DOI]
-
85. Yang TH, Shiue SC, Chen KY, Tseng YY, Wu WS. Identifying PiRNA targets on mRNAs in C. elegans using a deep multi-head attention network. BMC Bioinform. 2021;22:503.[DOI]
-
86. Wang Y, Wang X, Chen C, Gao H, Salhi A, Gao X, et al. RPI-CapsuleGAN: Predicting RNA-protein interactions through an interpretable generative adversarial capsule network. Pattern Recognit. 2023;141:109626.[DOI]
-
87. Lasantha D, Vidanagamachchi S, Nallaperuma S. CRIECNN: Ensemble convolutional neural network and advanced feature extraction methods for the precise forecasting of circRNA-RBP binding sites. Comput Biol Med. 2024;174:108466.[DOI]
-
88. Yuan L, Zhao L, Lai J, Jiang Y, Zhang Q, Shen Z, et al. iCRBP-LKHA: Large convolutional kernel and hybrid channel-spatial attention for identifying circRNA-RBP interaction sites. PLoS Comput Biol. 2024;20(8):e1012399.[DOI]
-
89. Du B, Liu Z, Luo F. Deep multi-scale attention network for RNA-binding proteins prediction. Inf Sci. 2022;582:287-301.[DOI]
-
90. Lan W, Dong Y, Chen Q, Zheng R, Liu J, Pan Y, et al. KGANCDA: Predicting circRNA-disease associations based on knowledge graph attention network. Brief Bioinform. 2022;23:bbab494.[DOI]
-
91. Han Y, Zhang SW. ncRPI-LGAT: Prediction of ncRNA-protein interactions with line graph attention network framework. Comput Struct Biotechnol J. 2023;21:2286-2295.[DOI]
-
92. Yuan L, Yang Y. DeCban: Prediction of circRNA-RBP interaction sites by using double embeddings and cross-branch attention networks. Front Genet. 2021;11:632861.[DOI]
-
93. Wekesa JS, Meng J, Luan Y. A deep learning model for plant lncRNA-protein interaction prediction with graph attention. Mol Genet Genomics. 2020;295(5):1091-1102.[DOI]
-
94. Dai Q, Liu Z, Wang Z, Duan X, Guo M. GraphCDA: A hybrid graph representation learning framework based on GCN and GAT for predicting disease-associated circRNAs. Brief Bioinform. 2022;23(5):bbac379.[DOI]
-
95. Zhao X, Wu J, Zhao X, Yin M. Multi-view contrastive heterogeneous graph attention network for lncRNA–disease association prediction. Brief Bioinform. 2023;24:bbac548.[DOI]
-
96. Li Z, Chen X, Zhang X, Jiang R, Chen S. Latent feature extraction with a prior-based self-attention framework for spatial transcriptomics. Genome Res. 2023;33(10):1757-1773.[DOI]
-
97. Yang W, Wang P, Xu S, Wang T, Luo M, Cai Y, et al. Deciphering cell–cell communication at single-cell resolution for spatial transcriptomics with subgraph-based graph attention network. Nat Commun. 2024;15:7101.[DOI]
-
98. Huang Y, Luo J, Jing R, Li M. Multi-model predictive analysis of RNA solvent accessibility based on modified residual attention mechanism. Brief Bioinform. 2022;23(6):bbac470.[DOI]
-
99. Gao M, Shang X. Identification of associations between lncRNA and drug resistance based on deep learning and attention mechanism. Front Microbiol. 2023;14:1147778.[DOI]
-
100. Hong J, Hou W, Sheng N, Zuo C, Wang Y. HATZFS predicts pancreatic cancer driver biomarkers by hierarchical reinforcement learning and zero-forcing set. Expert Syst Appl. 2025;260:125435.[DOI]
-
101. Asim MN, Ibrahim MA, Malik MI, Zehe C, Cloarec O, Trygg J, et al. EL-RMLocNet: An explainable LSTM network for RNA-associated multi-compartment localization prediction. Comput Struct Biotechnol J. 2022;20:3986-4002.[DOI]
-
102. Jiang JY, Ju CJ, Hao J, Chen M, Wang W. JEDI: Circular RNA prediction based on junction encoders and deep interaction among splice sites. Bioinformatics. 2021;37:i289-i298.[DOI]
-
103. Zhang X, Wei L, Ye X, Zhang K, Teng S, Li Z, et al. SiameseCPP: A sequence-based Siamese network to predict cell-penetrating peptides by contrastive learning. Brief Bioinform. 2023;24:bbac545.[DOI]
-
104. Wang Y, Sun H, Sheng N, He K, Hou W, Zhao Z, et al. ESMSec: Prediction of secreted proteins in human body fluids using protein language models and attention. Int J Mol Sci. 2024;25(12):6371.[DOI]
-
105. Zhao J, Yan W, Yang Y. DeepTP: A deep learning model for thermophilic protein prediction. Int J Mol Sci. 2023;24(3):2217.[DOI]
-
106. Wu Z, Guo M, Jin X, Chen J, Liu B. CFAGO: Cross-fusion of network and attributes based on attention mechanism for protein function prediction. Bioinformatics. 2023;39(3):btad123.[DOI]
-
107. Lin K, Quan X, Jin C, Shi Z, Yang J. An interpretable double-scale attention model for enzyme protein class prediction based on transformer encoders and multi-scale convolutions. Front Genet. 2022;13:885627.[DOI]
-
108. Zhang X, Wang Y, Wei Q, He S, Salhi A, Yu B. DRBPPred-GAT: Accurate prediction of DNA-binding proteins and RNA-binding proteins based on graph multi-head attention network. Knowl Based Syst. 2024;285:111354.[DOI]
-
109. Yu Q, Zhang Z, Liu G, Li W, Tang Y. ToxGIN: An In silico prediction model for peptide toxicity via graph isomorphism networks integrating peptide sequence and structure information. Brief Bioinform. 2024;25(6):bbae583.[DOI]
-
110. Zhang X, Guo H, Zhang F, Wang X, Wu K, Qiu S, et al. HNetGO: Protein function prediction via heterogeneous network transformer. Brief Bioinform. 2023;24(6):bbab556.[DOI]
-
111. Xia Z, Ma S, Li J, Guo Y, Jiang L, Tang J. RecGOBD: Accurate recognition of gene ontology related brain development protein functions through multi-feature fusion and attention mechanisms. Bioinform Adv. 2024;4:vbae163.[DOI]
-
112. Pan T, Li C, Bi Y, Wang Z, Gasser RB, Purcell AW, et al. PFresGO: An attention mechanism-based deep-learning approach for protein annotation by integrating gene ontology inter-relationships. Bioinformatics. 2023;39(3):btad094.[DOI]
-
113. Ding W, Nakai K, Gong H. Protein design via deep learning. Brief Bioinform. 2022;23(3):bbac102.[DOI]
-
114. Zhang B, Zheng M, Zhang Y, Quan L. DCMA: Faster protein backbone dihedral angle prediction using a dilated convolutional attention-based neural network. Front Bioinform. 2024;4:1477909.[DOI]
-
115. Jin X, Guo L, Jiang Q, Wu N, Yao S. Prediction of protein secondary structure based on an improved channel attention and multiscale convolution module. Front Bioeng Biotechnol. 2022;10:901018.[DOI]
-
116. Jin C, Shi Z, Kang C, Lin K, Zhang H. TLCrys: Transfer learning based method for protein crystallization prediction. Int J Mol Sci. 2022;23(2):972.[DOI]
-
117. Mahmud S, Guo Z, Quadir F, Liu J, Cheng J. Multi-head attention-based U-Nets for predicting protein domain boundaries using 1D sequence features and 2D distance maps. BMC Bioinform. 2022;23:283.[DOI]
-
118. Ma Q, Zou K, Zhang Z, Yang F. GLTM: A global-local attention LSTM model to locate dimer motif of single-pass membrane proteins. Front Genet. 2022;13:854571.[DOI]
-
119. Chen L, Jing XY, Hao Y, Liu W, Zhu X, Han W. A novel two-way rebalancing strategy for identifying carbonylation sites. BMC Bioinform. 2023;24:429.[DOI]
-
120. Song T, Yang Q, Qu P, Qiao L, Wang X. Attenphos: General phosphorylation site prediction model based on attention mechanism. Int J Mol Sci. 2024;25(3):1526.[DOI]
-
121. Zhu C, Zhang C, Shang T, Zhang C, Zhai S, Cao L, et al. GAPS: A geometric attention-based network for peptide binding site identification by the transfer learning approach. Brief Bioinform. 2024;25(4):bbae297.[DOI]
-
122. Zhu YH, Liu Z, Liu Y, Ji Z, Yu DJ. ULDNA: Integrating unsupervised multi-source language models with LSTM-attention network for high-accuracy protein–DNA binding site prediction. Brief Bioinform. 2024;25(2):bbae040.[DOI]
-
123. Zheng M, Sun G, Li X, Fan Y. EGPDI: Identifying protein–DNA binding sites based on multi-view graph embedding fusion. Brief Bioinform. 2024;25(4):bbae330.[DOI]
-
124. Sunny S, Prakash PB, Gopakumar G, Jayaraj PB. DeepBindPPI: Protein–protein binding site prediction using attention based graph convolutional network. Protein J. 2023;42(4):276-287.[DOI]
-
125. Abdin O, Nim S, Wen H, Kim PM. PepNN: A deep attention model for the identification of peptide binding sites. Commun Biol. 2022;5:503.[DOI]
-
126. Ko YS, Parkinson J, Liu C, Wang W. TUnA: An uncertainty-aware transformer model for sequence-based protein–protein interaction prediction. Brief Bioinform. 2024;25(5):bbae359.[DOI]
-
127. Alam R, Mahbub S, Bayzid MS. Pair-EGRET: Enhancing the prediction of protein–protein interaction sites through graph attention networks and protein language models. Bioinformatics. 2024;40(10):btae588.[DOI]
-
128. Tsukiyama S, Kurata H. Cross-attention PHV: Prediction of human and virus protein-protein interactions using cross-attention–based neural networks. Comput Struct Biotechnol J. 2022;20:5564-5573.[DOI]
-
129. Baranwal M, Magner A, Saldinger J, Turali-Emre ES, Elvati P, Kozarekar S, et al. Struct2Graph: A graph attention network for structure based predictions of protein–protein interactions. BMC Bioinform. 2022;23:370.[DOI]
-
130. Xiang Z, Gong W, Li Z, Yang X, Wang J, Wang H. Predicting protein–protein interactions via gated graph attention signed network. Biomolecules. 2021;11(6):799.[DOI]
-
131. Sun X, Wu Z, Su J, Li C. GraphPBSP: Protein binding site prediction based on graph attention network and pre-trained model ProstT5. Int J Biol Macromol. 2024;282:136933.[DOI]
-
132. Chen X, Huang J, Shen T, Zhang H, Xu L, Yang M, et al. DEAttentionDTA: Protein–ligand binding affinity prediction based on dynamic embedding and self-attention. Bioinformatics. 2024;40(6):btae319.[DOI]
-
133. Bian J, Lu H, Dong G, Wang G. Hierarchical multimodal self-attention-based graph neural network for DTI prediction. Brief Bioinform. 2024;25(4):bbae293.[DOI]
-
134. Luo Z, Wu W, Sun Q, Wang J. Accurate and transferable drug–target interaction prediction with DrugLAMP. Bioinformatics. 2024;40(12):btae693.[DOI]
-
135. Zhai H, Hou H, Luo J, Liu X, Wu Z, Wang J. DGDTA: Dynamic graph attention network for predicting drug–target binding affinity. BMC Bioinform. 2023;24:367.[DOI]
-
137. Bae H, Nam H. GraphATT-DTA: Attention-based novel representation of interaction to predict drug-target binding affinity. Biomedicines. 2023;11(1):67.[DOI]
-
138. Gim M, Choe J, Baek S, Park J, Lee C, Ju M, et al. ArkDTA: Attention regularization guided by non-covalent interactions for explainable drug–target binding affinity prediction. Bioinformatics. 2023;39:i448-i457.[DOI]
-
139. Rahman J, Newton MA, Ali ME, Sattar A. Distance plus attention for binding affinity prediction. J Cheminform. 2024;16:52.[DOI]
-
140. Wu J, Chen H, Cheng M, Xiong H. CurvAGN: Curvature-based adaptive graph neural networks for predicting protein-ligand binding affinity. BMC Bioinform. 2023;24:378.[DOI]
-
141. Kyro GW, Brent RI, Batista VS. HAC-net: A hybrid attention-based convolutional neural network for highly accurate protein–ligand binding affinity prediction. J Chem Inf Model. 2023;63(7):1947-1960.[DOI]
-
142. Seo S, Choi J, Park S, Ahn J. Binding affinity prediction for protein–ligand complex using deep attention mechanism based on intermolecular interactions. BMC Bioinform. 2021;22:542.[DOI]
-
143. Jin Z, Wu T, Chen T, Pan D, Wang X, Xie J, et al. CAPLA: Improved prediction of protein–ligand binding affinity by a deep learning approach based on a cross-attention mechanism. Bioinformatics. 2023;39(2):btad049.[DOI]
-
144. Shen LC, Liu Y, Song J, Yu DJ. SAResNet: Self-attention residual network for predicting DNA-protein binding. Brief Bioinform. 2021;22(5):bbab101.[DOI]
-
145. Wang N, Yan K, Zhang J, Liu B. iDRNA-ITF: Identifying DNA- and RNA-binding residues in proteins based on induction and transfer framework. Brief Bioinform. 2022;23(4):bbac236.[DOI]
-
146. Yin Z, Chen Y, Hao Y, Pandiyan S, Shao J, Wang L. FOTF-CPI: A compound-protein interaction prediction transformer based on the fusion of optimal transport fragments. iScience. 2024;27(1):108756.[DOI]
-
147. Campana PA, Nikoloski Z. Self- and cross-attention accurately predicts metabolite–protein interactions. NAR Genom Bioinform. 2023;5:lqad008.[DOI]
-
148. Koyama K, Hashimoto K, Nagao C, Mizuguchi K. Attention network for predicting T-cell receptor–peptide binding can associate attention with interpretable protein structural properties. Front Bioinform. 2023;3:1274599.[DOI]
-
149. Ni B, Kaplan DL, Buehler MJ. Generative design of de novo proteins based on secondary-structure constraints using an attention-based diffusion model. Chem. 2023;9(7):1828-1849.[DOI]
-
150. Wilman W, Wróbel S, Bielska W, Deszynski P, Dudzic P, Jaszczyszyn I, et al. Machine-designed biotherapeutics: Opportunities, feasibility and advantages of deep learning in computational antibody discovery. Brief Bioinform. 2022;23(4):bbac267.[DOI]
-
151. Choi JM, Chae H. moBRCA-net: A breast cancer subtype classification framework based on multi-omics attention neural networks. BMC Bioinform. 2023;24:169.[DOI]
-
152. Wang T, Xiang G, He S, Su L, Wang Y, Yan X, et al. DeepEnzyme: A robust deep learning model for improved enzyme turnover number prediction by utilizing features of protein 3D-structures. Brief Bioinform. 2024;25(5):bbae409.[DOI]
-
153. Dai Q, Chen H, Yi WJ, Zhao JN, Zhang W, He PA, et al. Precision DNA methylation typing via hierarchical clustering of Nanopore current signals and attention-based neural network. Brief Bioinform. 2024;25(6):bbae596.[DOI]
-
154. Consortium TU. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019;47(D1):D506-D515.[DOI]
-
155. Kouranov A. The RCSB PDB information portal for structural genomics. Nucleic Acids Res. 2006;34(90001):D302-D305.[DOI]
-
156. Varadi M, Bertoni D, Magana P, Paramval U, Pidruchna I, Radhakrishnan M, et al. AlphaFold Protein Structure Database in 2024: Providing structure coverage for over 214 million protein sequences. Nucleic Acids Res. 2024;52(D1):D368-D375.[DOI]
-
157. Perez G, Barber GP, Benet-Pages A, Casper J, Clawson H, Diekhans M, et al. The UCSC Genome Browser database: 2025 update. Nucleic Acids Res. 2025;53(D1):D1243-D1249.[DOI]
-
158. Snyder MP, Gingeras TR, Moore JE, Weng Z, Gerstein MB, Ren B, et al. Perspectives on ENCODE. Nature. 2020;583(7818):693-698.[DOI]
-
159. Sayers E, O’Sullivan C, Karsch-Mizrachi I. Using GenBank and SRA. Methods Mol Biol. 2022;2443:1-25.[DOI]
-
160. Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, et al. NCBI GEO: Archive for functional genomics data sets—Update. Nucleic Acids Res. 2012;41:D991-D995.[DOI]
-
161. Kim S, Chen J, Cheng T, Gindulyte A, He J, He S, et al. PubChem 2025 update. Nucleic Acids Res. 2025;53(D1):D1516-D1525.[DOI]
-
162. Kanehisa M. The KEGG database. Bock G, Goode JA, editors. p. 91-103.[DOI]
-
163. Islamaj R, Wei CH, Cissel D, Miliaras N, Printseva O, Rodionov O, et al. NLM-Gene, a richly annotated gold standard dataset for gene entities that addresses ambiguity and multi-species gene recognition. J Biomed Inform. 2021;118:103779.[DOI]
-
164. Szklarczyk D, Franceschini A, Kuhn M, Simonovic M, Roth A, Minguez P, et al. The STRING database in 2011: Functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011;39:D561-D568.[DOI]
-
165. Kalvari I, Nawrocki EP, Ontiveros-Palacios N, Argasinska J, Lamkiewicz K, Marz M, et al. Rfam 14: Expanded coverage of metagenomic, viral and microRNA families. Nucleic Acids Res. 2021;49(D1):D192-D200.[DOI]
-
166. Zhao Y, Li H, Fang S, Kang Y, Wu W, Hao Y, et al. NONCODE 2016: An informative and valuable data source of long non-coding RNAs. Nucleic Acids Res. 2016;44(D1):D203-D208.[DOI]
-
167. Dudekula DB, Panda AC, Grammatikakis I, De S, Abdelmohsen K, Gorospe M. CircInteractome: A web tool for exploring circular RNAs and their interacting proteins and microRNAs. RNA Biol. 2016;13(1):34-42.[DOI]
-
168. Xia S, Feng J, Chen K, Ma Y, Gong J, Cai F, et al. CSCD: A database for cancer-specific circular RNAs. Nucleic Acids Res. 2018;46(D1):D925-D929.[DOI]
-
169. Cui C, Zhong B, Fan R, Cui Q. HMDD v4.0: A database for experimentally supported human microRNA-disease associations. Nucleic Acids Res. 2024;52(D1):D1327-D1332.[DOI]
-
170. Ye P, Luan Y, Chen K, Liu Y, Xiao C, Xie Z. MethSMRT: An integrative database for DNA N6-methyladenine and N4-methylcytosine generated by single-molecular real-time sequencing. Nucleic Acids Res. 2017;45(D1):D85-D89.[DOI]
-
171. Sood AJ, Viner C, Hoffman MM. DNAmod: The DNA modification database. J Cheminform. 2019;11:30.[DOI]
-
172. Ye J, Zhang Y, Cui H, Liu J, Wu Y, Cheng Y, et al. WEGO 2.0: A web tool for analyzing and plotting GO annotations, 2018 update. Nucleic Acids Res. 2018;46(W1):W71-W75.[DOI]
-
173. Placzek S, Schomburg I, Chang A, Jeske L, Ulbrich M, Tillack J, et al. BRENDA in 2017: New perspectives and new tools in BRENDA. Nucleic Acids Res. 2017;45(D1):D380-D388.[DOI]
-
174. Kawashima S, Kanehisa M. AAindex: Amino acid index database. Nucleic Acids Res. 2000;28(1):374.[DOI]
-
175. Tomczak K, Czerwińska P, Wiznerowicz M. Review The Cancer Genome Atlas (TCGA): An immeasurable source of knowledge. Contemp Oncol. 2015;1:68-77.[DOI]
-
176. Yang X, Lian X, Fu C, Wuchty S, Yang S, Zhang Z. HVIDB: A comprehensive database for human–virus protein–protein interactions. Brief Bioinform. 2021;22(2):832-844.[DOI]
-
177. Higurashi M, Ishida T, Kinoshita K. PiSite: A database of protein interaction sites using multiple binding states in the PDB. Nucleic Acids Res. 2009;37:D360-D364.[DOI]
-
178. Zheng Y, Luo H, Teng X, Hao X, Yan X, Tang Y, et al. NPInter v5.0: NcRNA interaction database in a new era. Nucleic Acids Res. 2023;51(D1):D232-D239.[DOI]
-
179. Dunbar J, Krawczyk K, Leem J, Baker T, Fuchs A, Georges G, et al. SAbDab: The structural antibody database. Nucl Acids Res. 2014;42(D1):D1140-D1146.[DOI]
-
180. Wang R, Fang X, Lu Y, Yang CY, Wang S. The PDBbind database: methodologies and updates. J Med Chem. 2005;48(12):4111-4119.[DOI]
-
181. Chen L, Cruz A, Ramsey S, Dickson CJ, Duca JS, Hornak V, et al. Hidden bias in the DUD-E dataset leads to misleading performance of deep learning in structure-based virtual screening. PLoS One. 2019;14(8):e0220113.[DOI]
-
182. Alcántara R, Axelsen KB, Morgat A, Belda E, Coudert E, Bridge A, et al. Rhea: A manually curated resource of biochemical reactions. Nucleic Acids Res. 2012;40(D1):D754-D760.[DOI]
-
183. Wittig U, Kania R, Golebiewski M, Rey M, Shi L, Jong L, et al. SABIO-RK: Database for biochemical reaction kinetics. Nucleic Acids Res. 2012;40(D1):D790-D796.[DOI]
-
184. Hu QN, Deng Z, Hu H, Cao DS, Liang YZ. RxnFinder: Biochemical reaction search engines using molecular structures, molecular fragments and reaction similarity. Bioinformatics. 2011;27(17):2465-2467.[DOI]
-
185. Wang Y, Song C, Zhao J, Zhang Y, Zhao X, Feng C, et al. SEdb 2.0: A comprehensive super-enhancer database of human and mouse. Nucleic Acids Res. 2023;51(D1):D280-D290.[DOI]
-
186. Dinkel H, Chica C, Via A, Gould CM, Jensen LJ, Gibson TJ, et al. Phospho.ELM: A database of phosphorylation sites—Update 2011. Nucleic Acids Res. 2011;39:D261-D267.[DOI]
-
187. McDonald D, Jiang Y, Balaban M, Cantrell K, Zhu Q, Gonzalez A, et al. Greengenes2 unifies microbial data in a single reference tree. Nat Biotechnol. 2024;42(5):715-718.[DOI]
-
188. Zitnik M, Sosic R, Leskovec J. Biosnap datasets: Stanford biomedical network dataset collection. Stanford: Stanford University; 2018. Available from: https://snap.stanford.edu/biodata/
-
189. Caspi R, Billington R, Keseler IM, Kothari A, Krummenacker M, Midford PE, et al. The MetaCyc database of metabolic pathways and enzymes-a 2019 update. Nucleic Acids Res. 2020;48(D1):D445-D453.[DOI]
-
190. Zeng W, Chen S, Cui X, Chen X, Gao Z, Jiang R. SilencerDB: A comprehensive database of silencers. Nucleic Acids Res. 2021;49(D1):D221-D228.[DOI]
Copyright
© The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Publisher’s Note
Science Exploration remains a neutral stance on jurisdictional claims in published
maps
and institutional affiliations. The views expressed in this article are solely those
of
the author(s) and do not reflect the opinions of the Editors or the publisher.
Share And Cite



Science Exploration Style
Tang Y, Bao W. The application of attention mechanisms in biological sequence analysis. Comput Biomed. 2026;1:202602. https://doi.org/10.70401/cbm.2026.0014
Tips
Copy completed.
Submit a Manuscript
Author Instructions
Cite this Article
Article Metrics
0
View
0
Download
Cited
Article Updates

- Abstract
- Keywords
- 1. Introduction
- 2. Biological Databases
- 3. Application of Attention Mechanism in Biological Sequence Analysis
- 4. Conclusions
- Acknowledgements
- Authors contribution
- Conflicts of interest
- Ethical approval
- Consent to participate
- Consent for publication
- Availability of data and materials
- Funding
- References
- Copyright
Science Exploration Style
Tang Y, Bao W. The application of attention mechanisms in biological sequence analysis. Comput Biomed. 2026;1:202602. https://doi.org/10.70401/cbm.2026.0014
copy
Share Link
copy