Identification of potential associations between circRNAs and diseases based on meta relation aware
Xingyu Tan
1,#

,
Mengmeng Wei
1,#
,
Ziqi Xia
2
,
Xinfei Wang
3
,
Yuechao Li
4
,
Lei Wang
1,5,*
,
Zhuhong You
4,*
*Correspondence to:
Lei Wang, School of Computer Science and Technology/School of Artificial Intelligence, China University of Mining and Technology, Xuzhou 221116, Jiangsu, China.
E-mail: leiwang@cumt.edu.cn
Zhuhong You, School of Computer Science, Northwestern Polytechnical University, Xi’an 710129, Shaanxi, China E-mail: zhuhongyou@nwpu.edu.cn
Zhuhong You, School of Computer Science, Northwestern Polytechnical University, Xi’an 710129, Shaanxi, China E-mail: zhuhongyou@nwpu.edu.cn
Comput Biomed. 2026;1:202609. 10.70401/cbm.2026.0020
Received: March 10, 2026Accepted: June 22, 2026Published: June 22, 2026
Abstract
Aims: Circular RNAs (circRNAs) have been shown to be closely associated with the occurrence and progression of various diseases. However, most existing circRNA-disease association prediction methods are limited to homogeneous networks and are unable to effectively capture deep semantic associations through high-order meta-paths. This study aims to develop an efficient computational method for accurately predicting potential circRNA-disease associations.
Methods: We propose a meta-relation-aware heterogeneous graph learning framework for circRNA-disease association prediction. Specifically, known circRNA-disease associations are first used to compute Gaussian interaction profile kernel similarity and extract node attribute features, based on which a heterogeneous graph network is constructed. A graph neural network is then employed to perform multi-layer message passing on the heterogeneous graph, aggregating neighborhood information to achieve deep fusion of multi-source features and generate node embeddings that encode both local and global structural information. Finally, the learned embeddings are fed into a gradient boosting decision tree classifier, and an ensemble strategy is adopted to improve prediction accuracy. Five-fold cross-validation is used for performance evaluation.
Results: Experimental results on three benchmark datasets, CircR2Disease V2.0, circAtlas 3.0, and circRNADisease V2.0, show that the proposed model achieves area under the receiver operating characteristic curve (AUC) values of 92.17%, 91.83%, and 91.73%, respectively. The model outperforms traditional methods in terms of accuracy, precision, and recall. Furthermore, ablation studies validate the effectiveness of the meta-relation-aware strategy.
Conclusions: Overall, this work provides an efficient and reliable computational framework for molecular association prediction and biomarker discovery in the biomedical domain.
Keywords
circRNA-disease association prediction, meta-relation awareness, graph neural network, heterogeneous graph transformer network
1. Introduction
Circular RNAs (circRNAs) are a class of non-coding RNAs characterized by a covalently closed circular structure formed through alternative splicing[1,2]. Owing to their structural stability, circRNAs play critical roles in gene expression regulation, cellular signal transduction, and the progression of various diseases, providing new perspectives for research in fields such as cancer and neurodegenerative disorders[3]. With the rapid development of bioinformatics, the regulatory functions of circRNAs in disease occurrence and progression have been gradually uncovered[4,5]. These molecules not only exhibit strong potential as novel therapeutic targets but also serve as promising biomarkers for disease diagnosis[6]. Therefore, systematically elucidating the associations between circRNAs and diseases is of substantial scientific significance and practical value for disease prevention and treatment[7,8]. However, screening circRNA-disease associations using traditional biological experiments is time-consuming and costly, highlighting the urgent need for advanced computational methods to improve research efficiency and uncover the underlying complex mechanisms.
In recent years, numerous computational approaches have been developed to predict circRNA-disease associations, effectively alleviating the limitations of traditional experimental methods[9]. For example, the CRPGCN model integrates random walk with restart and graph convolutional networks[10]. It first measures node proximity on the similarity network using random walk with restart, then applies principal component analysis for dimensionality reduction and feature extraction, and finally employs a graph convolutional network for feature learning and score prediction. The MNMDCDA model constructs similarity networks for circRNAs and diseases by integrating circRNA functional similarity, disease semantic similarity, and Gaussian interaction profile kernel similarity, followed by high-order graph convolutional networks for feature extraction and a deep neural network for association prediction[11]. The deep matrix factorization with multi-source fusion (DMFMSF) method fuses multi-source data and combines singular value decomposition with matrix factorization techniques to simultaneously mine linear and nonlinear features for inferring potential associations[12]. The GATCDA model leverages a graph attention network to learn node representations within the graph, thereby effectively identifying potential circRNA-disease relationships[13]. In addition, the MDGF-MCEC framework is based on a multi-view dual-attention graph convolutional network and collaborative ensemble learning[14]. It first constructs multiple RNA-disease relationship graphs according to different similarity metrics, then performs representation learning via multi-view graph convolutional networks, and finally outputs prediction results through a multi-view cooperative ensemble classifier.
To fully explore the complex association patterns between circRNAs and diseases, this study constructs a meta-relation-aware prediction model. The model first introduces the Gaussian interaction profile kernel similarity of circRNAs as node attributes and builds a circRNA-disease heterogeneous relational network. Subsequently, a heterogeneous graph transformer is employed to deeply aggregate and learn high-order neighborhood information in the network, thereby obtaining fused features that characterize both local and global topological structures. Finally, the learned feature representations are fed into a gradient boosting decision tree classifier to achieve accurate inference of potential circRNA-disease associations. The overall workflow of the proposed model is illustrated in Figure 1.
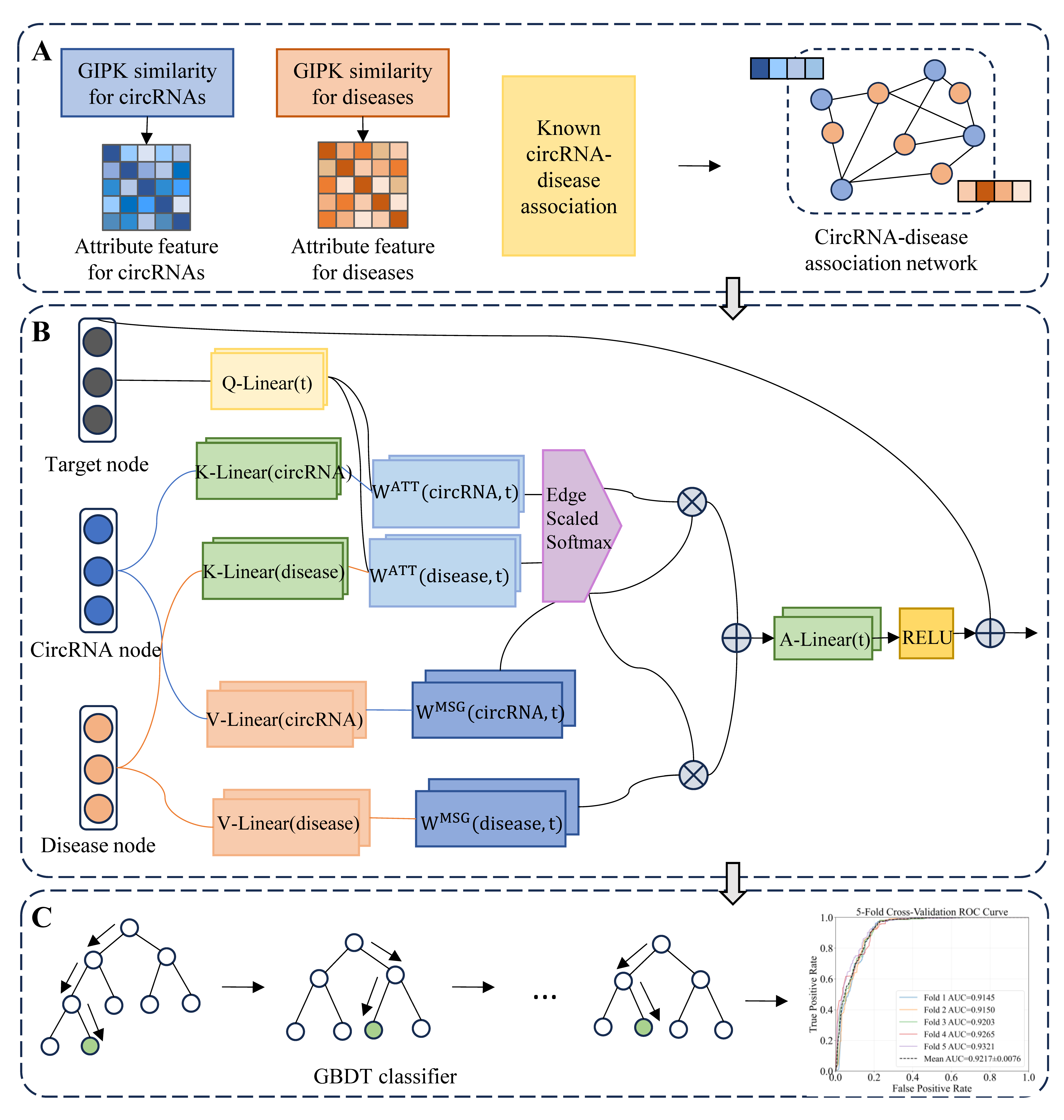{kind=link}
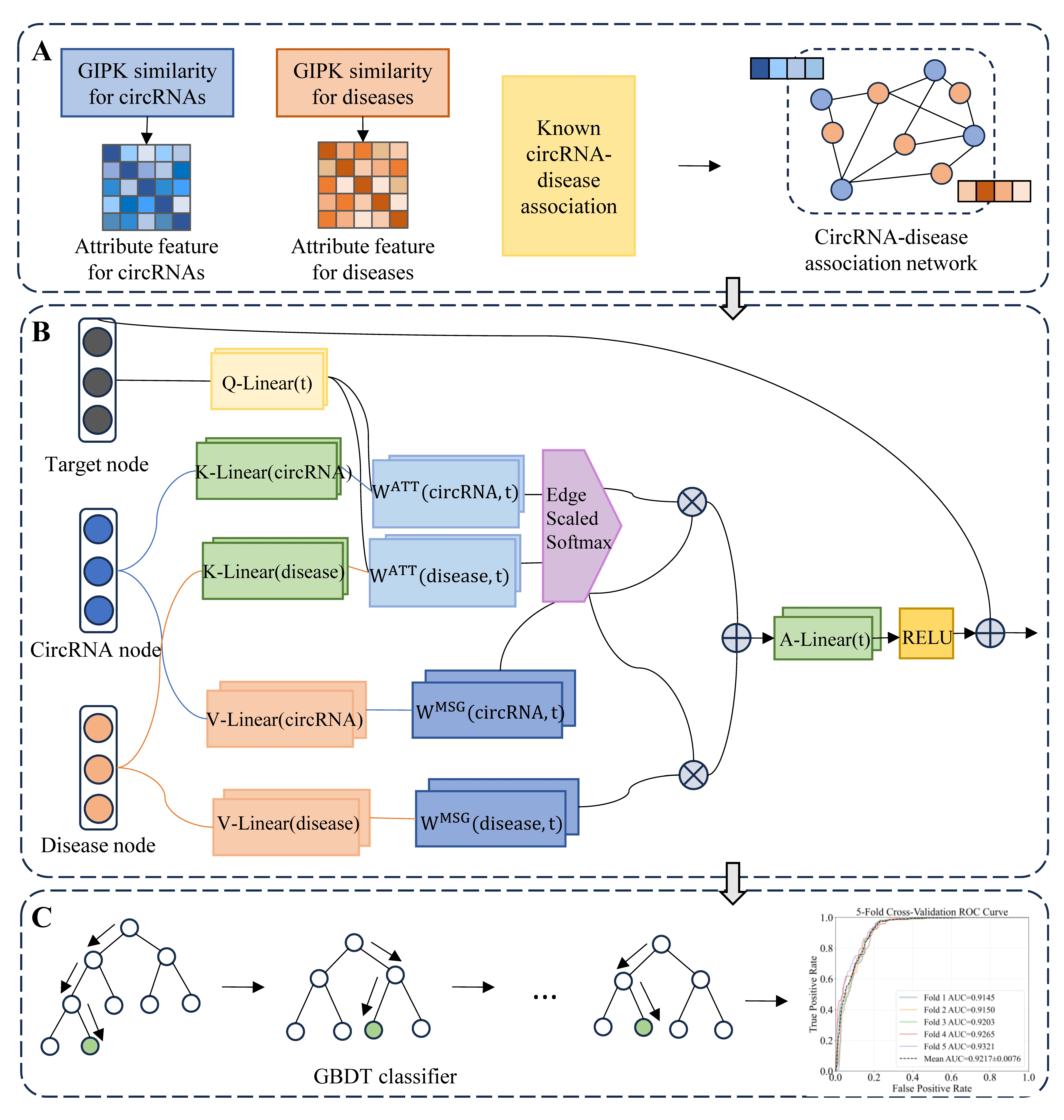
Figure 1. Overview of the proposed framework. circRNAs: circular RNAs; GIPK: Gaussian interaction profile kernel; ReLU: rectified linear unit; GBDT: Gradient boosting decision tree; ROC: receiver operating characteristic; AUC: area under the receiver operating characteristic curve.
2. Methods
2.1 Construction of Gaussian interaction profile kernel similarity
The Gaussian interaction profile kernel (GIPK) is a method for measuring entity similarity based on known association topological structures and is commonly applied in semi-supervised learning scenarios[15]. Its advantage lies in its ability to effectively characterize the potential degree of association among data instances. The fundamental rationale is that if two biological entities, such as circRNAs or diseases, exhibit similar patterns within known association profiles, they are more likely to demonstrate functional or phenotypic consistency. Based on this premise, the GIPK similarities of circRNAs and diseases are computed and incorporated as attribute features into the subsequent prediction framework. The detailed computational procedure is described as follows.
2.1.1 GIPK similarity calculation for circRNAs
For any two circRNAs ci and cj, the GIPK similarity Sc(ci, cj) is defined as:
Where Y(ci) denotes the -th row of matrix Y (i.e., the association vector of circRNA ci with all diseases), and γc is the bandwidth parameter that controls the decay rate of similarity.
The bandwidth parameter γc is calculated as:
2.1.2 GIPK Similarity calculation for diseases
Similarly, for any two diseases di and dj, the GIPK similarity Sd(di, dj) is defined as:
The bandwidth parameter γd is computed in a manner analogous to γc:
2.2 Construction of the circRNA-disease heterogeneous graph
A heterogeneous graph refers to a graph structure containing multiple types of nodes and edges. Compared with homogeneous graphs, where node and edge types are uniform, heterogeneous graphs can more naturally characterize multiple entities and their multi-dimensional relationships in complex systems[16]. Formally, a heterogeneous graph is defined as G = (V, E), where V denotes the node set and E denotes the edge set. Let the node mapping function be Φ: V→ A and the edge mapping function be ψ: E → R, where A represents the set of node types and R represents the set of edge types.
Based on this formulation, this study constructs a circRNA-disease heterogeneous graph consisting of two types of nodes and one type of bidirectional edge[17]. Specifically, for each known association, two directed edges are created to satisfy the subsequent model requirements for heterogeneous relation propagation.
2.3 Heterogeneous graph transformer
To capture the deep semantic associations embedded in high-order meta-paths between circRNAs and diseases, this study adopts and improves the heterogeneous graph transformer (HGT) to construct the model[18,19]. Compared with traditional graph convolutional networks that are primarily designed for homogeneous graphs with single-type nodes and edges, HGT is specifically tailored for heterogeneous graphs[20]. It employs a meta-relation-aware multi-head attention mechanism to dynamically aggregate information from heterogeneous neighbors[21]. In this work, the term meta-relation refers to the type-aware semantic interaction patterns determined by different combinations of source node types, target node types, and relation directions during heterogeneous message propagation within the HGT framework, rather than multi-hop biological paths involving additional molecular entities.
Specifically, the model takes the GIPK similarities of circRNAs and diseases together with the constructed heterogeneous network as input. According to the node types and edge types, independent attention weights are assigned, enabling the model to effectively capture the semantic specificity inherent in biological associations and to generate node embeddings that fuse high-order topological information. This mechanism significantly enhances the representation capability for complex heterogeneous graphs and better satisfies the requirements of circRNA-disease association prediction in this study.
The overall HGT module consists of four core steps: node feature projection, type-aware attention computation, multi-layer information aggregation, and output embedding generation. The detailed workflow and mathematical formulations are described as follows.
2.3.1 Node feature projection
Type-specific feature mappings are first applied to circRNA and disease nodes:
where Xc and Xd denote the initial feature matrices of circRNAs and diseases after principal component analysis (PCA)-based dimensionality reduction, respectively. Wc and Wd are learnable projection matrices, while h(0)c and h(0)d represent the projected initial node embeddings of circRNAs and diseases, respectively.
2.3.2 Type-aware attention mechanism
HGT performs heterogeneous message passing via type-specific attention weights. For a target node v under the meta-relation (t,r,s) (target node type t, edge type r, source node type s), the attention weight is computed as:
where α(l)t,r,s denotes the meta-relation-specific attention coefficient for circRNA-disease associations, α(t,r,s) is the corresponding attention vector, W(l)t and W(l)s are the projection matrices for the disease target node type and circRNA source node type at layer l, respectively, and h(l-1)v represents the (l-1)-th layer feature of the target disease node.
2.3.3 Multi-layer message aggregation
By stacking L HGT layers, the model achieves multi-scale feature learning from local structures to global topology. The representation of node v at layer l is updated as:
where T denotes the set of all meta-relation types, including circRNA-disease associations, circRNA self-loops, and disease self-loops, N(t,r,s)(v) represents the neighbor set of node v under the meta-relation (t,r,s), and σ denotes the rectified linear unit (ReLU) activation function.
2.3.4 Output embedding generation
In this study, a two-layer graph convolutional architecture is employed to learn the structural information of circRNAs and diseases. The first layer captures local neighborhood features, while the second layer further aggregates higher-order global topological information. Through iterative propagation and aggregation of neighboring node features, the representations of circRNA and disease nodes are progressively refined, ultimately producing embeddings that integrate both structural and semantic information.
The final embeddings are reduced via a linear transformation:
where zc and zd denote the final feature embeddings of circRNAs and diseases, respectively.
2.4 Gradient boosting decision tree
Gradient boosting decision tree (GBDT) is a powerful ensemble learning model that has demonstrated strong performance in both classification and regression tasks[22,23]. Its core mechanism iteratively trains a sequence of decision trees, where each subsequent tree fits the residual errors of the preceding model. The final prediction is obtained by a weighted combination of these weak learners, forming a strong learner. In the circRNA-disease association prediction task, GBDT is selected as the core classifier due to its strong nonlinear modeling capability and its ability to evaluate feature importance, which enables effective mining of latent patterns in complex biological data.
A decision tree performs classification by recursively partitioning the feature space. Each internal node represents a feature test, and each leaf node corresponds to a class label. For a given sample x, the predicted value
where Rj denotes the feature space region of the j-th leaf node, wj is the corresponding weight, and I(·) is the indicator function.
The GBDT classifier iteratively optimizes the loss function
The final prediction is the weighted sum of all trees:
where ρm is the learning rate controlling the contribution of each tree, and fm denotes the m-th decision tree.
For classification tasks, the logarithmic loss function is commonly adopted:
where
3. Results and Discussion
3.1 Dataset and evaluation
To validate the effectiveness of the proposed model, three publicly available benchmark datasets were employed in this study, namely CircR2Disease V2.0[24] curated by Fan et al., circAtlas 3.0[25] compiled by Wu et al., and circRNADisease V2.0[26] organized by Sun et al. Since these datasets contain only known circRNA-disease associations and lack explicit negative samples, it is necessary to construct a balanced dataset. Specifically, all possible circRNA-disease pairs were first generated. After removing the known positive samples, an equal number of samples were randomly selected from the remaining pairs as negative samples to ensure class balance. Detailed statistics of each dataset are presented in Table 1.
Table 1. Statistics of the benchmark datasets.
| Datasets | circRNAs | Diseases | Associations | Version |
| circAtlas | 1,968 | 223 | 2,576 | 3.0 |
| CircR2Disease | 2,175 | 236 | 2,793 | 2.0 |
| circRNADisease | 3,004 | 279 | 4,686 | 2.0 |
circRNAs: circular RNAs.
We define the circRNA-disease association matrix. Let the known association matrix be Y∈Rn×m, where n and m denote the numbers of circRNAs and diseases, respectively. Each element satisfies Y(i,j) = 1 if the i-th circRNA is known to be associated with the j-th disease; otherwise, Y(i,j) = 0. For disease similarity computation, the same association matrix is transposed as YT∈Rm×n. We further define the binary vector Y(ci) to represent the interaction profile of circRNA ci, whose values are derived from the adjacency relationships between circRNA ci and diseases in the benchmark datasets. To comprehensively evaluate the model performance and ensure fair comparison with other methods, this study adopts commonly used metrics for binary classification tasks, including Accuracy, Precision, Recall, and F1-score, to quantitatively assess the model[27]. In addition, to provide an intuitive evaluation of predictive capability, the receiver operating characteristic (ROC) curve and the precision-recall (PR) curve are plotted. The areas under these curves, namely area under the receiver operating characteristic curve (AUC) and area under the precision recall curve (AUPRC), are further calculated as overall performance indicators[28].
3.2 Model performance evaluation
To evaluate the predictive performance of the proposed model, five-fold cross-validation was conducted on three benchmark datasets. As shown in Table 2, the proposed model achieved an Acc. of 86.89%, Pre. of 80.54%, Rec. of 97.33%, and F1 of 88.14% on the CircR2Disease V2.0 dataset. On the circAtlas 3.0 dataset, the Acc., Pre., Rec., and F1 reached 86.00%, 79.75%, 96.50%, and 87.33%, respectively. On the circRNADisease V2.0 dataset, the corresponding metrics were 84.26%, 77.50%, 96.65%, and 86.00%.
Table 2. Five-fold cross-validation results of the proposed model on three benchmark datasets.
| Dataset | Test Set | Acc. (%) | Pre. (%) | Rec. (%) | F1 (%) |
| CircR2Disease V2.0 | 1 | 87.33 | 81.37 | 97.27 | 88.61 |
| 2 | 87.67 | 81.51 | 97.96 | 88.98 | |
| 3 | 86.87 | 80.11 | 98.16 | 88.22 | |
| 4 | 85.94 | 80.15 | 95.89 | 87.32 | |
| 5 | 86.62 | 79.57 | 97.38 | 87.58 | |
| Mean | 86.89 | 80.54 | 97.33 | 88.14 | |
| Std | 0.67 | 0.85 | 0.89 | 0.69 | |
| circAtlas 3.0 | 1 | 86.93 | 80.84 | 96.97 | 88.17 |
| 2 | 86.40 | 81.03 | 96.32 | 88.02 | |
| 3 | 84.88 | 78.99 | 95.19 | 86.34 | |
| 4 | 85.01 | 78.48 | 95.88 | 86.31 | |
| 5 | 86.79 | 79.41 | 98.16 | 87.79 | |
| Mean | 86.00 | 79.75 | 96.50 | 87.33 | |
| Std | 0.99 | 1.13 | 1.13 | 0.93 | |
| circRNADisease V2.0 | 1 | 83.74 | 77.73 | 96.49 | 86.10 |
| 2 | 84.65 | 79.11 | 94.55 | 86.14 | |
| 3 | 85.32 | 78.88 | 96.31 | 86.73 | |
| 4 | 85.06 | 77.61 | 97.97 | 86.61 | |
| 5 | 82.55 | 74.15 | 97.92 | 84.39 | |
| Mean | 84.26 | 77.50 | 96.65 | 86.00 | |
| Std | 1.13 | 1.99 | 1.41 | 0.94 |
In addition, Figure 2 illustrates the ROC and PR curves on each dataset. The proposed model achieved AUC values of 92.17%, 91.83%, and 91.73% on CircR2Disease V2.0, circAtlas 3.0, and circRNADisease V2.0, respectively. The corresponding AUPRC values were 88.07%, 83.89%, and 91.18%.
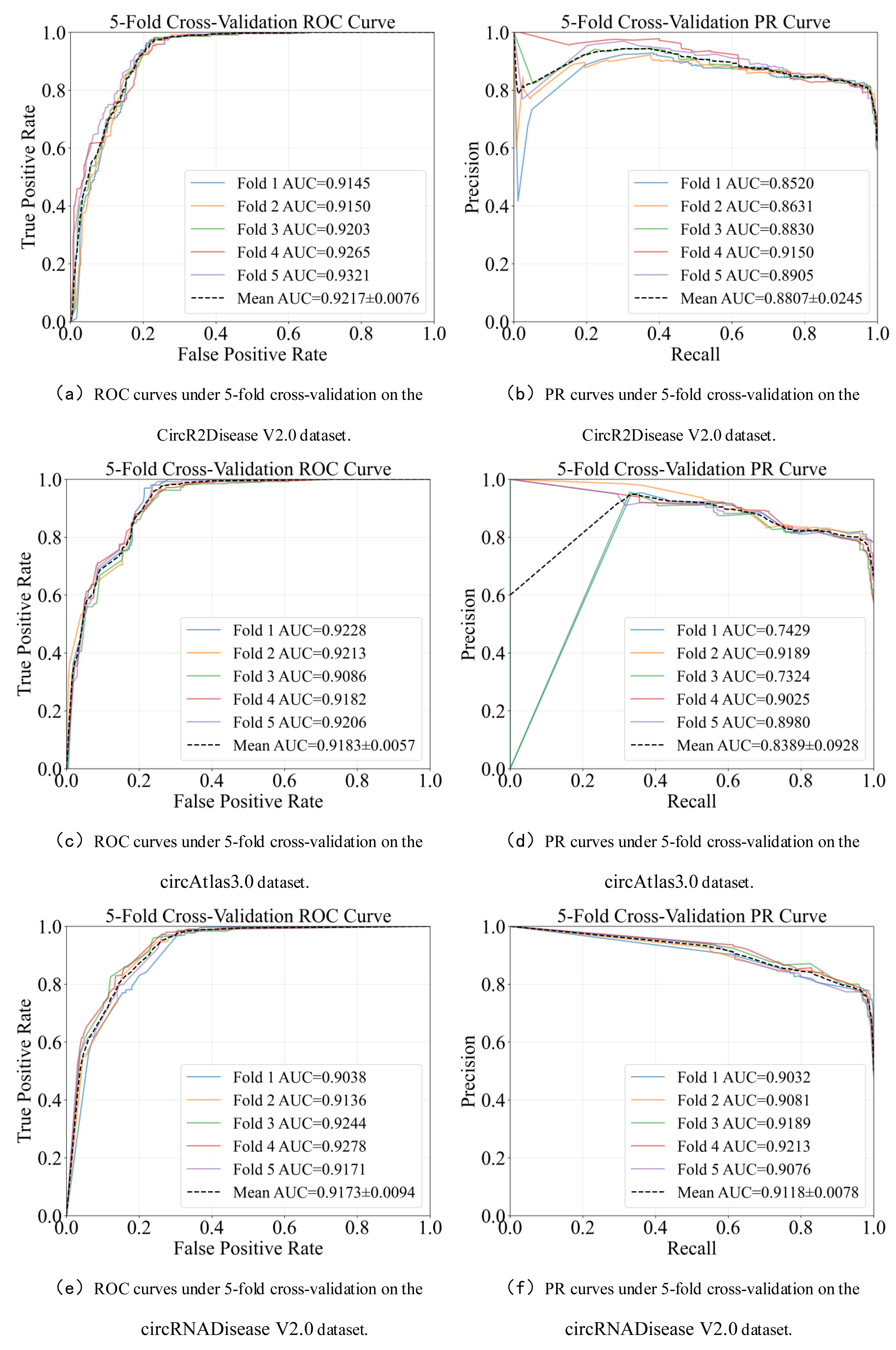{kind=link}
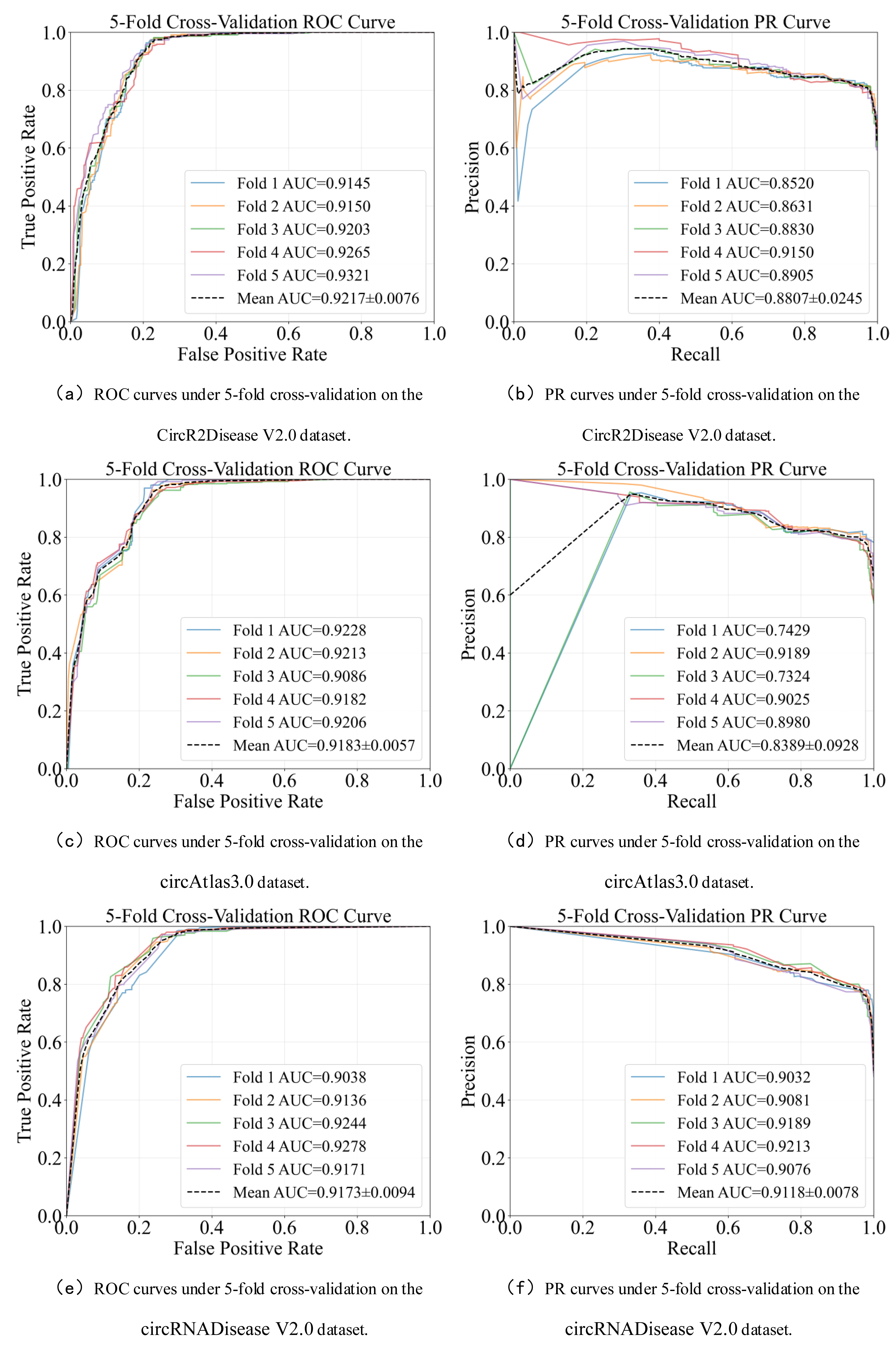
Figure 2. ROC and PR curves of the proposed model under five-fold cross-validation. ROC: receiver operating characteristic; PR: precision-recall; circRNAs: circular RNAs; AUC: area under the receiver operating characteristic curve.
3.3 Hyperparameter comparison
To investigate the impact of hyperparameters on the embedding quality of HGT and to determine the optimal configuration, this study systematically evaluates the effects of different combinations of learning rates and training epochs on the downstream association prediction performance. By generating multiple sets of embedding features and comparing the classification results, we aim to examine whether a smaller learning rate requires more epochs to converge and whether a larger learning rate tends to introduce training instability, thereby achieving a balance between underfitting and overfitting. The detailed performance of the model under different parameter settings is presented in Table 3.
Table 3. Mean AUC results of the three datasets under different hyperparameter combinations.
| Dataset | Learning Rate | Epochs | ||||
| 50 | 100 | 150 | 200 | 250 | ||
| CircR2Disease V2.0 | 0.002 | 0.9085 | 0.9096 | 0.9057 | 0.9217 | 0.9099 |
| 0.004 | 0.9089 | 0.9210 | 0.9032 | 0.9092 | 0.9041 | |
| 0.006 | 0.9140 | 0.9109 | 0.9103 | 0.9094 | 0.9022 | |
| 0.008 | 0.9161 | 0.9165 | 0.9140 | 0.9169 | 0.9052 | |
| 0.010 | 0.9212 | 0.9077 | 0.9098 | 0.9083 | 0.9094 | |
| circAtlas 3.0 | 0.002 | 0.9100 | 0.9091 | 0.9080 | 0.9105 | 0.9067 |
| 0.004 | 0.9058 | 0.9116 | 0.9183 | 0.9049 | 0.9134 | |
| 0.006 | 0.9101 | 0.9102 | 0.9098 | 0.9086 | 0.9042 | |
| 0.008 | 0.9101 | 0.9022 | 0.9096 | 0.9048 | 0.9076 | |
| 0.010 | 0.9035 | 0.9078 | 0.9044 | 0.9163 | 0.9059 | |
| circRNADisease V2.0 | 0.002 | 0.9150 | 0.9122 | 0.9115 | 0.9173 | 0.9082 |
| 0.004 | 0.9111 | 0.9048 | 0.9078 | 0.9090 | 0.9102 | |
| 0.006 | 0.9083 | 0.9098 | 0.9063 | 0.9068 | 0.9069 | |
| 0.008 | 0.9075 | 0.9036 | 0.9100 | 0.9081 | 0.9065 | |
| 0.010 | 0.9053 | 0.9097 | 0.9097 | 0.9079 | 0.9105 | |
AUC: area under the receiver operating characteristic curve.
The experimental results demonstrate that appropriate hyperparameter selection can effectively enhance feature representation capability and improve classification performance. On the CircR2Disease V2.0 dataset, the downstream GBDT classifier achieved the best performance when the learning rate was set to 0.002 and the number of training epochs was 200 (AUC = 0.9217, AUPRC = 0.8807). On the circAtlas 3.0 dataset, the optimal configuration was a learning rate of 0.004 with 150 training epochs (AUC = 0.9183, AUPRC = 0.8389). For the circRNADisease V2.0 dataset, the combination of learning rate 0.002 and 200 training epochs yielded the best performance (AUC = 0.9173, AUPRC = 0.9118).
3.4 Classifier comparison
In the circRNA-disease association prediction task, the choice of downstream classifier plays a crucial role in determining model stability and accuracy. Support vector machine (SVM) and random forest (RF) have been widely adopted in bioinformatics studies due to their robustness; however, they exhibit certain limitations when modeling complex nonlinear relationships. To evaluate the suitability of GBDT in this study, four alternative classifiers, SVM, RF, XGBoost, and LightGBM were introduced to replace GBDT under the same data fusion and feature extraction framework, thereby constructing four comparative prediction models.
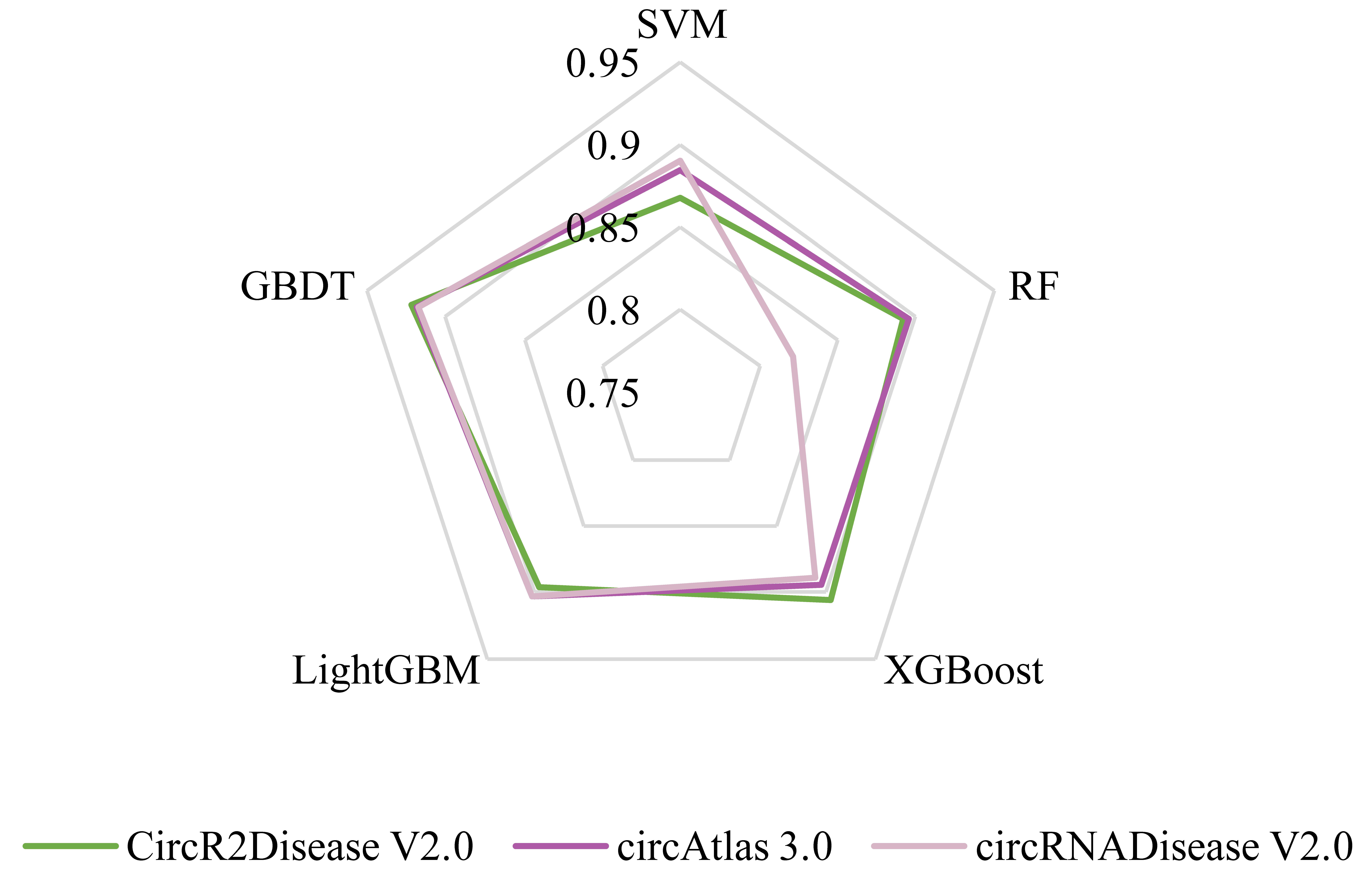
The mean AUC values of all models under five-fold cross-validation are illustrated in Figure 3. The results show that the GBDT classifier consistently achieves superior predictive performance across the three datasets compared with the other classifiers, demonstrating its effectiveness for circRNA-disease association identification. This finding indicates that GBDT can more accurately infer potential associations and provides useful guidance for classifier selection in biological network prediction tasks.
{kind=link}
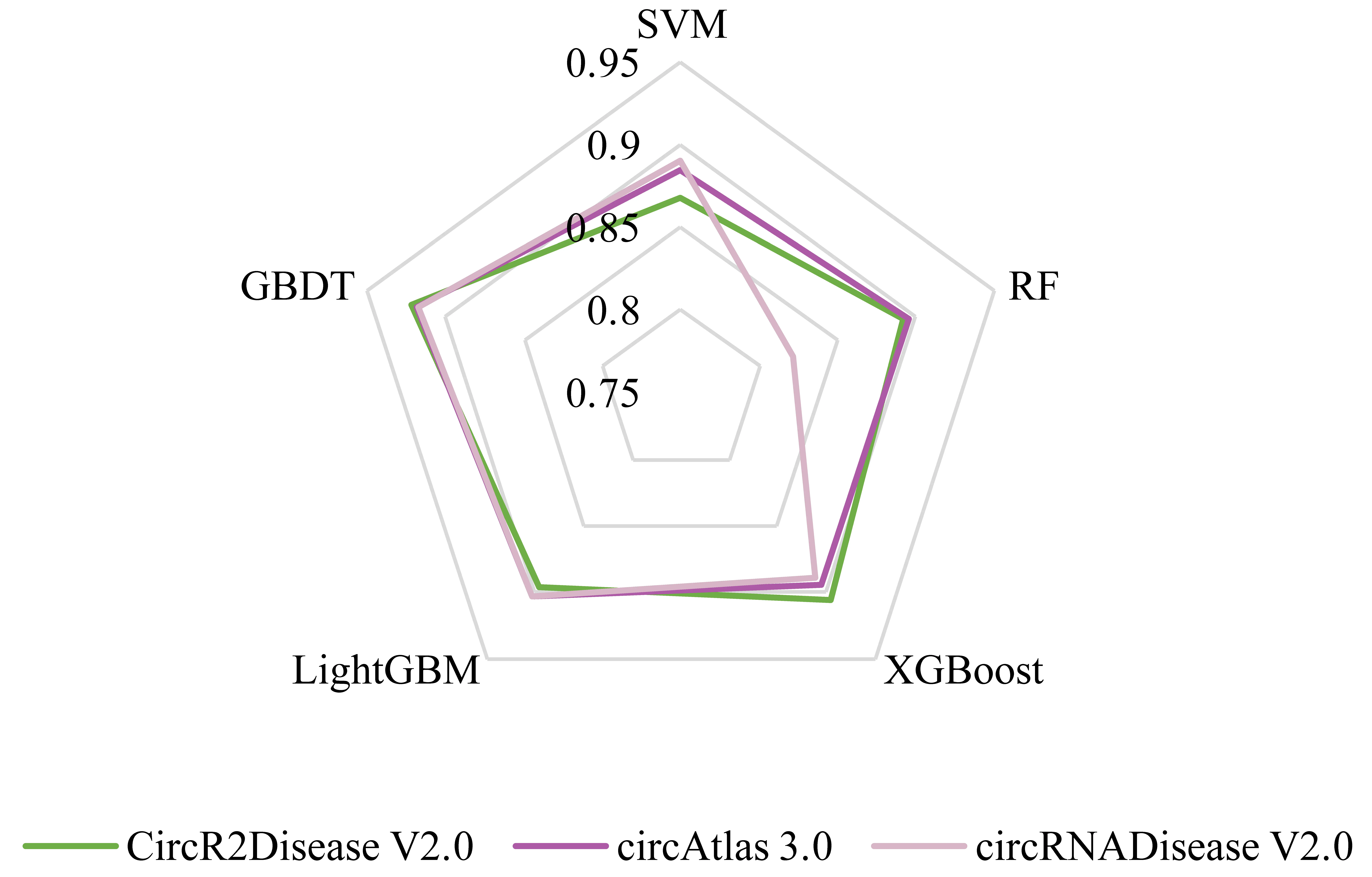
Figure 3. Comparison of AUC values across different classifiers. AUC: area under the receiver operating characteristic curve; RF: random forest; GBDT: Gradient boosting decision tree; SVM: support vector machine.
3.5 Ablation study
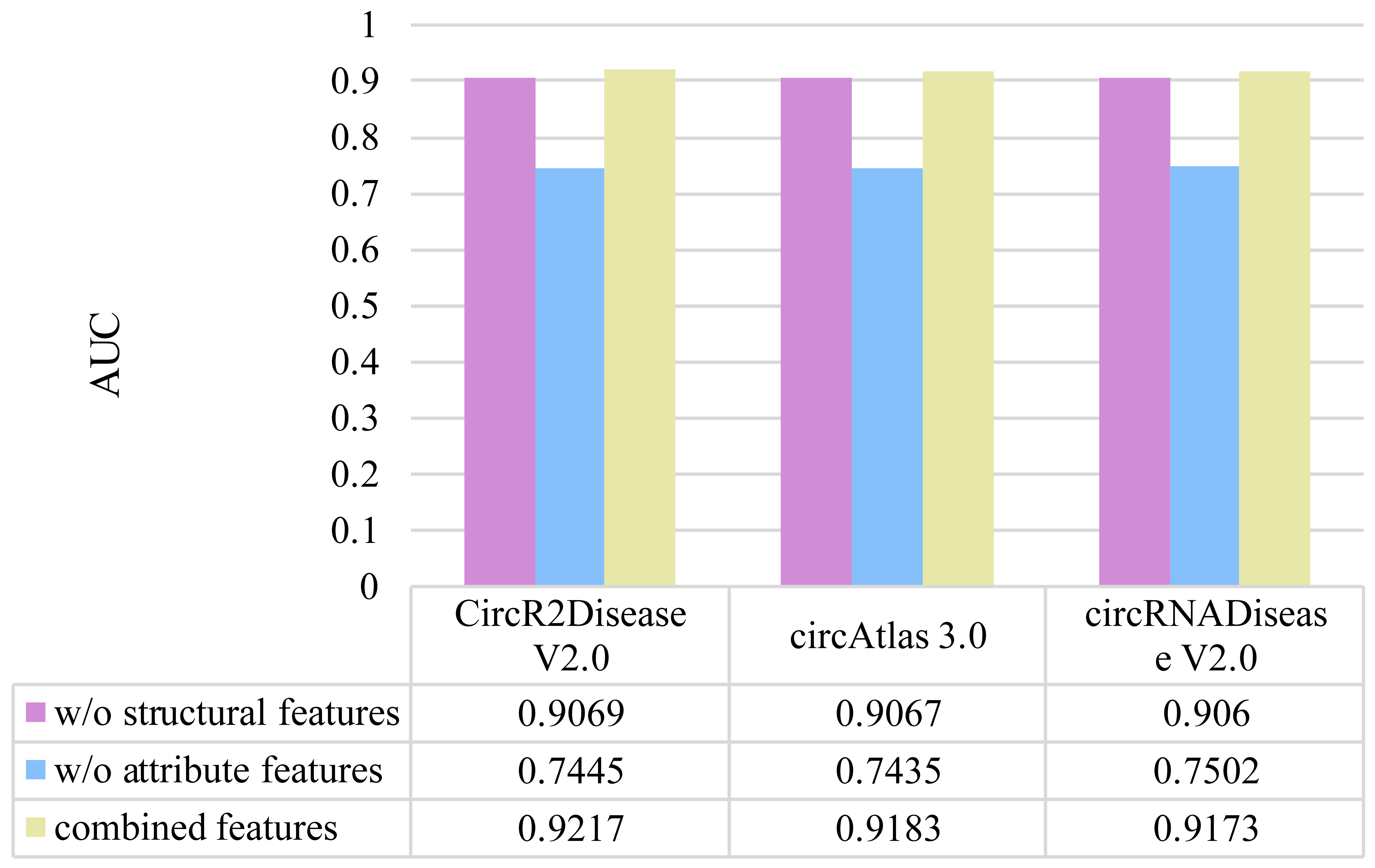
To investigate the contribution of multimodal feature fusion to circRNA-disease association prediction, ablation experiments were designed to compare the effectiveness of attribute features and structural features from different perspectives. Specifically, the attribute features include the GIPK similarities of circRNAs and diseases, whereas the structural features refer to the embeddings generated by HGT using only graph structure propagation without introducing node attribute inputs. Three feature configurations, attribute only, structure only, and their fusion, were evaluated under a fixed GBDT classifier and five-fold cross-validation setting.
The experimental results are illustrated in Figure 4. Compared with single-feature configurations, the model that integrates both attribute and structural features consistently achieves higher mean AUC values across all datasets. This observation indicates that attribute information and topological structure are complementary in characterizing entity semantics, and their effective fusion is a key factor in improving circRNA-disease association prediction performance.
{kind=link}
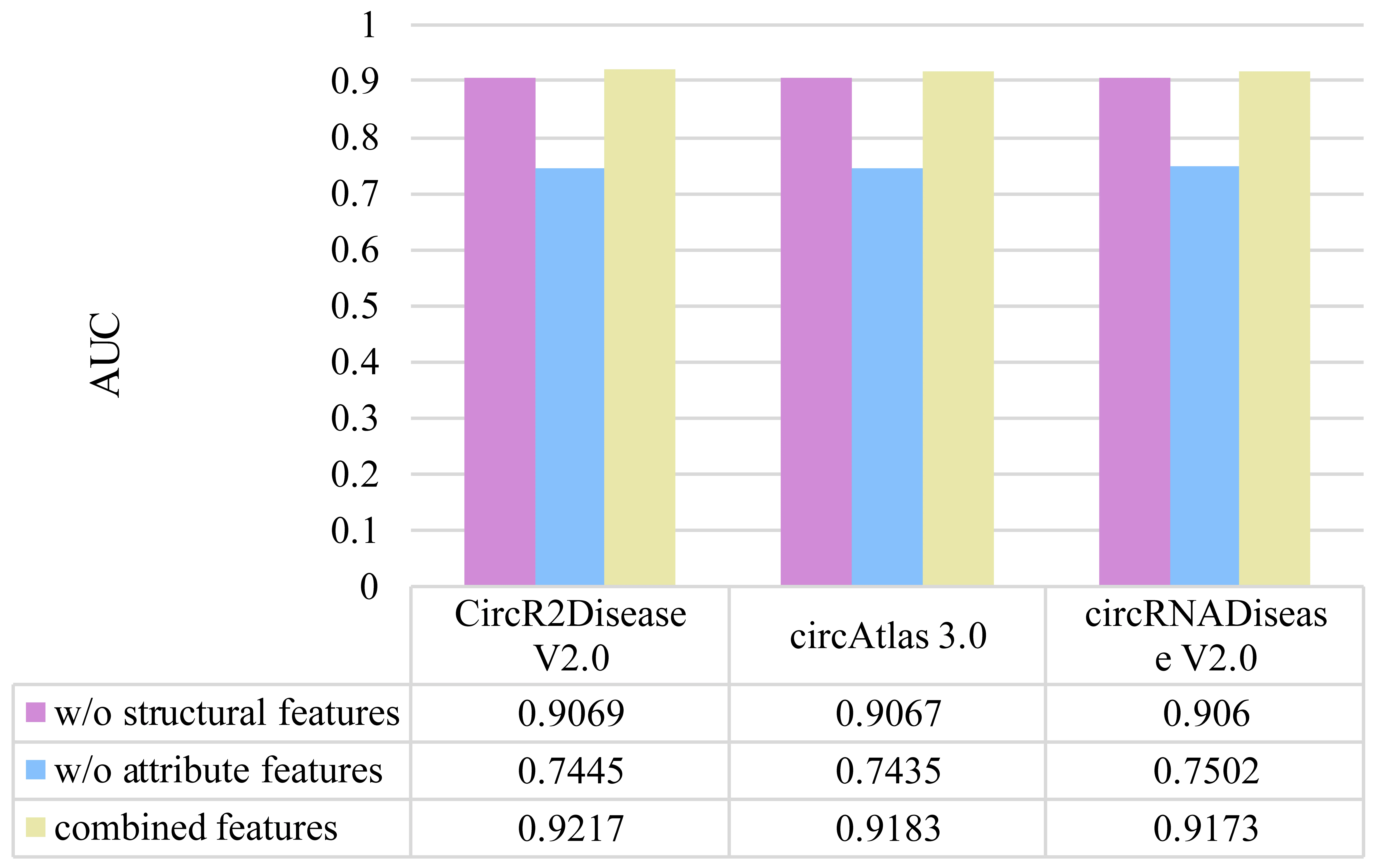
Figure 4. AUC comparison under different feature combinations across three datasets. AUC: area under the receiver operating characteristic curve.
3.6 Graph embedding method comparison
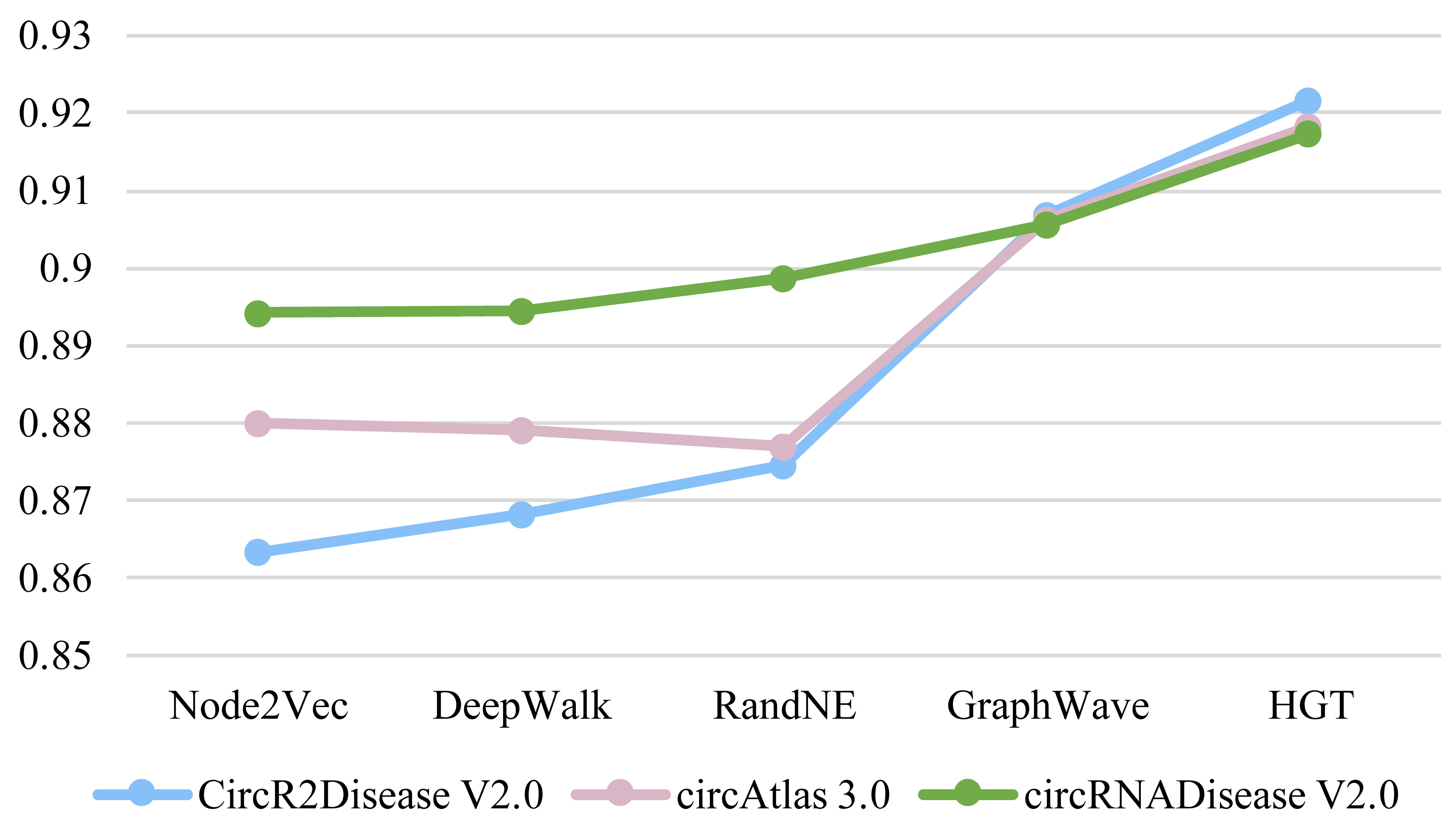
To systematically evaluate the performance of different graph embedding methods for circRNA-disease association prediction, this study employs the Karateclub graph embedding toolkit and compares HGT with four representative methods: Node2Vec[29], DeepWalk[30], RandNE[31], and GraphWave[32]. In this study, the downstream GBDT classifier is kept unchanged while only the graph embedding module is replaced to examine the impact of each method on predictive performance.
The selected methods span diverse technical routes. Node2Vec flexibly captures both local and global network structures by introducing the and parameters to control the random walk strategy. DeepWalk generates node sequences via truncated random walks and learns embeddings using the Skip-Gram model. RandNE preserves global network topology through an efficient random projection-based matrix factorization approach. GraphWave, from a graph signal processing perspective, characterizes local structural features of nodes via a heat kernel diffusion process.
The mean AUC comparison of all methods under five-fold cross-validation is shown in Figure 5. The experimental results demonstrate that HGT achieves the best performance across all three datasets, validating its capability to model complex relationships in heterogeneous biological networks through a dynamic attention mechanism and highlighting its significant advantage over traditional homogeneous graph embedding methods.
{kind=link}
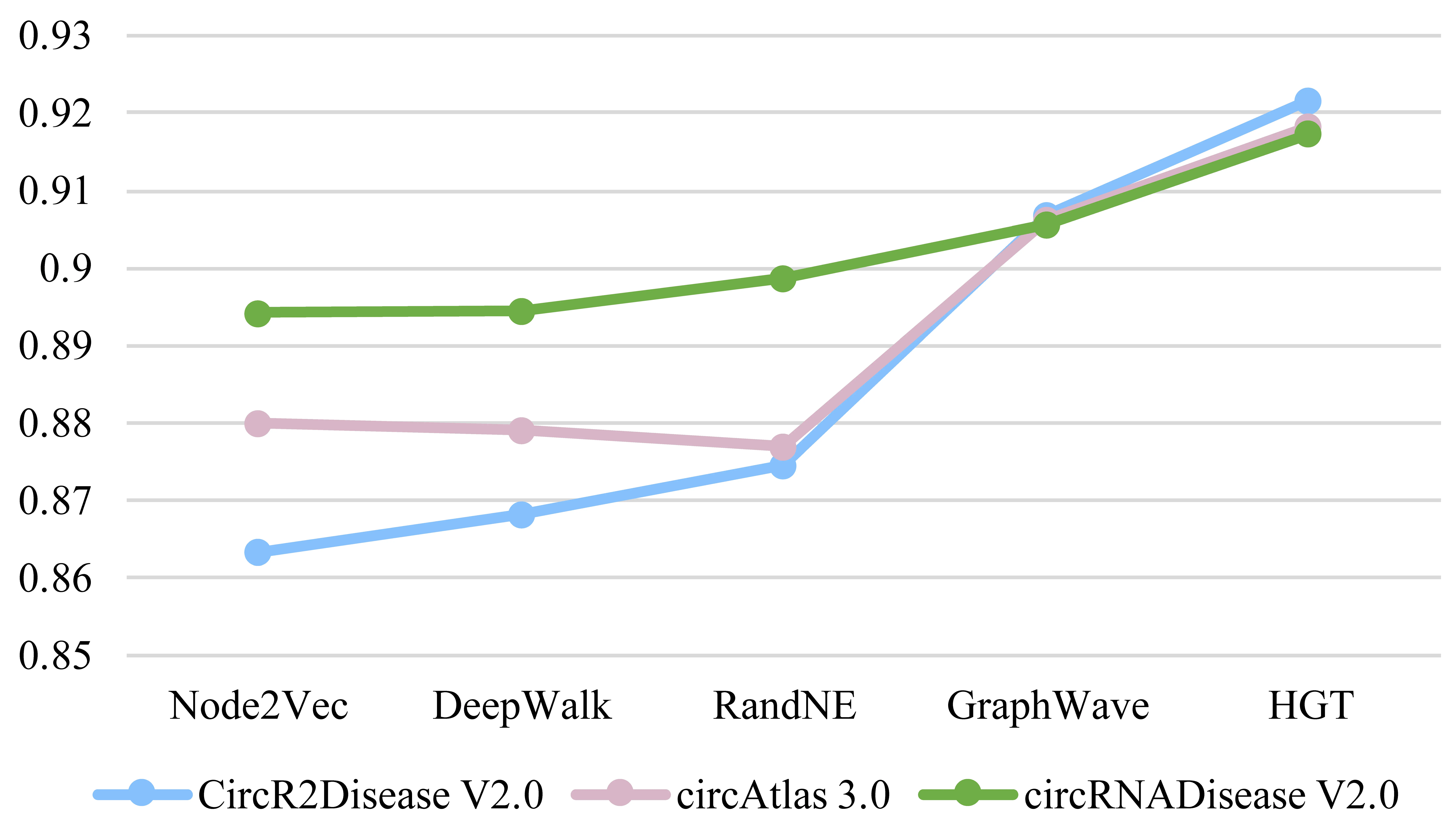
Figure 5. Comparison of AUC values across different graph embedding methods. AUC: area under the receiver operating characteristic curve.
3.7 Fusion strategy comparison
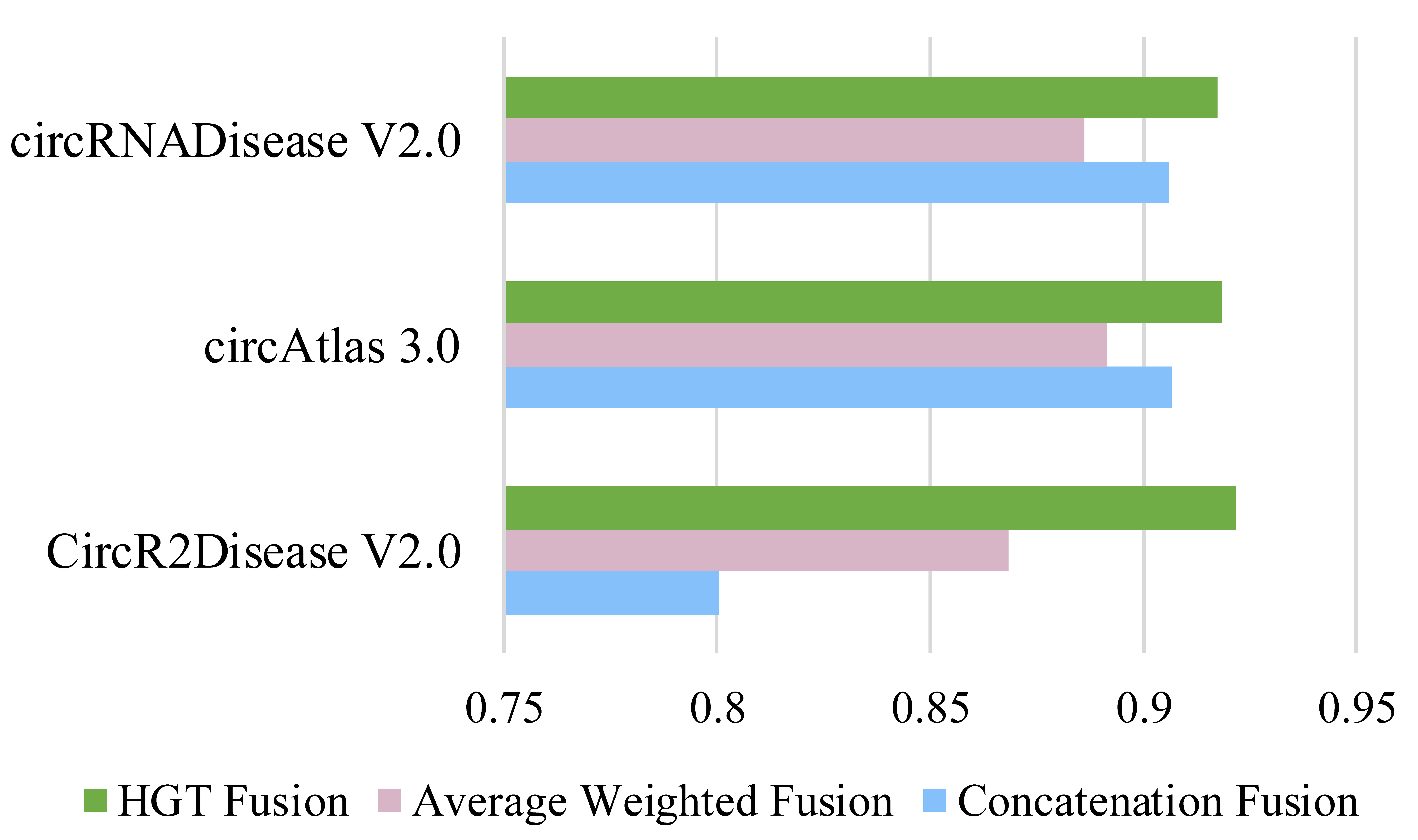
To validate the effectiveness of the heterogeneous graph neural network fusion strategy, comparative experiments were designed based on the preliminary framework[33]. By introducing two classical feature fusion methods, simple concatenation fusion and average weighted fusion, this study systematically evaluates the impact of different fusion strategies on circRNA-disease association prediction performance, aiming to provide practical guidance for feature fusion selection in this task.
The simple concatenation fusion directly performs horizontal concatenation of the circRNA attribute feature vector Xcirc and the structural feature vector Ycirc obtained solely from graph structure propagation. Similarly, the disease attribute feature vector Xdisease and structural feature vector Ydisease are concatenated to generate fused features Zcirc = (Xcirc, Ycirc), Zdisease = (Xdisease, Ydisease). The core assumption of this strategy is that the two feature types are naturally complementary in semantic space and that concatenation preserves complete information. The average weighted fusion generates fused features via weighted averaging Zcirc(disease) = αXcirc(disease) + (1-α)Ycirc(disease), where the weight α is set to 0.5 (equal-weight fusion). This strategy implicitly assumes that the two feature types contribute equally to the target task.
Figure 6 presents the performance comparison of the three fusion strategies in the form of bar charts. The results show that HGT-based fusion achieves the best performance across all evaluation metrics, particularly demonstrating a clear advantage in AUC compared with simple concatenation and average weighted fusion. Overall, HGT fusion more effectively captures the association patterns between circRNAs and diseases and provides superior feature representation capability compared with traditional fusion methods.
{kind=link}
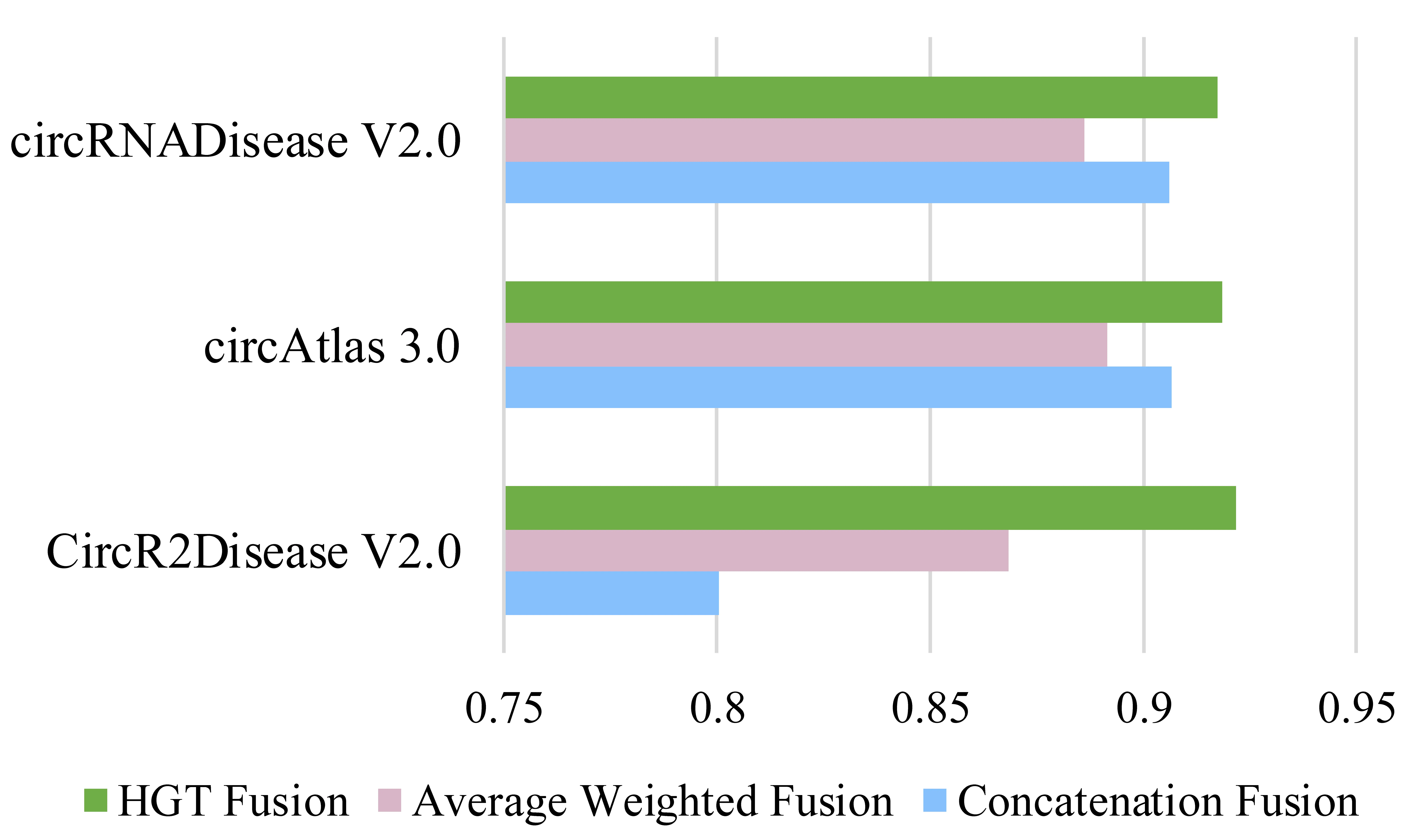
Figure 6. Comparison of AUC values across different fusion strategies. AUC: area under the receiver operating characteristic curve.
3.8 Comparison with representative state-of-the-art methods
To further validate the effectiveness and competitiveness of the proposed framework, we additionally compared our method with several representative circRNA-disease association prediction methods, including Lu’s model[34], Liu’s model[35], GATCL2CD[36], EELMCDA[37], and MuseCDA[38]. These methods cover different technical paradigms, including graph neural networks, ensemble learning, and multi-source feature fusion strategies.
The comparative results are presented in Table 4. As shown in the table, the proposed framework consistently achieves the best overall performance among all compared methods, obtaining an Accuracy of 86.89%, an F1-score of 88.14%, and an AUC of 92.17% on the benchmark dataset. In particular, compared with the strongest baseline MuseCDA, the proposed method improves the Accuracy, F1-score, and AUC by 1.61%, 2.66%, and 0.70%, respectively. These results demonstrate that the proposed meta-relation-aware heterogeneous graph learning framework can more effectively capture the complex interaction patterns between circRNAs and diseases, thereby achieving superior predictive performance compared with representative state-of-the-art methods.
Table 4. Performance comparison with state-of-the-art methods.
| Model | Acc. (%) | F1 (%) | AUC (%) |
| Lu’s model | 77.91 | 78.79 | 90.19 |
| Liu’s model | 70.52 | 70.11 | 73.16 |
| GATCL2CD | 53.98 | 23.61 | 59.18 |
| EELMCDA | 77.98 | 77.94 | 87.26 |
| MuseCDA | 85.28 | 85.48 | 91.47 |
| Ours | 86.89 | 88.14 | 92.17 |
AUC: area under the receiver operating characteristic curve.
3.9 Case study
To further evaluate the biological interpretability and practical applicability of the proposed framework, we conducted a case study on lung cancer based on the CircR2Disease V2.0 dataset. Specifically, the proposed model was used to predict potential circRNAs associated with lung cancer, and the top 20 candidate circRNAs ranked by prediction scores were selected for literature validation through published biomedical studies indexed in PubMed.
The prediction results are summarized in Table 5. As shown in the table, most of the top-ranked predicted circRNAs have been supported by previously published studies. For example, hsa_circ_0004214 (PMID: 28622299), circHIPK3 (PMID: 30352682), hsa_circ_0067934 (PMID: 33155212), and circZNF609 (PMID: 33459380) have all been experimentally reported to be associated with lung cancer progression, proliferation, or metastasis. Overall, 17 out of the top 20 predicted circRNAs were confirmed by existing literature evidence, demonstrating that the proposed framework can effectively identify biologically meaningful circRNA-disease associations with strong biological relevance and interpretability.
Table 5. Top 20 predicted circRNAs associated with lung cancer and supporting evidence.
| Num | circRNA | Predicted Score | PMID |
| 1 | hsa_circ_0004214 | 0.8265 | 28622299 |
| 2 | circHIPK3 | 0.8218 | 30352682 |
| 3 | hsa_circ_0067934 | 0.8213 | 33155212 |
| 4 | hsa_circ_0012673 | 0.7507 | 32141553 |
| 5 | Circ_100565 | 0.7416 | 32425695 |
| 6 | circ0006916 | 0.7410 | 29726904 |
| 7 | hsa_circ_0004350 | 0.7389 | 31197975 |
| 8 | hsa_circ_0008193 | 0.7364 | 31700878 |
| 9 | hsa_circ_0022812 | 0.7322 | 32511866 |
| 10 | hsa_circ_0008345 | 0.7200 | N/A |
| 11 | circDLGAP4 | 0.7194 | 32646340 |
| 12 | hsa_circ_0015278 | 0.7191 | 30176158 |
| 13 | circ_0032462 | 0.7189 | N/A |
| 14 | circZNF609 | 0.7185 | 33459380 |
| 15 | hsa_circ_0001073 | 0.7178 | 33475233 |
| 16 | hsa_circ_0007059 | 0.7137 | 31351967 |
| 17 | hsa_circ_0084003 | 0.7134 | N/A |
| 18 | circ-BANP | 0.7127 | 29969631 |
| 19 | hsa_circRNA_103809 | 0.7116 | 29698681 |
| 20 | hsa_circ_0000064 | 0.7056 | 29223555 |
circRNA: circular RNAs.
Despite its promising performance, several limitations remain. First, the current study is constrained by the scale and potential noise of known association data. Future work may incorporate multi-omics data, such as expression profiles and methylation information, to construct more comprehensive biological networks, or adopt semi-supervised learning strategies to alleviate data sparsity. Second, the existing model assumes a static biological network, whereas real biological processes are inherently dynamic. Future research could explore temporal graph neural networks to capture the dynamic evolution of associations. In future work, we will focus on integrating richer biological information and further optimizing graph neural network architectures to improve predictive accuracy and generalization capability.
4. Conclusion
This study proposes a meta-relation-aware circRNA-disease association prediction model. By integrating neighborhood structural information of biomolecules, the model employs a heterogeneous graph neural network to effectively learn high-order neighborhood features of circRNAs and diseases, generating fused embedding representations. A gradient boosting decision tree is further introduced to perform association inference. The five-fold cross-validation results on three benchmark datasets demonstrate the superior overall predictive performance of the proposed model. Moreover, across multiple comparative studies, including hyperparameter analysis, classifier selection, ablation experiments, graph embedding methods, and feature fusion strategies, the proposed model consistently achieves the best performance, further validating its stability and effectiveness. Overall, the model can reliably identify potential circRNA-disease associations and provide valuable candidate molecules for subsequent wet-lab validation.
Acknowledgements
The authors declare that Gemini 3.1 was used solely for language polishing during the manuscript preparation process. All research content, including study design, data analysis, interpretations, figures, and tables, is original and was not generated using AI tools. The authors take full responsibility for the integrity, originality, and accuracy of the work.
Authors contribution
Tan X: Conceptualization, methodology, software, formal analysis, investigation, data curation, writing-original draft.
Wei M: Conceptualization, methodology, validation, formal analysis, writing-original draft, writing-review & editing.
Xia Z: Data curation, validation, investigation.
Wang X: Software, data curation, visualization.
Li Y: Formal analysis, validation, investigation.
Wang L: Conceptualization, supervision, funding acquisition, project administration, writing-review & editing.
You Z: Supervision, funding acquisition, project administration, writing-review & editing.
Conflicts of interest
Lei Wang is an Executive Editor of Computational Biomedicine. The other authors declare no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
Data supporting the findings of this study are available from the corresponding author upon reasonable request.
Funding
This work was supported by the Guangxi Science and Technology Program (Grant No. 2024-102-3); the Natural Science Foundation of Guangxi (Grant Nos. 2024GXNSFAA010283 and 2023GXNSFDA026031); Natural Science Foundation of Shandong (Grant No. ZR2024MF042); National Natural Science Foundation of China (Grant Nos. 62573419 and 62172355); National Science Foundation for Distinguished Young Scholars of China (Grant No. 62325308).
Copyright
© The Author(s) 2026.
References
-
1. Vo JN, Cieslik M, Zhang Y, Shukla S, Xiao L, Zhang Y, et al. The landscape of circular RNA in cancer. Cell. 2019;176(4):869-881.e13.[DOI]
-
2. Slack FJ, Chinnaiyan AM. The role of non-coding RNAs in oncology. Cell. 2019;179(5):1033-1055.[DOI]
-
4. Li B, Bai WW, Guo T, Tang ZY, Jing XJ, Shan TC, et al. Statins improve cardiac endothelial function to prevent heart failure with preserved ejection fraction through upregulating circRNA-RBCK1. Nat Commun. 2024;15:2953.[DOI]
-
5. Conn VM, Chinnaiyan AM, Conn SJ. Circular RNA in cancer. Nat Rev Cancer. 2024;24(9):597-613.[DOI]
-
6. Lei X, Tang X, Zhang Y. Drug-target affinity prediction based on multi-source information and graph convolutional network. Comput Biomed. 2026;1(2).[DOI]
-
7. Rahmani-Kukia N, Abbasi A. New insights on circular RNAs and their potential applications as biomarkers, therapeutic agents, and preventive vaccines in viral infections: With a glance at SARS-CoV-2. Mol Ther Nucleic Acids. 2022;29:705-717.[DOI]
-
8. Liu X, Chen F, Pan J, Ai C, Guo F, Tang J. MediHerb: A multi-modal enhanced framework for disease inference via herbal knowledge. Comput Biomed. 2026;1(1).[DOI]
-
9. Huang YA, Li YC, You ZH, Hu L, Hu PW, Wang L, et al. Consensus representation of multiple cell–cell graphs from gene signaling pathways for cell type annotation. BMC Biol. 2025;23(1):23.[DOI]
-
10. Ma Z, Kuang Z, Deng L. CRPGCN: Predicting circRNA-disease associations using graph convolutional network based on heterogeneous network. BMC Bioinform. 2021;22(1):551.[DOI]
-
11. Li Y, Hu XG, Wang L, Li PP, You ZH. MNMDCDA: Prediction of circRNA–disease associations by learning mixed neighborhood information from multiple distances. Brief Bioinform. 2022;23(6):bbac479.[DOI]
-
12. Xie G, Chen H, Sun Y, Gu G, Lin Z, Wang W, et al. Predicting circRNA-disease associations based on deep matrix factorization with multi-source fusion. Interdiscip Sci Comput Life Sci. 2021;13(4):582-594.[DOI]
-
14. Wu Q, Deng Z, Pan X, Shen HB, Choi KS, Wang S, et al. MDGF-MCEC: A multi-view dual attention embedding model with cooperative ensemble learning for CircRNA-disease association prediction. Brief Bioinform. 2022;23(5):bbac289.[DOI]
-
16. Yi HC, You ZH, Huang DS, Kwoh CK. Graph representation learning in bioinformatics: Trends, methods and applications. Brief Bioinform. 2022;23:bbab340.[DOI]
-
17. Wang L, Li S, Su X, Li Z, Wei M, Zhao B, et al. Prediction of Budd-Chiari syndrome based on attention mechanisms of high-risk factors in multi-hop graph learning. Sci China Inf Sci. 2025;68(7):179102.[DOI]
-
20. Yao L, Mao C, Luo Y. Graph convolutional networks for text classification. Proc AAAI Conf Artif Intell. 2019;33(1):7370-7377.[DOI]
-
21. Qiao Y, Hu L, Zhang J, Hu P, Luo X. Identifying novel therapeutic targets of natural compounds in traditional Chinese medicine herbs with hypergraph representation learning. Brief Bioinform. 2025;26(4):bbaf399.[DOI]
-
22. Wei M, Wang L, Su X, Zhao B, You Z. Multi-hop graph structural modeling for cancer-related circRNA-miRNA interaction prediction. Pattern Recognit. 2026;170:112078.[DOI]
-
23. Du L, Song H, Xu Y, Dai S. An architecture as an alternative to gradient boosted decision trees for multiple machine learning tasks. Electronics. 2024;13(12):2291.[DOI]
-
26. Sun ZY, Yang CL, Huang LJ, Mo ZC, Zhang KN, Fan WH, et al. circRNADisease v2.0: An updated resource for high-quality experimentally supported circRNA-disease associations. Nucleic Acids Res. 2024;52(D1):D1193-D1200.[DOI]
-
27. Wang L, You ZH, Huang DS, Li JQ. MGRCDA: Metagraph recommendation method for predicting CircRNA–disease association. IEEE Trans Cybern. 2023;53(1):67-75.[DOI]
-
28. Li Y, Liu XZ, You ZH, Li LP, Guo JX, Wang Z. A computational approach for predicting drug–target interactions from protein sequence and drug substructure fingerprint information. Int J Intell Syst. 2021;36(1):593-609.[DOI]
-
29. Grover A, Leskovec J. node2vec: Scalable feature learning for networks. In: Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining; 2016 Aug 13; San Francisco California, USA. New York: Association for Computing Machinery; 2016. p. 855-864.[DOI]
-
30. Perozzi B, Al-Rfou R, Skiena S. DeepWalk: Online learning of social representations. In: In: Proceedings of the 20th ACM SIGKDD international conference on Knowledge discovery and data mining; 2014 Aug 24-27; New York, USA. New York: Association for Computing Machinery; 2014. p. 701-710.[DOI]
-
31. Zhang Z, Cui P, Li H, Wang X, Zhu W. Billion-scale network embedding with iterative random projection. In: 2018 IEEE International Conference on Data Mining (ICDM); 2018 Nov 17-20; Singapore. Piscataway: IEEE; 2018. p. 787-796.[DOI]
-
32. Lee J, Amornpaisannon B, Mitra T, Carlson TE. GraphWave: A highly-parallel compute-at-memory graph processing accelerator. In: 2022 Design, Automation & Test in Europe Conference & Exhibition (DATE); 2022 Mar 14-23; Antwerp, Belgium. Piscataway: IEEE; 2022. p. 256-261.[DOI]
-
33. Wei M, Wang L, Li Y, Li Z, Zhao B, Su X, et al. BioKG-CMI: A multi-source feature fusion model based on biological knowledge graph for predicting circRNA-miRNA interactions. Sci China Inf Sci. 2024;67(8):189104.[DOI]
-
34. Lu M, Wang L, Sun J, Li Y, Wei M, Wei Y, et al. Predicting CircRNA-disease associations through non-negative matrix factorization and adversarially regularized variational graph autoencoder. In: 2024 IEEE International Conference on Bioinformatics and Biomedicine (BIBM); 2024 Dec 3-6; Lisbon, Portugal. Piscataway: IEEE; 2024. p. 1060-1065.[DOI]
-
35. Liu C, Wang L, Zhao B, Wei M, Li Y, Lu M, et al. Collaborative framework for circRNA-disease associations prediction using dual variational graph. IEEE Trans Big Data. 2025;11(6):2884-2895.[DOI]
-
37. Wang Z, Wang L, You ZH, Wang L, Li Y, Wang Z. EELMCDA: Combining evolutionary ensemble learning with matrix feature decomposition for predicting circRNA-disease associations. In: 2024 IEEE International Conference on Bioinformatics and Biomedicine (BIBM); 2024 Dec 3-6; Lisbon, Portugal. Piscataway: IEEE; 2024. p. 1199-1206.[DOI]
-
38. Wei MM, Li SL, Li YC, Wang XF, Ruan CW, Li RR, et al. MuseCDA: Predicting CircRNA-disease associations via multi-scale structure embedding. In: 2025 IEEE International Conference on Bioinformatics and Biomedicine (BIBM); 2025 Dec 15-18; Wuhan, China. Piscataway: IEEE; 2025. p. 1313-1318.[DOI]
Copyright
© The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Publisher’s Note
Science Exploration remains a neutral stance on jurisdictional claims in published
maps
and institutional affiliations. The views expressed in this article are solely those
of
the author(s) and do not reflect the opinions of the Editors or the publisher.
Share And Cite



Science Exploration Style
Tan X, Wei M, Xia Z, Wang X, Li Y, Wang L, et al. Identification of potential associations between circRNAs and diseases based on meta relation aware. Comput Biomed. 2026;1:202609. https://doi.org/10.70401/cbm.2026.0020
Tips
Copy completed.
Submit a Manuscript
Author Instructions
Cite this Article
Article Metrics
0
View
0
Download
Cited
Article Updates

Science Exploration Style
Tan X, Wei M, Xia Z, Wang X, Li Y, Wang L, et al. Identification of potential associations between circRNAs and diseases based on meta relation aware. Comput Biomed. 2026;1:202609. https://doi.org/10.70401/cbm.2026.0020
copy
Share Link
copy