Advances in database resources and computational methods for predicting antibody polyreactivity
Haoxiang Tang
1
,
Zixuan Zhang
1
,
Wenzhi Li
1
,
Xianrun Pan
1

,
Yuwei Zhou
2,*
,
Jian Huang
1,3,*
*Correspondence to:
Jian Huang, The Clinical Hospital of Chengdu Brain Science Institute, School of Life Science and Technology, University of Electronic Science and Technology of China, Chengdu 611731, Sichuan, China.
E-mail: hj@uestc.edu.cn
Yuwei Zhou, Key Laboratory of Non-coding RNA and Drug Discovery at Chengdu Medical College of Sichuan Province, School of Basic Medical Sciences, Chengdu Medical College, Chengdu 610500, Sichuan, China. E-mail: zyw@cmc.edu.cn
Yuwei Zhou, Key Laboratory of Non-coding RNA and Drug Discovery at Chengdu Medical College of Sichuan Province, School of Basic Medical Sciences, Chengdu Medical College, Chengdu 610500, Sichuan, China. E-mail: zyw@cmc.edu.cn
Comput Biomed. 2026;1:202616. 10.70401/cbm.2026.0021
Received: April 29, 2026Accepted: July 13, 2026Published: July 14, 2026
Abstract
Antibody polyreactivity refers to the ability of a monoclonal antibody to non-specifically bind to a diverse range of antigens. While this property may be an intrinsic mechanism of the immune response, it poses significant challenges in therapeutic antibody development, often leading to off-target effects, poor pharmacokinetics, and potential toxicity. This review compiles the data resources related to polyreactive antibodies and places a particular emphasis on computational models for predicting antibody polyreactivity. The latter includes empirical models based on physicochemical properties, traditional machine learning models, deep learning networks, and protein language models. Through delineating the complexity of antibody polyreactivity, this review emphasizes the critical role and growing potential of computational prediction tools in selecting and engineering antibody drug candidates at early stages, thereby reducing development risks and accelerating the development of safer and better therapeutic antibodies.
Keywords
Antibody polyreactivity, machine learning, protein language models, antibody polyspecificity
1. Introduction
Antibody-antigen interactions were previously believed to strictly follow a “lock-and-key” model of precise complementarity[1]. However, a growing number of antibodies have been found capable of binding to multiple structurally unrelated antigens. This phenomenon was initially regarded as a harmful pathological occurrence[2], yet subsequent studies found that these antibodies capable of binding multiple antigens can also be generated under normal physiological conditions[3-5]. At present, it is generally accepted that antibody specificity is not a binary property, but rather resembles a continuous spectrum[6].
In therapeutic antibody development, polyreactive antibodies can bind diverse antigens in a nonspecific manner, thereby perturbing pharmacokinetics and giving rise to off-target effects, deposition in non-target tissues, and rapid clearance, ultimately reducing efficacy while increasing toxicity[7-10]. They may even trigger immune complex deposition and autoimmune reactions through cross reactivity with endogenous antigens, and have been implicated in a range of diseases and graft rejection[11-18]. Therefore, it is necessary to evaluate the polyreactivity of candidate therapeutic antibodies in order to mitigate these adverse impacts.
1.1 Polyreactivity and polyspecificity
The terms polyreactive antibodies and polyspecific antibodies (also known as multispecific antibodies) are often used interchangeably in the field[6,10,19]. Although both phenomena manifest as a single antibody being able to bind multiple antigens, their molecular bases differ substantially. Many earlier studies treated binding affinity as the key criterion for distinguishing polyreactivity from polyspecificity[10,20]. However, polyspecific antibodies can exhibit enormous differences in stability when binding different antigens, while polyreactive antibodies can also achieve a marked increase in “apparent affinity” through assistance from the membrane-surface environment or heterogeneous ligands[21,22]. Therefore, affinity should not be used as the sole criterion to classify antibodies as polyreactive or polyspecific. Accordingly, this review distinguishes polyspecificity from polyreactivity by whether binding relies on a paratope-epitope binding mode. Polyspecific antibodies are typically mediated by classical paratope-epitope interactions, displaying higher specificity and generally higher affinity; by contrast, polyreactive antibodies do not rely on a single fixed binding mode, and therefore exhibit nonspecificity and relatively weaker affinity. Based on these considerations, we define polyreactive antibodies as a class of antibodies that lack specific and stable paratope-epitope interactions and are primarily characterized by nonspecific hydrophobic and electrostatic stickiness. In practice, however, this binding-mode-based criterion is not readily applicable to most existing high-throughput datasets due to the lack of epitope-level or structural information required to infer binding mechanisms.
Polyspecific antibodies can further be divided into two categories based on their heavy-light (HL) chain composition (Figure 1A). Each antibody in the first category contains different HL chain pairs, with antigen-binding fragment (Fab) arms derived from different parental antibodies. This can happen naturally, for example, as a result of Fab-arm exchange in immunoglobulin G4 (IgG4)[23]. These antibodies can also be engineered products made by DuoBody or CrossMab technology[24,25]. Although each antibody in the first category can bind to two or more different antigens, each Fab arm still follows the conventional “one Fab, one epitope” recognition mode. In the second category, HL chain pairs of each antibody are identical. However, each antibody can still bind to two or more different epitopes on the same or different antigens[26]. That is to say, they exhibit a “one Fab, two or more epitopes” recognition mode. Nevertheless, the binding still depends on specific paratope-epitope recognition. This type of polyspecificity can happen naturally due to conformational diversity and SPE-7 immunoglobulin E (IgE) is a typical example[27,28]. This also reflects that the complementarity-determining regions (CDRs) of only one Fab are sufficient to form split paratopes or overlapping paratopes, such as DutaFabs or Dual Action Fabs[29-31].
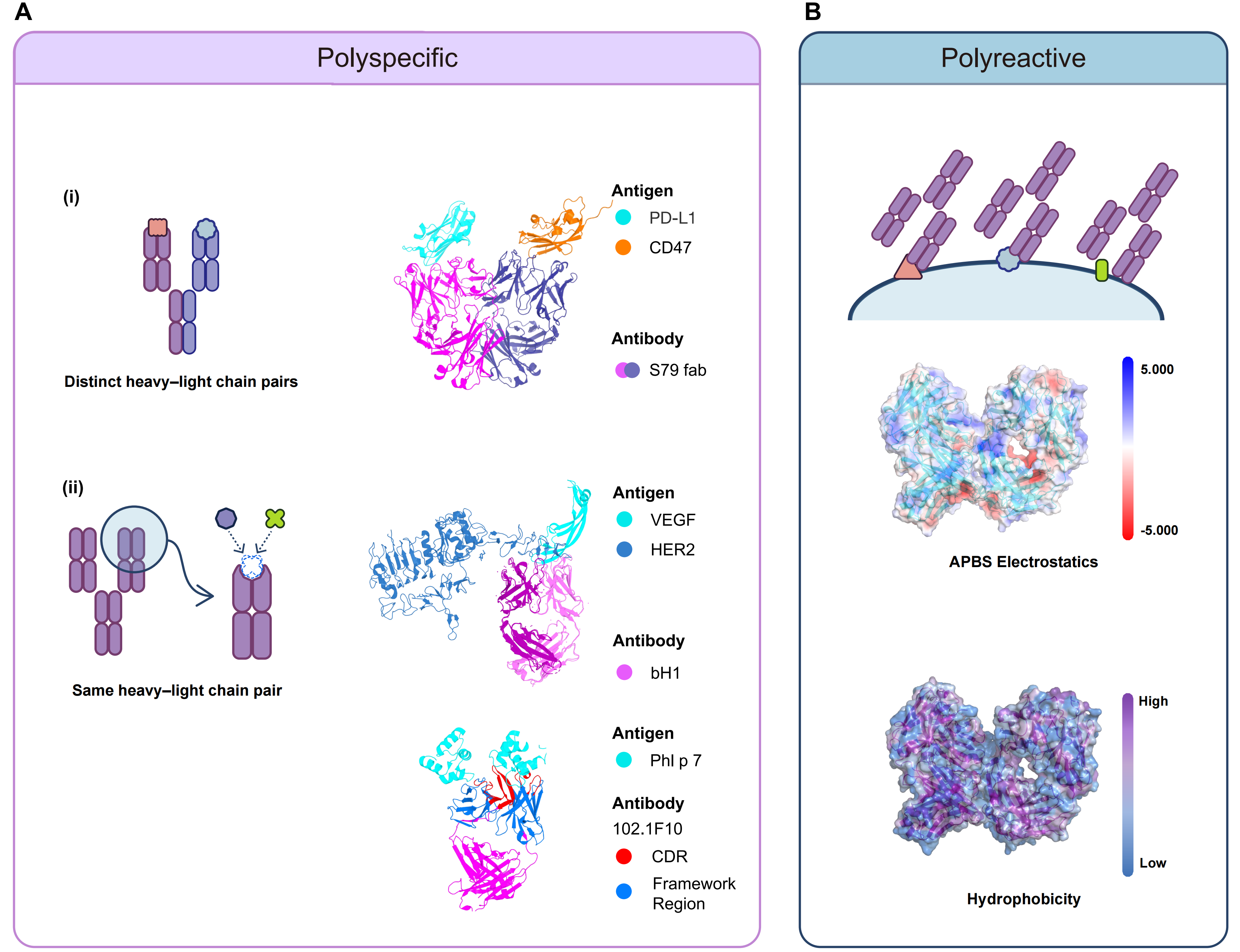{kind=link}
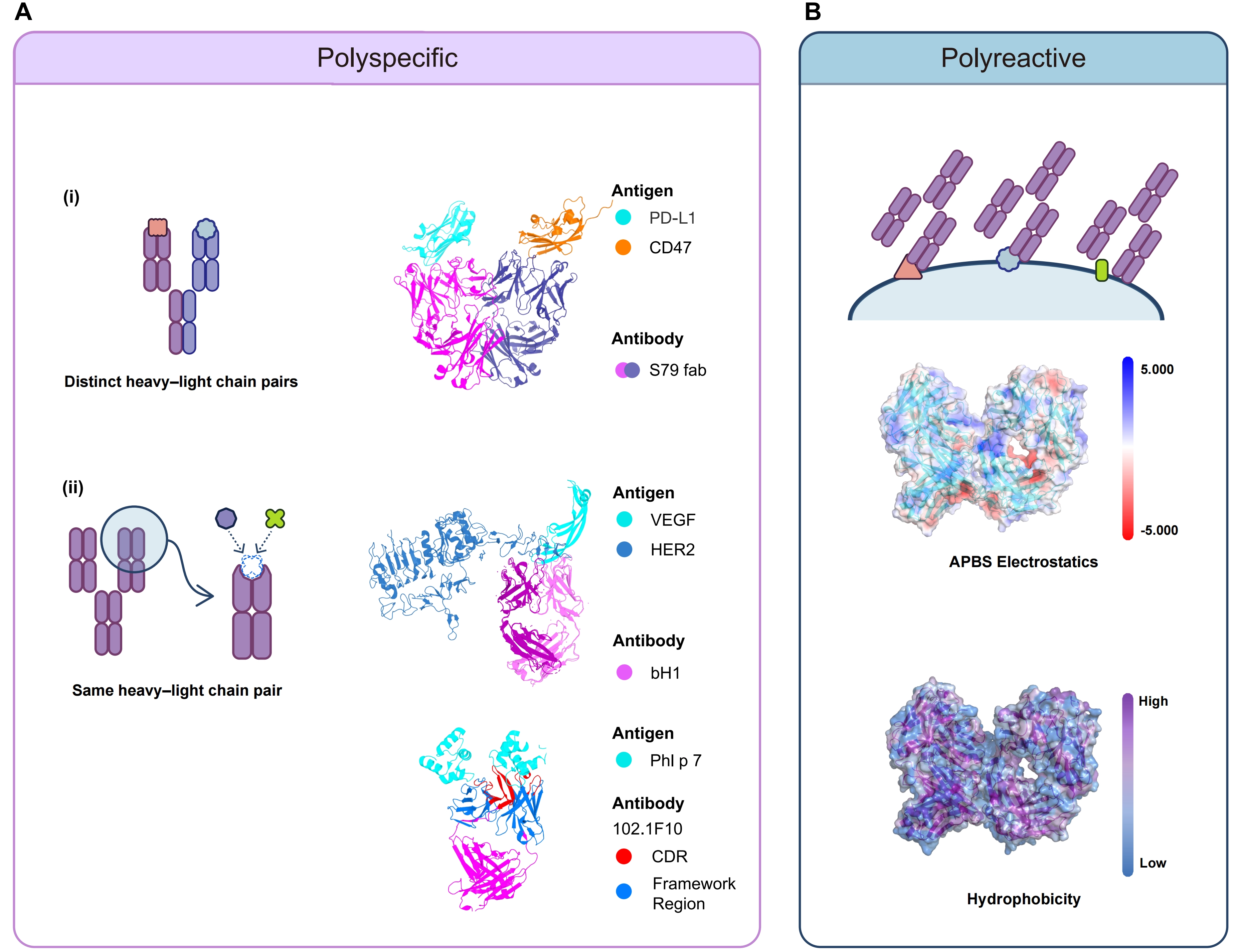
Figure 1. Distinct recognition modes of polyspecific and polyreactive antibodies. (A) Polyspecific antibodies bind to multiple antigens through two ways: (i) distinct HL chain pairs that form different paratopes (e.g., S79 Fab recognizing CD47 and PD-L1 through two distinct light chains paired with a common heavy chain); and (ii) the same HL chain pair with conformational diversity or multiple paratopes (e.g., bH1 binding both HER2 and VEGF; and 102.1F10 binding two different epitopes on Phl p 7); (B) Polyreactive antibodies exhibit promiscuous, nonspecific binding via surface physicochemical stickiness and distributed weak interactions rather than discrete paratope-defined recognition. HL: heavy-light; CD47: cluster of differentiation 47; PD-L1: programmed death-ligand 1; HER2: human epidermal growth factor receptor 2; VEGF: vascular endothelial growth factor; CDR: complementarity-determining region; APBS: adaptive Poisson-Boltzmann solver.
Compared with polyspecific antibodies, which often retain high specificity toward multiple antigens and can achieve relatively high affinity, the binding of polyreactive antibodies to antigens is typically not based on precise recognition of a defined epitope[10], but more commonly manifests as low affinity, distributed, multipoint weak interactions (Figure 1B)[32]. This behavior is thought to be driven primarily by physicochemical properties such as surface hydrophobicity and the distribution of charged residues, and is mediated predominantly by the variable region (Fv). In particular, an increased number of positively charged residues in the variable heavy-chain (VH) domain is positively correlated with enhanced polyreactivity[33]. Beyond the Fv, the heavy-chain constant region may also contribute to regulation. For example, CH1 can modulate the “tightness” of the variable-region binding site by altering the structural coupling between VH and CH1, thereby influencing antibody affinity, specificity, and polyreactivity propensity without changing the contact residues[34]. Overall, antibody polyreactivity is more likely to be co-encoded by broadly distributed sequence features with individually modest effects; although each feature contributes only limitedly on its own, their synergistic accumulation can support functional polyreactive binding[35], and phenotypically gives rise to a nonspecific, low-affinity “chemical stickiness”[10]. Taken together, polyreactivity is better understood as a distributed physicochemical binding propensity rather than a classical paratope-defined recognition mode.
1.2 Assays for antibody polyreactivity
Given the adverse impact of antibody polyreactivity on therapeutic antibody development, various experimental methods have been used to evaluate antibody polyreactivity or nonspecific binding risk and to provide phenotypic labels for subsequent computational modeling. From the perspective of experimental workflow and label generation, these methods can be broadly grouped into three technical routes. The first route is based on enzyme-linked immunosorbent assay (ELISA) or ELISA-like solid-phase binding assays, which are typically used to measure the binding signals of purified antibodies against predefined antigens, complex multicomponent reagents, or biologically derived materials. The second route involves high-throughput screening using display systems coupled with flow cytometry, such as yeast or phage display combined with fluorescence-activated cell sorting (FACS)[36,37]. In these approaches, antibody fragments are displayed on the surface of cells or phages and sorted according to their fluorescence binding signals to polyspecificity reagent (PSR), baculovirus particle (BVP), Chinese hamster ovary solubilized membrane protein (CHO-SMP), or other polyreactivity-related reagents. The third route is based on panning/next-generation sequencing enrichment, in which sequencing is performed after selection with target antigens or polyantigen mixtures, and labels are assigned according to whether clones are detected or enriched under different selection conditions[35]. These three routes correspond to solid-phase binding signals, flow-cytometric sorting signals, and enrichment-source signals, respectively, and therefore differ in signal readout, threshold definition, enrichment strategy, and sources of noise.
In addition to differences in experimental workflow, different assays may also produce distinct readouts because of differences in the detection substrate or bait system. The biological signals captured by different substrates are not fully equivalent. Multi-antigen assays, including multi-antigen ELISA and protein/peptide arrays, typically measure antibody binding signals against a series of predefined and often structurally unrelated antigens or surfaces, thereby characterizing the binding profile of antibodies toward a specific antigen set and evaluating their level of polyreactivity[21,38]. PSR mainly reflects nonspecific binding of antibodies to complex multicomponent reagents; BVP reflects binding to viral particle surfaces; and CHO-SMP reflects binding to host-cell proteins or complex components in cell culture supernatants. It should be noted that the same detection platform can use different substrates; for example, ELISA or FACS can both be used to measure antibody binding to PSR, BVP, CHO-SMP, or multiple predefined antigens. Conversely, the same substrate can also be read out using different platforms. Therefore, although these assays are all related to polyreactivity, nonspecific binding, or developability risk, their detection substrates, signal backgrounds, and mechanistic origins are not identical and should not be simply regarded as measuring the same biological property.
Although these experimental assays provide critical risk-screening information for candidate antibodies, they are often constrained by high cost, long turnaround time, and limited throughput, and therefore cannot fully meet the demands of high-throughput antibody design and screening[39,40]. With the development of bioinformatics, computational approaches have been widely applied to the prediction and optimization of antibody developability attributes[41,42]. Meanwhile, extensive polyreactivity assay data accumulated from previous research and development efforts have provided quantifiable phenotypic labels for database and dataset construction, enabling systematic extraction of determinants and statistical regularities underlying polyreactivity. Therefore, computational methods have important potential for assisting polyreactivity risk screening in antibody development pipelines. However, the construction and evaluation of computational models are highly dependent on the quality and consistency of experimental labels. As discussed above, polyreactivity-related data generated from different platforms are not necessarily equivalent, which may affect dataset integration, label interpretation, cross-study comparison of model performance, and model generalization. Therefore, when using existing experimental data for computational modeling, assay context and label heterogeneity should be explicitly considered, and experimental readouts from different sources should not be simply treated as fully equivalent polyreactivity labels.
In this review, we systematically summarize antibody polyreactivity databases and datasets, commonly used computational approaches, and predictive models. Their limitations and future study directions are also discussed.
2. Polyreactivity-Related Data Resources
Antibody polyreactivity is an important developability-related property and arises from complex underlying mechanisms[10,43], making systematic polyreactivity data highly valuable both for antibody development and for understanding antibody behavior itself. At the same time, polyreactive antibody sequences with labels at a sufficient scale can support computational modeling and machine-learning approaches, thereby improving the overall efficiency of antibody screening and design. Most polyreactivity-related data currently come from primary experimental results reported in individual studies and are dispersed across different articles and supplementary materials. These scattered datasets are often relatively small, typically containing only hundreds to thousands of antibodies. However, with the continued accumulation of data, publicly curated polyreactive antibody sequence datasets have gradually become more abundant, and some now reach the scale of tens of thousands of entries. In general, these datasets quantify polyreactivity by measuring binding strength across multiple background conditions and then link the results to antibody sequences obtained by deep sequencing, thereby forming “sequence–polyreactivity label” datasets suitable for modeling and analysis.
2.1 Polyreactivity datasets and database
As shown in Table 1, we summarize the currently accessible data resources relevant to polyreactivity and compare them in terms of sequence type, data size, experimental assays, data type, and other characteristics. Based on differences in experimental readouts and data processing strategies, existing polyreactivity data can be broadly categorized into two types. The first type comprises continuous quantitative data, which directly reflect the binding strength of antibodies to multiple non-target antigens or cellular components. The second type consists of discrete qualitative data, in which antibodies are classified into groups such as polyreactive versus non-polyreactive or low versus high polyreactive. These two types of labels are not fundamentally distinct, as discrete labels are typically derived from experimental signal intensities through thresholding or normalization procedures to assign binary labels[38,44].
Table 1. Polyreactivity-related data resources.
| Sequence type | Data Size | Measurement | Label | label generation Strategy | Raw Data Availability | Data Resource | Reference |
| Fv fragments | 137 antibodies | Multi-antigen ELISA | Continuous | Direct measurement | Processed only | https://www.pnas.org/doi/10.1073/pnas.1616408114#acknowledgments | [2] |
| Fv fragments | 197 antibodies | CHO SMP/Ova polyreactivity assay | Continuous | Direct measurement | Processed only | https://huggingface.co/datasets/ginkgo-datapoints/GDPa1 | [44] |
| Fv fragments | 43 antibodies | Yeast display + PSR/FACS | Continuous | Direct measurement | Processed only | https://oss.sciexplor.com/web/files/KMAB_A_2384104_SM4152.xlsx | [45] |
| Fv fragments | 524 non-polyreactive/529 polyreactive | Multi-antigen ELISA | Binary | Direct measurement | Processed only | https://github.com/ctboughter/AIMS | [38] |
| VHH sequences | Main: 65.1k low-polyreactive/69.1k high-polyreactive Extended: 1,221.8k low-polyreactive/1,058.8k high-polyreactive | Yeast display + PSR/FACS | Binary | Direct measurement | Available upon request | https://github.com/debbiemarkslab/nanobody-polyreactivity | [46] |
| scFv sequences | Library #1: 131,255 low/115,038 high; Library #2: 34,137 low/93,080 high | Yeast display + multi-reagent FACS | Binary | Enrichment-based datasets | Processed only | https://zenodo.org/records/14735846 | [33] |
| VH sequences | 1.34M non-polyreactive/15.0M polyreactive | Polyantigen panning | Binary | Enrichment-based datasets | Raw available | https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA1052988 | [35] |
| Multiple types | million-scale records | Multiple assays | Both | Curated/mixed labels | Processed only | https://i.uestc.edu.cn/DOTAD/ | [47] |
Fv: variable region; ELISA: enzyme-linked immunosorbent assay; CHO SMP: Chinese hamster ovary solubilized membrane protein; Ova: ovalbumin; PSR: polyspecificity reagent; FACS: fluorescence-activated cell sorting; DOTAD: database of therapeutic antibody developability.
For continuous quantitative data, representative resources include datasets reported by Wittrup et al.[2] and the GDPa1 dataset from Ginkgo[44]. Wittrup et al. quantified the polyreactivity of 137 therapeutic antibodies using yeast display in combination with CHO SMP assays, establishing an early framework for quantitative evaluation of therapeutic antibody polyreactivity. The GDPa1 dataset contains developability-related data for 242 IgG antibodies and assesses polyreactivity using CHO SMP and ovalbumin (Ova), yielding 198 antibodies with data relevant to polyreactivity. The advantage of such datasets lies in their ability to preserve detailed signal intensity information, enabling regression modeling of polyreactivity and developability prediction, and facilitating the characterization of subtle differences among antibodies. However, their sample sizes usually are very limited.
In contrast, binary classification datasets are often larger in scale and better suited for supervised classification tasks. Adams et al.[38] measured 1,053 human and murine Fv fragments using ELISA and classified them into polyreactive and non-polyreactive categories, representing a classical binary dataset. Subsequently, Kruse et al.[46], Tessier et al.[33], and Ros et al.[35] expanded dataset sizes across nanobody, single-chain Fv, and VH sequence levels. The dataset from Ros et al. reached tens of millions of sequences. In particular, the incorporation of high-throughput techniques such as yeast display, deep sequencing, and B cell panning has enabled the generation of polyreactivity labels at massive sequence scales, significantly advancing the application of deep learning models in this field. It should be noted that these binary labels are not uniformly derived from thresholding a single continuous measurement, but instead reflect heterogeneous labeling strategies across studies, including antigen-count-based ELISA classification, PSR-based FACS enrichment with extremal selection across multiple reagents, and sequence-level enrichment under polyreactivity- or polyspecificity-related selection pressures. Therefore, these labels represent a mixture of discretization, enrichment-based selection, and weak supervision rather than simple binarization of continuous signals. Nevertheless, such datasets still suffer from important limitations: threshold definitions are often empirically determined without standardized criteria, leading to potential label instability for samples near decision boundaries, while most datasets lack sample-level confidence scores or uncertainty estimates, preventing explicit quantification of label reliability. Future studies should therefore retain raw or normalized experimental signals whenever possible and clearly report thresholding or selection criteria to enable more transparent handling of label uncertainty.
Beyond individual studies with relatively dispersed data sources, polyreactivity, as a key developability-related property, has been included in special databases. Database of therapeutic antibody developability (DOTAD)[47] represents a notable example, with version 1.0 including over 100 records related to antibody polyreactivity and integrating data from multiple experimental sources through manual curation. Its current version 2.0 further expands polyreactivity-related data and provides a more comprehensive aggregation of information across studies. Compared with individual datasets, DOTAD offers greater integration and includes both qualitative labels and continuous quantitative measurements, making it particularly suitable as a foundational resource for multi-parameter developability analysis. By organizing polyreactivity data across diverse sources, DOTAD also facilitates cross-dataset integration and supports multi-task modeling approaches.
2.2 Issues in polyreactivity datasets and database
Existing polyreactivity data are generated through diverse experimental routes, which influence both label definition and cross-study comparability, and also shape the overall composition of available datasets. Therefore, the current limitations of antibody polyreactivity datasets can be summarized in three main aspects.
2.2.1 Imbalanced data distribution
The generation of polyreactivity data is highly dependent on the source of candidate molecules, the throughput of screening platforms, and the practicality of downstream validation systems. As a result, existing datasets do not represent a balanced sampling of different antibody formats and application scenarios, but instead exhibit strong technological-path dependence. Most currently available large-scale datasets are derived from display-library screening and high-throughput sequencing platforms, and therefore mainly consist of fragment antibody sequences such as Fv, scFv, VH, or VHH[33,35,46], whereas polyreactivity-annotated data for full-length IgG antibodies, especially bona fide therapeutic antibodies, remain relatively limited. Importantly, this gap is not merely quantitative. Fragment formats such as scFv, VH, and VHH lack structural features present in full-length IgG, including native heavy–light chain pairing, Fab–Fc interactions, and constant-region contributions that can modulate variable-region binding behavior[34], which may themselves influence polyreactivity. Models trained predominantly on fragments therefore risk learning determinants that do not fully transfer to the intact IgG context. This imbalance is reflected not only in antibody format, but also in the composition of positive and negative samples across datasets and in the types of data produced by different experimental platforms. Consequently, existing models are often more likely to learn patterns specific to particular libraries or antibody formats, rather than features that can be directly transferred to full-length therapeutic antibody systems with greater practical relevance for developability assessment.
2.2.2 Limited comparability across studies
Polyreactivity typically manifests as low affinity, distributed, and weak interactions. Thus, its experimental readout is highly sensitive to antigen composition, surface immobilization strategies, buffer conditions, detection platforms, and signal normalization procedures. In addition, different studies often adopt distinct thresholding criteria, normalization strategies, and label-construction schemes to transform raw experimental signals into binary, ordinal, or continuous labels, which further amplifies discrepancies across platforms, laboratories, and studies. Continuous quantitative data, although potentially more informative, are often even more difficult to integrate than binary labels because their scales, baselines, and experimental contexts are rarely standardized. For example, studies have shown that polyreactivity measurements obtained from yeast-display systems correlate only weakly with binding results measured for full-length IgG antibodies[44]. This discrepancy may arise from several factors, including the absence of in vivo selection pressure in display systems[48], the fact that such platforms often cover only antibody fragments rather than full-length IgG molecules, and system-specific effects such as yeast surface glycosylation. Therefore, although datasets from different sources are all “polyreactivity data”, the biological meaning they capture and their transferability are not necessarily equivalent.
2.2.3 Lack of a unified database
Although many polyreactivity studies report model performance or key biological findings, the underlying raw data, complete labels, experimental metadata, and standardized processing pipelines are often not fully disclosed. This limits data reusability and hinders fair comparison and external validation across studies. At the same time, dedicated data platforms specifically designed for antibody polyreactivity remain scarce. Publicly accessible resources are often scattered across supplementary materials, code repositories, or broader developability databases, and there is still no special database that provides relatively unified coverage of antibody formats, experimental systems, label definitions, and metadata annotations. This situation not only increases the difficulty of data integration and benchmark construction, but also constrains the systematic advancement of model training, generalization assessment, and cross-study reproducibility.
3. Computational Methods for Polyreactivity
3.1 Conventional computational approaches for polyreactivity evaluation
Before machine learning methods became widely used, computational assessment of antibody polyreactivity mainly relied on empirical physicochemical risk indicators. These methods are based on the basic assumption that nonspecific antibody interactions are influenced by intrinsic physicochemical properties of the Fv, including surface charge distribution, hydrophobicity, isoelectric point, and the enrichment of specific amino acid classes such as aromatic or charged residues[49-51]. For example, Kelley et al.[52] used charge and hydrophobicity descriptors of the antibody Fv region to estimate nonspecific clearance of IgG1 antibodies in cynomolgus monkeys, showing that global physicochemical properties are associated with unfavorable in vivo behavior. Although this model was designed for nonspecific clearance rather than antibody polyreactivity in the strict sense, the two phenotypes are closely related in the broader context of antibody developability. Similarly, Wittrup et al.[2] performed biophysical profiling of clinical-stage antibodies, and Tessier et al.[50] further showed from a sequence-feature perspective that CDR charge and other sequence-derived physicochemical features are closely associated with antibody specificity, self-interaction, and nonspecific binding propensity. These empirical indicators remain practically useful because they are computationally inexpensive, easy to implement, and physically interpretable.
However, these empirical indicators are better regarded as risk indicators rather than standardized predictive models for antibody polyreactivity[32,33]. While they provided an important foundation for subsequent machine-learning approaches by identifying useful features and offering mechanistic clues, their predictive scope is limited by assay systems, antibody formats, species background, and other factors, and polyreactivity itself is not determined by a single physicochemical parameter. Structure-based molecular dynamics (MD) simulations have also been used to explore how conformational flexibility, binding-surface properties, and interfacial interactions influence polyreactivity. However, their high computational cost and sensitivity to system setup make them difficult to apply to large-scale prediction. Therefore, in this review, MD methods are not considered a major class of scalable predictive approaches; instead, they are discussed in the perspective section as complementary tools for mechanistic interpretation and experimental hypothesis generation.
3.2 Machine learning based approaches
In early-stage antibody discovery, the number of candidate molecules is extremely large while the throughput of experimental validation remains limited, making machine learning an essential computational tool for antibody engineering and developability prediction. However, antibody polyreactivity, as a complex phenotype jointly governed by sequence, physicochemical properties, and structural context, is difficult for traditional computational approaches to effectively integrate across these multidimensional factors, thereby limiting predictive performance and cross-system generalization ability. With the continuous accumulation of polyreactivity-related experimental data and the increasing diversity of data modalities, machine learning methods have gradually been introduced into this field and are capable of automatically extracting informative patterns from large-scale experimental datasets and multi-level antibody features, while capturing nonlinear relationships among features, thereby improving predictive performance[53,54].
Extensive biochemical and structural studies have shown that the key physicochemical determinants of polyreactivity can be broadly categorized into three major groups: charge distribution (e.g., positively charged clusters in CDR regions and HCDR3 length-dependent effects, where arginine exhibits pronounced position- and sequence-context dependence)[35], surface hydrophobicity (spatially clustered hydrophobic patches and exposed aromatic residues, rather than overall hydrophobic content)[33], and conformational dynamics (CDR loop flexibility, transient exposure of buried residues, and antigen-induced conformational adaptation)[6]. Together, these factors constitute the core biophysical basis of polyreactivity; therefore, existing predictive models can be uniformly interpreted as different forms of encoding the same set of physicochemical determinants.
Different machine-learning approaches differ in how these features are represented. Traditional machine learning methods rely on hand-crafted feature engineering to explicitly encode these properties into descriptors such as net charge, hydrophobicity indices, and solvent accessibility, resulting in a near one-to-one correspondence between mechanistic interpretation and features, and therefore offering strong interpretability[55]. Deep learning models, in contrast, learn these factors implicitly from sequences in an end-to-end manner, with interpretation typically relying on permutation importance or ablation analyses; recurrent architectures (e.g., recurrent neural networks (RNNs)) can partially capture long-range dependencies and cooperative effects among multiple residues[35,46]. Protein language models (PLMs) further encode physicochemical properties into distributed representations through large-scale self-supervised pretraining, forming more abstract statistical representations in latent space[56].
Based on this framework, Figure 2 outlines the workflow of machine learning-based polyreactivity prediction: candidate antibodies (VH/VL or VHH formats) are transformed into model inputs via sequence- or structure-based encoding, while experimentally determined measurements (e.g., polyreactivity ELISA, BVP assays, and PSR assays based on CHO/HEK293 systems) provide supervised labels, jointly enabling model training and prediction. Table 2 further summarizes representative studies in terms of input features, model types, and performance differences. The following sections will sequentially introduce three categories of methods: traditional machine learning, deep learning, and PLMs.
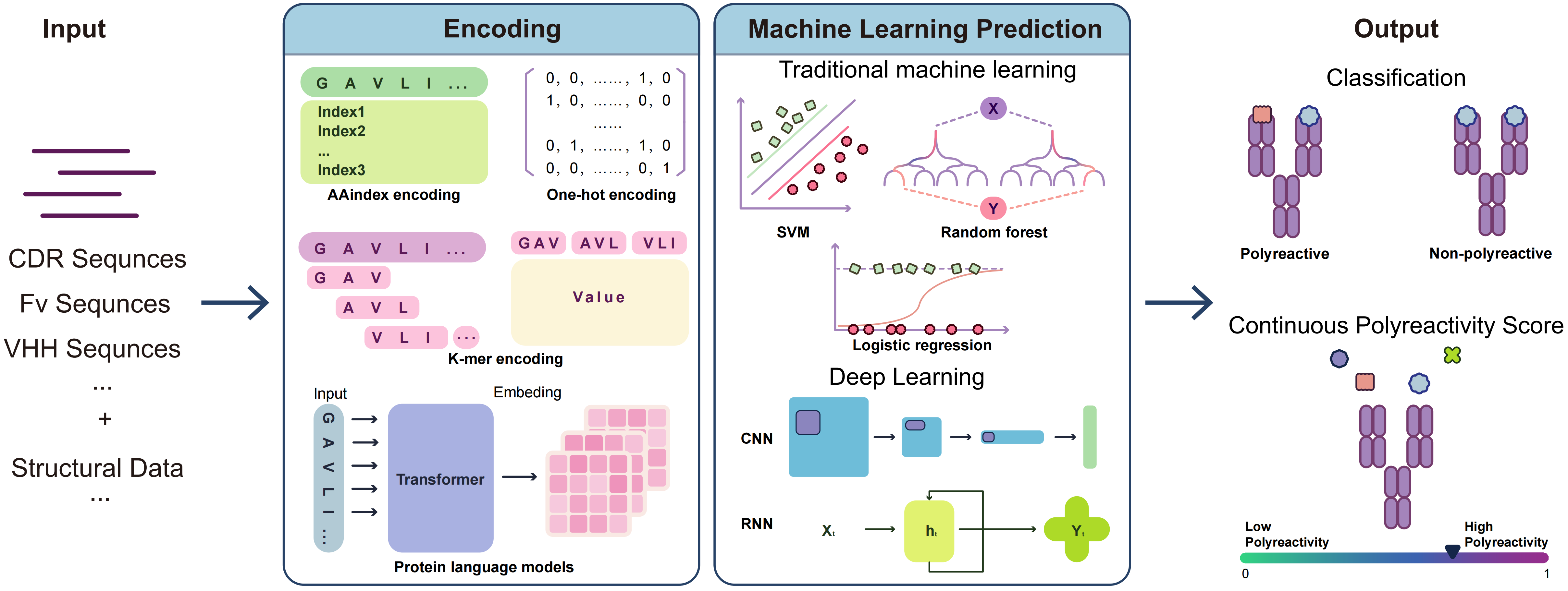{kind=link}
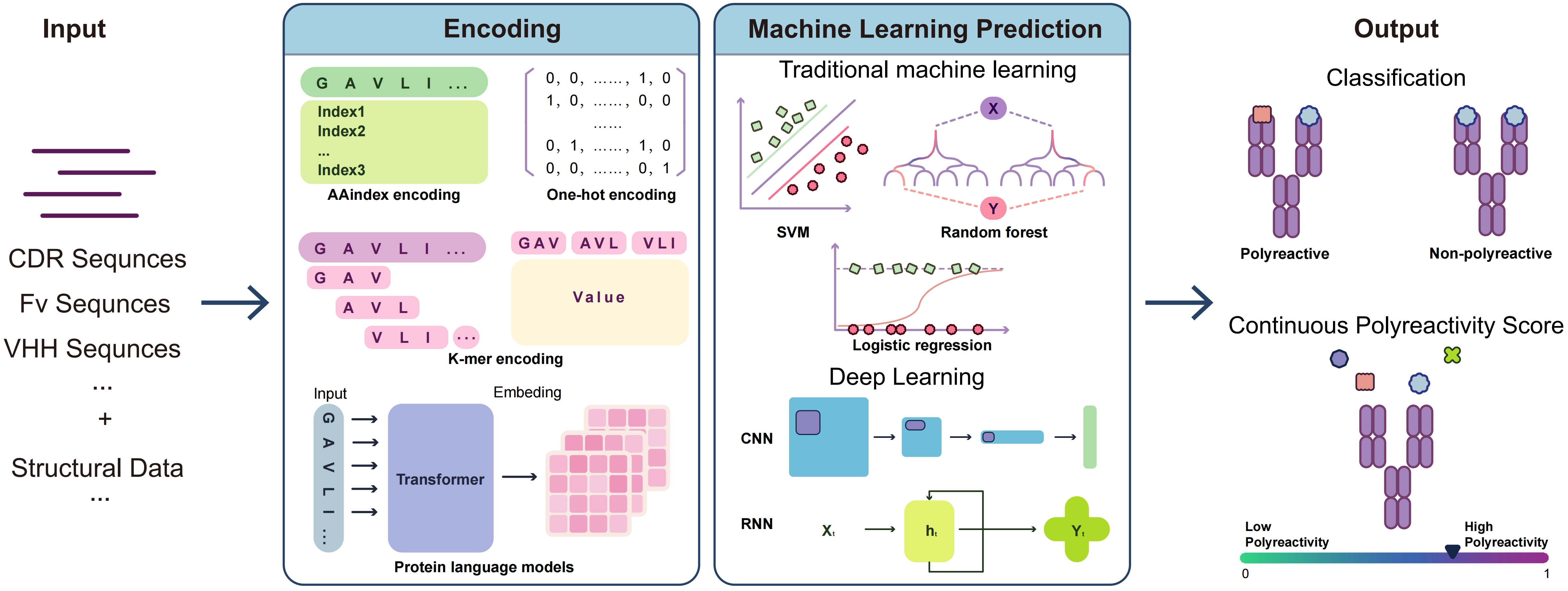
Figure 2. Workflow of machine-learning-based antibody polyreactivity prediction. CDR: complementarity-determining region; Fv: variable region; SVM: support vector machines; CNN: convolutional neural network; RNN: recurrent neural network.
Table 2. Overview of ML-based methods for antibody polyreactivity prediction.
| ML Method | Input Type | Output | Performance# | Data Resource | Reference |
| AAindex-encoded CDR features + LDA/SVM classifier | CDR sequences | polyreactive/non-polyreactive and LDA projection score | AAC ≈ 0.75 | https://github.com/ctboughter/AIMS | [38] |
| One-hot Logistic Regression, k-mer Logistic Regression model | CDR sequences | polyreactivity risk score | AUC: 0.85 for the one-hot logistic regression model | https://github.com/debbiemarkslab/nanobody-polyreactivity | [46] |
| AAindex-encoded VH sequence + neural network | VH sequences | polyreactive/non-polyreactive | AUC: 0.835 | https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA1052988 | [35] |
| Biochemical & structural features and PLM embeddings + ML classifier | Biochemical & structural features + scFv sequences NLP embeddings | polyreactive/non-polyreactive | AUC:0.84 for ensemble model | https://github.com/hclim0213/Pred_nonspecificity_scFvs | [57] |
| Sequence-derived physicochemical features + random forest classifier | Heavy-chain CDR sequences + physicochemical properties | polyreactive/non-polyreactive and polyreactivity risk score | AUC > 0.8 | https://zenodo.org/records/14735846 | [33] |
| PLM embeddings + CNN models | VH/VL/scFv/nanobody sequences + experimental conditions (normalized antibody concentration, coating type, and location matrix) | polyreactive/non-polyreactive | AUC = 0.863 on the expanded dataset | https://github.com/xinyu-dev/bvp_manuscript | [58] |
| ESM-2 end-to-end fine-tuning | scFv sequences | polyreactive/non-polyreactive and polyreactivity risk score | AUC = 0.967 | https://github.com/zzyywww/PolyXpert | [59] |
#: Reported performance metrics are taken from the original publications. Given the heterogeneity in datasets, assay platforms, label definitions, and evaluation protocols, these values should be interpreted as study-specific results rather than directly comparable benchmarks. CDR: complementarity-determining region; LDA: linear discriminant analysis; SVM: support vector machine; AAC: average area under the ROC curve; AUC: area under the curve; PLM: protein language model; ML: machine learning; NLP: natural language processing; CNN: convolutional neural network; VH: variable heavy-chain; ESM-2: evolutionary scale modeling 2.
3.2.1 Traditional machine learning models for polyreactivity prediction
Traditional machine-learning approaches established the foundational paradigm for antibody polyreactivity prediction and demonstrated the feasibility of sequence-based prediction. The core idea of these methods is to transform amino acid sequences into numerical representations with explicit biophysical meaning through feature engineering, and then apply statistical learning algorithms to capture the physicochemical determinants of antibody polyreactivity.
In terms of data and modeling targets, most existing studies use VHH or VH+VL sequences as input and focus on the CDRs with an emphasis on heavy chain CDR3. Feature construction is generally centered on amino acid composition, net charge, hydrophobicity, sequence entropy, and K-mer frequency. Based on these features, classical algorithms such as logistic regression (LR), support vector machines (SVM), random forests (RF), and gradient boosting decision trees are commonly employed to perform classification or regression tasks.
At the level of individual studies, differences in dataset size and modeling strategies are evident. In early explorations of traditional machine-learning approaches, Adams et al.[38] performed a systematic analysis using a dataset of approximately one thousand conventional antibody sequences curated from multiple published studies. By encoding position-specific biophysical features across the six CDR loops and combining linear discriminant analysis for feature selection with SVM classification, the model achieved an accuracy of approximately 75% on the parsed dataset using leave-one-out cross-validation. Importantly, this study provided the first systematic investigation into the interpretable determinants of antibody polyreactivity, revealing that the major discriminative signals were concentrated in the germline-encoded HCDR1 and HCDR2 regions, while polyreactive antibodies exhibited a shift toward a more neutral binding interface rather than simply increased charge or hydrophobicity. Furthermore, mutual information analysis revealed enhanced heavy-chain CDR loop crosstalk in polyreactive antibodies, suggesting that polyreactivity may arise from coordinated interactions among multiple residues rather than the cumulative effects of a few isolated key residues. These findings established an initial performance baseline for the field and provided mechanistic insights for subsequent interpretable models of polyreactivity prediction.
With the expansion of dataset size, both model performance and applicability have improved. Kruse et al.[46] constructed a LR model based on a dataset of approximately 130,000 VHH sequences with a balanced positive-to-negative ratio (~1:1). Sequence-based encodings, including one-hot and k-mer (k = 3) representations, were applied to CDR sequences, enabling LR models to extract discriminative patterns associated with antibody polyreactivity directly from sequence information, without relying on explicit structural features. Importantly, the study employed a sequence clustering and edit-distance–constrained splitting strategy to construct training and test sets, ensuring low sequence homology between the two partitions. In addition, prospective validation was performed through directed evolution experiments and functional antibody optimization, enabling a systematic evaluation of the model’s cross-sequence generalization ability and its practical engineering relevance beyond standard in-distribution benchmarks. The model achieved an area under the curve (AUC) greater than 0.8 in distinguishing high and low polyreactive nanobodies on an internal test set from a synthetic nanobody library. Furthermore, although trained within a binary classification framework, the model outputs continuous scores that exhibit some degree of quantitative interpretability. Specifically, on an independently constructed experimental validation set, the predicted scores showed a significant rank correlation with experimentally measured polyreactivity levels (Spearman ρ > 0.77), indicating it successfully captures sequence features underlying polyreactivity. Prospective experimental validation further demonstrated that the model could accurately predict mutational enrichment directions during directed evolution and guide rational mutagenesis design in independent nanobody scaffolds. In particular, it enabled the reduction of polyreactivity while preserving antigen-binding functionality, highlighting its potential for engineerable optimization of antibody properties.
In addition, the interpretability of the linear model enabled residue- and motif-level analysis of amino acid contributions to polyreactivity: basic residues such as Arg were generally associated with increased polyreactivity, but their effects were strongly position-dependent; Lys, Tyr, and Trp mainly showed promoting effects in CDR3, whereas acidic residues such as Asp/Glu in CDR2 and CDR3 were associated with reduced polyreactivity. Notably, the Y29R mutation in AT118i4h32 introduced an additional Arg into CDR1 and formed an RRR motif, but instead reduced polyreactivity, suggesting that the effect of Arg is not determined solely by residue identity but is jointly regulated by position and sequence context. However, because this study was based on synthetic VHH libraries, both model training and validation relied on nanobody-specific sequence frameworks and CDR features, thereby limiting direct applicability to conventional IgG antibodies involving HL chain pairing effects.
Ensemble learning methods further expanded the application scope of polyreactivity prediction. Tessier et al.[33] developed a RF model based on more than 370,000 human scFv sequences. In this framework, Library #1 was used for model training and cross-validation, whereas Library #2 served as an independent external validation set, enabling robust cross-dataset evaluation. The model was further evaluated across multiple orthogonal antibody datasets, including clinical-stage antibodies, engineered mutational libraries, and natural antibody repertoires, thereby providing additional evidence for its strong cross-dataset generalization ability. In addition to sequence-derived features, this study incorporated structural features with clear physical meaning, such as charge distribution, hydrophobic composition, and solvent accessibility-weighted descriptors, forming a manually designed feature set that balanced interpretability and predictive performance. Further interpretability analysis showed that antibody polyreactivity was primarily driven by the heavy-chain complementarity-determining regions, with HCDR2 showing the strongest ability to distinguish high- and low-polyreactivity antibodies. The effect of net positive charge in HCDR3 was also strongly length-dependent, showing greater discriminatory power in antibodies with longer HCDR3 sequences. Compared with earlier studies that mainly focused on residue composition associated with polyreactivity, this study advanced the understanding of molecular determinants of polyreactivity from the level of key residues to the level of regional contributions, thereby providing a theoretical basis for prioritizing specific CDR regions in subsequent antibody engineering. The model achieved receiver-operating characteristic (ROC)-AUC values greater than 0.8 on the independent test set (Library #2) as well as on multiple external datasets from diverse sources, consistently discriminating polyreactive differences among clinical-stage antibodies, engineered variants, and natural repertoires, further demonstrating its robust cross-system generalization capability.
Overall, traditional machine learning methods offer advantages such as low computational cost, ease of implementation, and strong interpretability. They perform well in data-limited settings and can provide both predictive results and mechanistic insights. As such, they are well suited for early-stage polyreactivity risk screening, pre-engineering assessment, and as baseline models for more complex approaches. However, their predictive performance is highly dependent on feature engineering and the distribution of training data, leading to limited transferability across antibody types and experimental systems. Moreover, they struggle to fully capture complex polyreactivity mechanisms driven by spatial hydrophobic patches, charge clusters, and long-range residue interactions[35,57]. In addition, current polyreactivity datasets are generally limited in size, heterogeneous in labeling schemes, and often exhibit class imbalance due to the relatively low prevalence of polyreactive antibodies in real samples, further constraining model generalization[35,60]. These limitations collectively highlight the need for more expressive modeling approaches and provide the context for the adoption of deep learning and PLMs in polyreactivity prediction, which have demonstrated improved performance in recent studies[42,61,62].
3.2.2 Deep learning models for polyreactivity prediction
With the rapid expansion of antibody sequence data driven by high-throughput sequencing technologies, deep learning algorithms have increasingly been applied to antibody polyreactivity prediction, emerging as a new paradigm for modeling complex biological properties[63,64]. Unlike traditional machine-learning approaches that rely on manual feature engineering, deep learning models typically adopt an end-to-end learning framework, directly extracting latent biophysical patterns from amino acid sequences and learning complex mappings between sequence features and polyreactivity phenotypes[65-67]. From a modeling perspective, current deep learning frameworks are mainly sequence-driven[68,69]. Fv, VH, or CDR regions of antibody sequences are encoded using one-hot or learned embeddings and fed into neural network architectures that capture local sequence motifs, long-range dependencies, and nonlinear feature interactions.
For example, Ros et al.[35] developed a deep learning model using a dataset comprising approximately 15 million VH sequences, thereby systematically evaluating the feasibility of sequence-based polyreactivity prediction. In this study, VH sequences were first aligned using the WolfGuy numbering scheme, and physicochemical properties from the AAindex database were reduced via principal component analysis (PCA). These features were then used to train a multilayer feedforward neural network for binary classification of antibody polyreactivity. The evaluation was performed in a multi-stage framework. First, standard in-distribution performance was assessed using random 10-fold cross validation and repeated subsampling. Second, group-wise cross validation stratified by animal species and antigen identity was applied to evaluate cross-individual and cross-immunization generalization. Third, a hold-out set comprising pre-immune clones and cross-group shared sequences was constructed to simulate out-of-distribution scenarios. Finally, external validation was conducted using a small independent ELISA dataset and publicly available antibody datasets, further supporting model robustness across experimental and distributional shifts. The model achieved an AUC of approximately 0.8 under random 10-fold cross validation; however, its performance dropped substantially under more stringent group-based evaluation schemes (AUC ≈ 0.67 when split by animal, and AUC ≈ 0.63 when split by antigen). These results indicate that sequence features associated with polyreactivity exhibit pronounced distribution shifts across biological contexts (individual donors and immunization antigens), suggesting that part of the predictive signal depends on experimental and immunological background rather than being fully determined by generalizable VH sequence rules. To address the limited interpretability of deep learning models, the study incorporated permutation-based feature importance analysis, quantifying the contribution of each physicochemical principal component via a “dropout loss ratio”, and combined this with PCA loadings to identify key features. The results confirmed that charge distribution and hydrophobicity remain dominant determinants of polyreactivity, consistent with established biophysical understanding.
Another line of work employs fully sequence-driven neural network architectures. Notably, in the same study in which LR models were developed as traditional machine learning baselines, Kruse et al.[46] further used concatenated VHH CDR sequences as input and compared convolutional neural networks (CNNs) and RNNs with traditional machine learning methods within a unified dataset and evaluation framework. The results showed that different architectures capture distinct levels of sequence information. CNNs are more effective at identifying local sequence motifs formed by specific residues, whereas RNNs, due to their memory mechanisms, are better suited for modeling long-range dependencies. In distinguishing high versus low polyreactivity nanobodies, the RNN model achieved the best performance (AUC ≈ 0.84), while CNN performed slightly lower (AUC ≈ 0.78). With the expansion of training data to approximately 2.28 million sequences, CNN performance further improved to AUC ≈ 0.83. In addition, the correlation between model predictions and experimentally measured polyreactivity also increased (Spearman ρ ≈ 0.88), suggesting that larger-scale data enables deep models to capture higher-order dependencies beyond local sequence motifs. Although deep learning models provide greater flexibility in capturing local and contextual sequence patterns, their performance improvement over LR remains limited. This suggests that in VHH CDR-based polyreactivity prediction, relatively simple and interpretable models are already able to capture most of the discriminative information.
Overall, the main advantages of deep learning methods in antibody polyreactivity prediction lie in their ability to automatically extract features and model complex patterns, making them particularly suitable for large-scale, high-dimensional, or weakly labeled datasets. These models are well suited for early-stage developability assessment and preliminary candidate screening, rather than serving as definitive functional predictors. Existing sequence-based deep learning models typically achieve AUC values in the range of 0.70-0.85, indicating that even without explicit structural input, they can effectively capture the complex relationships between antibody sequences and polyreactivity phenotypes.
Nevertheless, deep learning approaches also exhibit notable limitations. On one hand, model performance is highly dependent on the scale and diversity of the training data, making it difficult to maintain stable predictive performance when transferring across antibody formats (e.g., from nanobodies to full-length monoclonal antibodies (mAbs)). On the other hand, although architectures such as RNNs can partially model long-range dependencies, sequence-based neural networks still struggle to accurately capture the complex spatial effects arising from three-dimensional protein folding. In addition, the relatively limited interpretability of deep learning models constrains their applicability in scenarios that require mechanistic insight for rational antibody engineering[46].
This limitation can be partly mitigated by integrating model-attribution analyses with experimental or biophysical validation. For example, attention- or gradient-based attribution, in silico mutational scanning[70], and comparison of model-prioritized residues with established physicochemical liabilities, such as charge clusters and hydrophobic patches[46], may help identify sequence positions or motifs that drive model predictions. Importantly, these interpretability outputs should be interpreted with caution. Even advanced saliency maps, attention-based visualizations, or feature attribution methods primarily identify statistical associations between sequence-derived features and polyreactivity phenotypes, rather than establishing causal determinants of polyreactive binding. Therefore, residues or motifs highlighted by such methods should be regarded as hypothesis-generating signals rather than direct engineering rules. Rational antibody engineering and optimization based on computational predictions still require rigorous experimental validation, including targeted mutagenesis and orthogonal developability assays. These challenges have also motivated the recent adoption of PLMs, which leverage large-scale self-supervised pretraining to learn more expressive and biologically informative sequence representations.
3.2.3 PLMs for antibody polyreactivity prediction
With the rapid development of natural language processing (NLP), protein sequences have been modeled with NLP-based approaches, opening broad opportunities in protein research[71,72]. By leveraging self-supervised learning on massive protein sequence datasets, PLMs can capture complex patterns and contextual dependencies within amino acid sequences, representing residues or entire proteins as dense vector embeddings[72,73]. General-purpose PLMs, such as UniRep[65], TAPE[74], ESM-1b[68], and ESM-1v[75], encode unlabeled protein sequences into high-dimensional representations that provide rich informational bases for downstream modeling tasks[71]. In antibody polyreactivity prediction, PLMs are mainly applied along two technical routes: (i) as general feature extractors that provide high dimensional embeddings for downstream predictive models[72], and (ii) as end-to-end models fine-tuned on task-specific data to jointly optimize representation learning and prediction performance[59,76].
For the first route, PLMs are typically used as static embedding generators, with the primary goal of evaluating how well different pretrained representations adapt to polyreactivity prediction tasks. No et al.[57] trained a model using approximately 19,000 scFv polyreactivity records. This dataset was primarily integrated from records in the ProtaBank database[77], and high throughput nonspecificity assay results originally reported by Wittrup et al.[78]. The study adopted a stratified random splitting strategy, using 80% of the data for model training and 20% as a held-out test set. During training, hyperparameters were optimized via 10-fold cross validation combined with grid search, and final performance was reported on the held-out test set. With this dataset, the authors systematically compared the performance of general-purpose PLM embeddings, including UniRep, TAPE, ESM-1b, and ESM-1v, in the classification task. The results showed that UniRep combined with tree-based classifiers achieved an AUC of 0.82-0.83 on an independent test set, confirming the feasibility of PLM embeddings for this task. When concatenating PLM embeddings with traditional physicochemical features, the AUC of the model was improved to 0.83-0.84, indicating that PLM embeddings provide partially complementary information to traditional physicochemical features, albeit with a relatively small incremental gain. It should be noted that both training and test sets were derived from random splits of the same dataset; therefore, no cross-dataset or independent experimental cohort validation was performed in this study, and the true out-of-distribution generalization ability remains to be further evaluated. Similar trends have been observed in subsequent studies, further supporting the effectiveness of PLMs as general-purpose representations for predicting antibody nonspecific interactions[79].
PLMs also demonstrate strong applicability in more complex antibody formats. Huang et al.[58] evaluated general-purpose PLMs, including ESM-2 and ProtT5, as well as the antibody-specific model AntiBERTy, on a proprietary multi-format dataset comprising mAbs, bispecific antibodies, and VHH-Fc constructs. Using variable-region sequences as input and incorporating experimental conditions, such as antibody concentration and coating type, the study applied a 2D CNN to learn residue-level features and constructed a binary classifier for polyreactivity risk prediction. During model development, 1,664 data points, defined by combinations of antibody sequences and assay conditions, were randomly split into training, validation, and test sets at an 80:10:10 ratio. In this setting, ESM-2 achieved the best test-set performance, with a ROC AUC of 0.838, slightly higher than the AntiBERTy-based model (ROC AUC = 0.826). The authors further incorporated newly collected antibodies over an approximately two-month period, expanding the dataset to 2,998 data points, and again applied an 80:10:10 split for blinded evaluation. On this enlarged dataset, ESM-2, ProtT5, and AntiBERTy showed highly comparable test performance, with ROC AUC values of 0.863, 0.862, and 0.862, respectively. This finding challenges the intuitive assumption that antibody-specific PLMs are inherently superior and suggests that key determinants of polyreactivity, such as clustered surface charge and exposed hydrophobic patches, may reflect general physicochemical principles of proteins rather than antibody-specific sequence signals.
Although the embedding-based feature extraction strategy offers advantages in simplicity and low engineering cost, it relies on frozen pretrained representations, which limits its adaptability to specific tasks. When experimental conditions or data distributions shift, model performance may deteriorate due to insufficient task-specific adaptation. To address this limitation, recent studies have adopted end-to-end fine-tuning strategies, in which PLM parameters are directly optimized using polyreactivity labels, enabling joint refinement of representation learning and predictive modeling.
A representative example is the PolyXpert model proposed by Huang et al.[59]. Built upon a high-throughput scFv polyreactivity dataset constructed by Tessier et al., this model performed full-parameter fine-tuning of ESM-2, adapting general-purpose protein representations to the task of antibody polyreactivity prediction. The study first divided a dataset containing approximately 246,000 scFv sequences into training, validation, and internal test sets at a ratio of 60%/20%/20%, and further used another independently sourced dataset containing approximately 127,000 scFv sequences as an external test set to evaluate cross-dataset generalization. By integrating contextual sequence encoding with a task-specific classification head, the model enabled end-to-end binary classification of antibody polyreactivity. On the external independent test set, PolyXpert achieved an AUC of approximately 0.97 and exhibited greater robustness to dataset distribution shift than conventional machine learning models. Furthermore, when evaluated on a set of 131 therapeutic antibodies spanning different clinical stages reported by Wittrup et al.[2], the model predictions showed good concordance with experimentally measured polyreactivity indicators (PSR SMP scores), supporting its potential utility in antibody developability assessment.
Overall, PLMs provide a high-throughput approach for antibody polyreactivity prediction with minimal requirement for manual feature engineering. Using only variable region sequences, they enable rapid screening of large candidate libraries in early discovery stages. In addition, PLM-derived representations can be integrated with physicochemical features and experimental variables to further enhance predictive performance.
However, PLM-based approaches also have important limitations and should not be interpreted as definitive mechanistic predictors of antibody polyreactivity. First, their performance remains strongly dependent on the labeling scheme and experimental platform used to generate the training data. Because polyreactivity labels may be derived from ELISA, PSR binding, panning enrichment, or other nonspecificity assays, a model trained on one label definition may not generalize well to another. Such dataset dependence can introduce domain shift and make high reported performance difficult to compare across studies. Reported metrics are further confounded by test set construction: random splits that leave near-identical sequences in both training and test sets can substantially inflate performance relative to clustered or out-of-distribution splits. Consequently, very high reported performance should be interpreted as platform- and split-dependent rather than as direct evidence of mechanistic generalization. Critically, this implies that absolute performance values reported across different studies are not directly comparable. Because models are evaluated on different datasets under task settings of varying difficulty, ranking them by their reported AUC values would be statistically misleading. Establishing standardized, publicly available benchmark datasets with harmonized labels and predefined evaluation splits is therefore essential for fair model comparison. Second, PLMs typically learn statistical sequence representations rather than explicit structural or dynamical determinants. Although their embeddings may indirectly encode statistical correlates of physicochemical and structural features, they do not explicitly model the three-dimensional and dynamical origins of these properties, such as surface charge organization, conformational flexibility, transient exposure of hydrophobic patches, charge redistribution, or antigen-dependent binding environments, all of which may influence antibody polyreactivity. Third, the interpretability of PLM-based predictors depends strongly on the modeling framework. When PLM embeddings are combined with relatively interpretable models, such as LR or tree-based methods, attribution analyses can provide insights into sequence features associated with polyreactivity. However, the biological meaning of embedding dimensions often remains unclear[80]. Moreover, interpretability decreases when PLMs are coupled with complex nonlinear classifiers or fine-tuned end-to-end. Consequently, although PLMs can support residue-level attribution analyses, they generally do not directly reveal the physicochemical and structural mechanisms underlying antibody polyreactivity.
Therefore, PLMs are best viewed as high-throughput prioritization tools rather than standalone mechanistic models. Their limitations can be partly addressed by integrating PLM predictions with interpretable physicochemical descriptors, structure-based features, model-attribution analyses, and targeted experimental validation. For example, candidates prioritized by PLMs can be further examined using net charge, isoelectric point, hydrophobic patch analysis, solvent-accessible surface properties, in silico mutational scanning, attention- or gradient-based attribution, and, where appropriate, MD simulations for selected high-risk cases. Such a combined workflow would allow PLMs to support large-scale screening while retaining mechanistic interpretability for downstream antibody optimization.
4. Outlook
As an intrinsic property of antibodies, polyreactivity is of importance both in immune defense and in therapeutic antibody development. In this review, we defined polyreactivity and emphasized that polyreactivity is different from polyspecificity in terms of binding modes. Specifically, polyspecificity is the result of different paratope-epitope pairs, whereas polyreactivity more often manifests as low affinity, distributed, nonspecific weak interactions that are primarily driven by hydrophobic and electrostatic interactions. We also summarized the current data resources relevant to antibody polyreactivity and recent progress in antibody polyreactivity prediction.
Building on this synthesis, several directions are likely to shape the future of antibody polyreactivity prediction, each accompanied by its own technical challenges and corresponding solutions. At the level of data infrastructure, polyreactivity data remain highly fragmented: studies differ substantially in reagent and matrix sources, buffer systems, coating strategies, concentration ranges, detection platforms, and thresholding criteria, which makes readouts difficult to compare or integrate across studies; moreover, many existing datasets are assembled by simply pairing deep-sequencing outputs with polyreactivity measurements and lack the experimental parameters and condition metadata needed to support developability assessment. Addressing this will require, on one hand, standardized data reporting in which each polyreactivity annotation is accompanied by key experimental metadata (e.g., antibody format; expression and purification system; reagent/matrix source; coating and detection platform; concentration and buffer conditions; batch and control settings; normalization and thresholding strategies) to improve cross-study comparability and reusability; and, on the other hand, the construction of cross-platform and cross-format bridging datasets, built from paired measurements of the same antibodies across different assay systems and from joint annotation of polyreactivity together with other developability readouts, to provide a basis for cross-system calibration. In particular, because existing datasets are dominated by fragments (e.g., scFv, VHH) while full-length IgG data remain scarce, there is a pressing need to expand polyreactivity datasets for full-length IgG and to develop strategies that bridge fragment-level and IgG-level data, so that models can be calibrated and validated for the clinically relevant therapeutic antibody formats.
At the level of model capability, as data infrastructure matures, the research focus is expected to shift from performance optimization within a single dataset toward transferable modeling across antibody formats and assay systems. The central challenge here is that polyreactivity phenotypes are themselves shaped by platform differences, threshold definitions, and batch effects, and that models trained on fragments often fail when transferred to full-length IgG owing to heavy–light chain pairing effects; in response, future models may incorporate experimental information explicitly as covariates and exploit bridging datasets to learn mappings and calibrations between assay systems, so that scores produced under different measurement systems can be reconciled within a unified scoring framework that supports cross-platform comparison and decision-making. From an application perspective, models are likewise expected to evolve from tools that merely output a risk score into tools that support engineering decisions, an evolution constrained, however, by the inherent decline in interpretability as models grow more complex or are fine-tuned end-to-end, and by the fact that localized risk drivers are not necessarily causal. A practical path is to combine attribution analyses with in silico mutational scanning, localizing major risk drivers such as charge clusters, hydrophobic patches, key CDR motifs, and framework-region contributions while also indicating actionable intervention sites and their potential trade-offs (e.g., effects on affinity or stability). In addition, as the dimensionality of developability data continues to expand, polyreactivity is likely to be modeled jointly with properties such as aggregation, self-interaction, viscosity, and nonspecific clearance; the challenge that these properties differ in data source and scale and often trade off against one another motivates the use of multi-task learning and multi-objective optimization frameworks that more faithfully reflect the decision logic of overall developability optimization.
Beyond prediction, computational methods can also add value at the level of mechanistic understanding. Although structure-derived descriptors have already been used as input features for polyreactivity prediction, their reliance on predicted structures makes them inherently lower-throughput than sequence-based encodings, and they remain descriptive rather than mechanistic. Structure-based MD simulations can complement this gap by addressing not whether a sequence is polyreactive but how polyreactivity arises, through explicit modeling of three-dimensional conformation, interfacial physicochemistry, and interaction dynamics; in particular, MD provides a way to test whether the risk drivers localized by the interpretable models discussed above are mechanistically real, for example, whether a flagged charge cluster or hydrophobic patch actually forms an exposed, conformationally accessible sticky surface, rather than merely a sequence-level correlate. At present, however, there is still no unified simulation framework for antibody polyreactivity, and studies often reach inconsistent conclusions owing to differences in system setup, antibody type, sampling scale, and phenotype definition. For example, while conformational flexibility of the Fab, particularly CDR3[81], has traditionally been associated with polyreactivity, more recent work suggests that polyreactive Fabs may instead be relatively rigid and accommodate diverse antigens through a comparatively “neutral” binding surface[32]; the relative contributions of sequence-level physicochemical properties versus structure-level dynamics likewise remain under debate[35]. These discrepancies, together with the high computational cost of MD and its sensitivity to sampling and convergence, mean that MD is best positioned not as a scalable predictor but as a mechanistic adjunct, applied selectively to representative or high-risk candidates to generate and test hypotheses about the origins of polyreactivity, thereby guiding targeted experimental validation and rational engineering.
Overall, as data resources, modeling capability, and mechanistic understanding advance together, antibody polyreactivity prediction is expected to develop from performance competition on individual datasets into a unified framework spanning measurement, prediction, and mechanistic interpretation, providing a more reliable foundation for the developability assessment and rational engineering of therapeutic antibodies.
Authors contribution
Tang H: Conceptualization, writing-original draft.
Zhang Z, Li W: Data curation.
Pan X, Zhou Y, Huang J: Writing-review & editing.
Conflicts of interest
The authors declare no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
This work was supported by the National Natural Science Foundation of China (Grant Nos. 62371112 and 62071099) and the Sichuan Science and Technology Program (Grant No. 2024NSFSC0635).
Copyright
© The Author(s) 2026.
References
-
1. Cramer F. Emil fischer’s lock-and-key hypothesis after 100 years-towards a supracellular chemistry. In: Behr JP, editior. Perspectives in supramolecular chemistry: The lock-and-key principle. Chichester: John Wiley & Sons; 1994. p. 1-23.[DOI]
-
3. Ichiyoshi Y, Casali P. Analysis of the structural correlates for antibody polyreactivity by multiple reassortments of chimeric human immunoglobulin heavy and light chain V segments. J Exp Med. 1994;180(3):885-895.[DOI]
-
4. Tsuiji M, Yurasov S, Velinzon K, Thomas S, Nussenzweig MC, Wardemann H, et al. A checkpoint for autoreactivity in human IgM+ memory B cell development. J Exp Med. 2006;203(2):393-400.[DOI]
-
5. Dighiero G, Lymberi P, Mazié JC, Rouyre S, Butler-Browne GS, Whalen RG, et al. Murine hybridomas secreting natural monoclonal antibodies reacting with self antigens. J Immunol. 1983;131(5):2267-2272.[DOI]
-
7. Starr CG, Tessier PM. Selecting and engineering monoclonal antibodies with drug-like specificity. Curr Opin Biotechnol. 2019;60:119-127.[DOI]
-
9. Sigounas G, Harindranath N, Donadel G, Notkins AL. Half-life of polyreactive antibodies. J Clin Immunol. 1994;14(2):134-140.[DOI]
-
15. Zhang J, Jacobi AM, Wang T, Berlin R, Volpe BT, Diamond B, et al. Polyreactive autoantibodies in systemic lupus erythematosus have pathogenic potential. J Autoimmun. 2009;33(3-4):270-274.[DOI]
-
16. Amara K, Steen J, Murray F, Morbach H, Fernandez-Rodriguez BM, Joshua V, et al. Monoclonal IgG antibodies generated from joint-derived B cells of RA patients have a strong bias toward citrullinated autoantigen recognition. J Exp Med. 2013;210(3):445-455.[DOI]
-
20. Mouquet H, Nussenzweig MC. Polyreactive antibodies in adaptive immune responses to viruses. Cell Mol Life Sci. 2012;69(9):1435-1445.[DOI]
-
23. Broug E, Bland-Ward PA, Powell J, Johnson KS. Fab-arm exchange. Nat Biotechnol. 2010;28(2):123-125.[DOI]
-
24. Bajaj G, Shchelokov D, Demin O Jr, Adams HC, Feng Y, Gibiansky L, et al. Dose selection for DuoBody®-CD40x4-1BB (GEN1042/BNT312) using a mPBPK/RO model leveraging preclinical and clinical data. Clin Pharma Ther. 2025;118(2):418-427.[DOI]
-
31. Wymann S, Morales RAV, Toh WH, Remlinger J, Guse K, Ghai R, et al. CSL305: A dual functional therapeutic antibody targeting complement C2 and FcRn. Int J Mol Sci. 2026;27(5):2383.[DOI]
-
34. Torres M, Casadevall A. The immunoglobulin constant region contributes to affinity and specificity. Trends Immunol. 2008;29(2):91-97.[DOI]
-
36. Xu Y, Roach W, Sun T, Jain T, Prinz B, Yu TY, et al. Addressing polyspecificity of antibodies selected from an in vitro yeast presentation system: A FACS-based, high-throughput selection and analytical tool. Protein Eng Des Sel. 2013;26(10):663-670.[DOI]
-
37. Almagro JC, Pedraza-Escalona M, Arrieta HI, Pérez-Tapia SM. Phage display libraries for antibody therapeutic discovery and development. Antibodies. 2019;8(3):44.[DOI]
-
39. Frese K, Eisenmann M, Ostendorp R, Brocks B, Pabst S. An automated immunoassay for early specificity profiling of antibodies. MAbs. 2013;5(2):279-287.[DOI]
-
43. Arakawa T, Akuta T. Mechanistic insight into poly-reactivity of immune antibodies upon acid denaturation or arginine mutation in antigen-binding regions. Antibodies. 2023;12(4):64.[DOI]
-
46. Harvey EP, Shin JE, Skiba MA, Nemeth GR, Hurley JD, Wellner A, et al. An in silico method to assess antibody fragment polyreactivity. Nat Commun. 2022;13(1):7554.[DOI]
-
48. Gai SA, Wittrup KD. Yeast surface display for protein engineering and characterization. Curr Opin Struct Biol. 2007;17(4):467-473.[DOI]
-
53. Marks C, Deane CM. How repertoire data are changing antibody science. J Biol Chem. 2020;295(29):9823-9837.[DOI]
-
61. Yang X, Wu C, Liu W, Fu K, Tian Y, Wei X, et al. A clinical-information-free method for early diagnosis of lung cancer from the patients with pulmonary nodules based on backpropagation neural network model. Comput Struct Biotechnol J. 2024;24:404-411.[DOI]
-
62. Zhou Y, Liu W, Luo C, Huang Z, Samarappuli Mudiyanselage Savini G, Zhao L, et al. Ab-Amy 2.0: Predicting light chain amyloidogenic risk of therapeutic antibodies based on antibody language model. Methods. 2025;233:11-18.[DOI]
-
63. LeCun Y, Bengio Y, Hinton G. Deep learning. Nature. 2015;521(7553):436-444.[DOI]
-
69. Zhang Z, Xu M, Jamasb A, Chenthamarakshan V, Lozano A, Das P, et al. Protein representation learning by geometric structure pretraining. arXiv:2203.06125v5 [Preprint]. 2022.[DOI]
-
70. Ruffolo JA, Sulam J, Gray JJ. Antibody structure prediction using interpretable deep learning. Patterns. 2022;3(2):100406.[DOI]
-
71. Ofer D, Brandes N, Linial M. The language of proteins: NLP, machine learning & protein sequences. Comput Struct Biotechnol J. 2021;19:1750-1758.[DOI]
-
75. Meier J, Rao R, Verkuil R, Liu J, Sercu T, Rives A, et al. Language models enable zero-shot prediction of the effects of mutations on protein function. bioRxiv [Preprint]. 2021.[DOI]
-
77. Robin G, Sato Y, Desplancq D, Rochel N, Weiss E, Martineau P, et al. Erratum to “restricted diversity of antigen binding residues of antibodies revealed by computational alanine scanning of 227 antibody-antigen complexes”. J Mol Biol. 2014;426(22):3729-3743.[DOI]
-
79. Sakhnini LI, Beltrame L, Fulle S, Sormanni P, Henriksen A, Lorenzen N, et al. Prediction of antibody non-specificity using protein language models and biophysical parameters. mAbs. 2026;18(1):2678000.[DOI]
Copyright
© The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Publisher’s Note
Science Exploration remains a neutral stance on jurisdictional claims in published
maps
and institutional affiliations. The views expressed in this article are solely those
of
the author(s) and do not reflect the opinions of the Editors or the publisher.
Share And Cite



Science Exploration Style
Tang H, Zhang Z, Li W, Pan X, Zhou Y, Huang J. Advances in database resources and computational methods for predicting antibody polyreactivity. Comput Biomed. 2026;1:202616. https://doi.org/10.70401/cbm.2026.0021
Tips
Copy completed.
Submit a Manuscript
Author Instructions
Cite this Article
Article Metrics
0
View
0
Download
Cited
Article Updates

- Abstract
- Keywords
- 1. Introduction
- 2. Polyreactivity-Related Data Resources
- 3. Computational Methods for Polyreactivity
- 4. Outlook
- Authors contribution
- Conflicts of interest
- Ethical approval
- Consent to participate
- Consent for publication
- Availability of data and materials
- Funding
- References
- Copyright
Science Exploration Style
Tang H, Zhang Z, Li W, Pan X, Zhou Y, Huang J. Advances in database resources and computational methods for predicting antibody polyreactivity. Comput Biomed. 2026;1:202616. https://doi.org/10.70401/cbm.2026.0021
copy
Share Link
copy