ZBP1-mediated sensing of genomic stress in cancer therapy
Xiao Zhong
1

,
Siddharth Balachandran
2,*
,
Ting Zhang
1,3,4,*
*Correspondence to:
Siddharth Balachandran, Center for Immunology, Fox Chase Cancer Center, Philadelphia, PA 19111-2497, USA.
E-mail: Siddharth.balachandran@fccc.edu
Ting Zhang, Laboratory of Hepatic AI Translation, Frontiers Science Center for Disease-Related Molecular Network, West China Hospital, Sichuan University, Chengdu 610041, Sichuan, China. E-mail: tingzhang923@wchscu.cn
Ting Zhang, Laboratory of Hepatic AI Translation, Frontiers Science Center for Disease-Related Molecular Network, West China Hospital, Sichuan University, Chengdu 610041, Sichuan, China. E-mail: tingzhang923@wchscu.cn
Ferroptosis Oxid Stress. 2026;2:202621. 10.70401/fos.2026.0035
Received: May 18, 2026Accepted: July 08, 2026Published: July 08, 2026
Abstract
Z-nucleic acid-binding protein 1 (ZBP1) is a cytosolic innate immune sensor that detects left-handed Z-DNA and Z-RNA, structures arising from endogenous retroelements, splicing stress, R-loops, viruses, or viral mimicry. Originally viewed as an antiviral receptor, ZBP1 has emerged as a sentinel of genomic and transcriptomic instability. Upon ligand binding, ZBP1 recruits receptor-interacting serine/threonine-protein kinase 1 (RIPK1) and receptor-interacting serine/threonine-protein kinase 3 (RIPK3) via RIP homotypic interaction motif domains; in humans, RIPK1 acts as an essential scaffold to form the necrosome, which phosphorylates mixed lineage kinase domain-like pseudokinase (MLKL) and triggers necroptosis, a lytic, immunogenic cell death. This pathway is intimately regulated by reactive oxygen species (ROS): oxidative stress promotes RIPK1 activation, and necroptosis further drives a mitochondrial ROS burst, establishing a feed-forward amplification loop. In cancer, therapeutic induction of viral mimicry (e.g., using curaxins or splicing inhibitors) combined with tumor-localized ROS generation activates ZBP1-driven necroptosis, leading to the release of damage-associated molecular patterns and tumor antigens. This process converts immunosuppressive “cold” tumors into inflamed “hot” tumors, enhancing dendritic cell maturation, CD8+ T cell infiltration, and sensitivity to immune checkpoint blockade. Integrating epigenetic priming, nanomedicine, and patient stratification according to ZBP1/RIPK3/MLKL pathway status holds promise for overcoming therapeutic resistance. Thus, ZBP1-mediated nucleic acid surveillance represents a central innate checkpoint bridging genomic stress to antitumor immunity.
Keywords
Necroptosis, Z-nucleic acid-binding protein 1, Z-RNA, reactive oxygen species, viral mimicry
1. Introduction
1.1 ZBP1 in innate immune surveillance
The capacity of the innate immune system to discriminate between self and non-self is essential for maintaining cellular integrity and organismal survival[1]. At the center of this surveillance network are pattern recognition receptors, which detect molecular features associated with infection or tissue injury[2,3]. Among them, Z-nucleic acid-binding protein 1 (ZBP1) has emerged as an intracellular sensor that governs key innate immune checkpoints[4,5]. Its defining property is the ability to recognize an unusual nucleic acid topology, the left-handed double-helical structures Z-DNA and Z-RNA[6,7]. This specificity is conferred by the Zα domain, an evolutionarily conserved module found across most life domains and in partial viruses, highlighting the deep evolutionary origin of this sensing system[8].
1.2 Endogenous Z-RNA and viral mimicry
What, then, gives rise to the ligands that engage ZBP1? Early work positioned ZBP1 primarily within antiviral immunity, where it functions as a sensor of pathogen-associated molecular patterns (PAMPs)[9,10]. During viral replication, ZBP1 detects viral Z-RNA and initiates inflammatory signaling programs that limit pathogen spread[11,12]. More recent studies, however, have broadened this framework considerably by showing that Z-RNA is not uniquely derived from microbes[13]. A large fraction of the host genome is composed of endogenous retroelements and other repetitive sequences[14,15]. Under normal conditions, these regions are largely silenced through epigenetic repression, but they can become transcriptionally derepressed during cellular stress, virus-induced transcriptional perturbation, or experimental epigenetic reprogramming[16]. Once expressed, these host-derived transcripts may adopt the Z conformation and function as bona fide damage-associated molecular patterns (DAMPs)[17,18]. Their recognition by ZBP1 elicits an innate immune response that, even when triggered in non-viral scenarios, closely resembles that is activated during actual virus infections, a process widely described as viral mimicry[19,20]. In this broader context, ZBP1 is no longer viewed solely as a sensor of invading viral pathogens; rather, it appears to function as a more general intracellular sentinel of genomic instability and transcriptomic stress[4,21].
1.3 Signaling consequences: Necroptosis and ROS amplification
Following engagement by Z-form nucleic acids, ZBP1 initiates a signaling cascade that culminates in necroptosis, a regulated lytic type of cell death distinct from apoptosis[22,23]. Unlike apoptosis, which is typically immunologically silent, necroptosis promotes inflammatory signaling through the release of intracellular contents into the surrounding tissue[24]. This pathway depends on the recruitment of receptor-interacting serine/threonine-protein kinase 3 (RIPK3), followed by phosphorylation of mixed lineage kinase domain-like pseudokinase (MLKL) and its translocation to cellular membranes, ultimately causing membrane rupture[25,26]. Within this signaling architecture, reactive oxygen species (ROS) serve as both metabolic effectors and regulatory amplifiers[27]. Mitochondrial and cytosolic ROS influence necrosome assembly and activation, establishing a feed-forward circuit that reinforces necroptotic commitment while reshaping the oxidative state of the local microenvironment[26,28,29].
1.4 Translational potential in cancer immunotherapy
These mechanisms are particularly relevant in oncology, where resistance to apoptosis and a profoundly immunosuppressive tumor microenvironment (TME) remain major barriers to effective immune checkpoint blockade[30,31]. Could ZBP1-driven necroptosis and inflammatory gene activation help overcome these limitations? Growing evidence suggests that the functional interplay among ZBP1, ROS, and downstream receptor-interacting serine/threonine-protein kinase 1 (RIPK1)/3-driven signaling pathways provides a promising route to do so[32]. Because necroptosis is lytic, it enables the release of a broad spectrum of DAMPs and tumor-associated antigens[33]. In experimental tumor settings, this form of immunogenic cell death promotes dendritic cell maturation and enhances antigen cross-presentation[34]. The downstream consequence is increased infiltration of cytotoxic CD8+ T cells, facilitating the conversion of poorly inflamed “cold” tumors into immune-responsive “hot” tumors[35,36]. Moreover, noncytolytic signaling by ZBP1 or RIPK3 is also potently immunogenic[37].
In this review, we comprehensively examine how ZBP1 serves as a sentinel of innate immune surveillance, initiating necroptosis in stressed cells during infection or other pathological conditions to remove hazardous stimuli and triggering inflammatory gene expression programs to mobilize host immune defenses[38]. We further discuss the reciprocal relationship between ROS generation and ZBP1-driven signaling, with particular emphasis on how redox signaling shapes necroptotic pathway activation, as well as gene transcription programs[39]. We then highlight recent advances in translational research, including strategies that induce viral mimicry and the development of tumor-responsive nanotherapeutic platforms, both of which harness ROS-associated necroptosis to reprogram the TME and, ultimately, strengthen systemic antitumor immunity[40-43].
2. Z-RNA Triggers Checkpoints of Innate Immune Responses in Pathogenesis
A clear understanding of the sources of Z-RNA is necessary before considering how ZBP1-initiated viral mimicry responses might be therapeutically exploited. Innate immunity relies on germline-encoded pattern recognition receptors to detect structural features associated with pathogens as well as signals generated by pathological stress[1,44,45]. One such defense mechanism is the sensing of Z-RNA, a left-handed nucleic acid conformation with unusual biological significance. Although classical models of innate immune recognition emphasize direct detection of pathogen-derived molecular patterns, Z-RNA arises from a more complex set of origins. It can be produced not only by invading microorganisms but also, and in some contexts abundantly, by host cells themselves. Depending on the physiological setting and stage of stress, these Z-RNAs may function as either damage-associated or PAMPs[3,17,44].
2.1 Three checkpoints of innate immune responses provoked by pathogen infection
In eukaryotic cells, transcription termination of protein-coding genes is tightly coordinated with 3′-end cleavage and mRNA polyadenylation. This process is executed by a large macromolecular machinery centered on the cleavage and polyadenylation specificity factor (CPSF) complex. When CPSF-dependent recognition or cleavage is impaired, transcription fails to terminate properly, and readthrough transcripts accumulate. Because CPSF is a core component of the host transcriptional and mRNA 3′-end processing machinery, it represents a strategic target for viral interference. By hijacking or inhibiting CPSF, viruses can efficiently suppress host gene expression, including a large number of antiviral transcripts. In doing so, they redirect the cellular RNA-processing landscape to favor viral replication and expression. Loss of normal CPSF activity also readily induces host disruption of transcription termination (DoTT)[46].
Because many pathogens manipulate host CPSF machinery to support their replication, they inadvertently trigger the production of DoTT-derived Z-RNA[47,48]. ZBP1 appears to function as a sentinel at this early checkpoint. When these signals are sufficiently abundant or accessible, ZBP1 activation can drive the death of infected cells before productive infection is fully established, thereby limiting viral dissemination.
What happens if this early checkpoint is not strong enough to eliminate infected cells? In that setting, the infected tissue progresses to a second phase dominated by interferon signaling. Cellular stress and widespread transcriptional dysregulation promote cytosolic accumulation of multiple nucleic acid species, including single-stranded RNA, double-stranded RNA in the right-handed conformation (A-RNA), and double-stranded DNA, all of which are sensed by distinct pattern recognition pathways[49]. Within the RIG-I-like receptor family, RIG-I preferentially binds short double-stranded RNAs (dsRNAs) bearing 5′-triphosphate ends, whereas MDA5 assembles on long double-stranded A-RNA molecules[50]. Ligand binding induces oligomerization of these receptors and activation of the mitochondrial antiviral signaling adaptor, mitochondrial antiviral signaling protein (MAVS), which forms prion-like aggregates on the outer mitochondrial membrane[51]. In parallel, cytosolic double-stranded DNA is detected by cyclic GMP-AMP synthase (cGAS), which produces the second messenger 2′3′-cGAMP[52]. Binding of cGAMP activates the endoplasmic reticulum adaptor, stimulator of interferon genes (STING) and drives its trafficking to the Golgi apparatus. Both MAVS and STING recruit TBK1 and IKKε, which phosphorylate IRF3 and IRF7. The activated transcription factors then dimerize and translocate to the nucleus, inducing robust expression of type I interferons and type III interferons[53,54]. Secreted interferons act through autocrine and paracrine signaling via IFNAR1-IFNAR2 and IFNLR1-IL10Rβ receptor complexes, respectively, to activate the JAK-STAT pathway[55]. STAT1 and STAT2, together with IRF9, form the ISGF3 complex, which enters the nucleus and binds interferon-stimulated response elements, thereby inducing hundreds of interferon-stimulated antiviral genes[56,57]. This interferon response is essential for host antiviral defense, but it is not without risk. Many interferon-stimulated transcripts harbor Z-conformation-prone sequences within their 3′ untranslated regions, most of which are derived from endogenous retroelements. As these transcripts accumulate, they can give rise to abundant endogenous Z-RNA, which is subsequently sensed by ZBP1 and can trigger necroptotic cell death. This process constitutes an additional ZBP1-governed innate immune checkpoint. Its protective function, however, is accompanied by clear pathological potential: when interferon signaling and ZBP1-driven necroptosis (or inflammatory gene transcription) become mutually reinforcing, excessive inflammatory activation may ensue, ultimately contributing to autoimmune disease[32].
To prevent this transition from antiviral defense to systemic autoinflammation, mammals rely heavily on the constitutive editing activity of adenosine deaminase acting on RNA 1 (ADAR1). The p150 isoform of ADAR1, itself an interferon-stimulated gene product, contains a Zα domain that allows selective recognition of Z-RNA[58-60]. Once bound to these duplex structures, ADAR1 catalyzes adenosine-to-inosine editing, destabilizing the Watson-Crick interactions that support the Z-helical conformation[32]. In functional terms, this editing process unwinds or neutralizes immunogenic Z-RNA, converting it into a form less likely to engage innate sensors[61]. Yet this protective capacity is not unlimited. When interferon signaling becomes sustained and endogenous Z-RNA production rises sharply, ADAR1-mediated editing can be outpaced. Mutations in the ADAR1 Zα domain that prevent Z-RNA binding abolish this control mechanism, allowing unedited dsRNAs to accumulate and provoke a spontaneous MDA5-MAVS-dependent type I interferon response. In both humans and mice, this failure underlies the severe autoinflammatory disorder Aicardi-Goutières syndrome[62].
A third checkpoint becomes prominent during acute infection or periods of intense viral replication. Under such conditions, high replicative stress and the intrinsically limited fidelity of many viral polymerases promote polymerase slippage and template switching, generating abundant double-stranded RNA species and defective viral genomes (DVGs)[63]. Because DVGs often contain internal deletions and inverted repeat elements, they readily fold into stable higher-order RNA structures in the crowded intracellular milieu. In certain sequence and topological contexts, notably alternating purine-pyrimidine tracts and conditions favoring negative supercoiling, these duplexes may transition from the more common A-RNA form into left-handed Z-RNA. Influenza A virus provides a well-characterized example: as a segmented negative-strand RNA virus that replicates in the nucleus using an error-prone polymerase lacking proofreading capacity, it frequently generates internally deleted DVGs, especially from long genomic segments. The conserved terminal complementarity of these RNAs facilitates formation of stable long double-stranded structures[64]. Although SARS-CoV-2 encodes the proofreading exonuclease Nsp14, high viral burden still promotes discontinuous transcription and polymerase derailment at structural hotspots, resulting in substantial DVG production[65,66]. In poxviruses, convergent transcription of neighboring genes late in infection, together with inefficient termination and extensive readthrough, generates complementary transcripts that anneal into long double-stranded RNA networks within cytoplasmic viral factories[67]. The vaccinia virus E3 protein, through its Z-DNA-binding domain, has also been implicated in recognition and stabilization of Z-RNA-containing structures during infection[68,69]. At later stages of infection, virus-derived Z-RNAs act as the terminal trigger of the innate immune checkpoint monitored by ZBP1. Extensive viral replication generates large amounts of these ligands within infected cells, thereby promoting ZBP1 engagement and the death of infected cells.
Taken together, these observations indicate that many viruses are capable of generating immunogenic Z-RNA through several converging mechanisms. In this context, ZBP1 exerts its most direct antimicrobial function by restricting further viral spread. Z-RNA therefore serves not only as an indicator of infection but also as a critical molecular cue through which ZBP1 surveys innate immune checkpoint activation.
2.2 Stress-driven checkpoints of innate immune responses in non-infectious pathophysiological states
The generation of Z-RNA, however, is not confined to antiviral defense. It is also closely linked to cellular stress and disordered RNA metabolism. As noted above, mammalian cells are capable of producing substantial amounts of Z-RNA from endogenous genomic templates[70,71]. The principal source of these endogenous ligands is the large reservoir of noncoding repetitive elements dispersed throughout the genome[14,72]. Among them, endogenous retroelements make up the largest fraction, accounting for nearly 45% of the human genome[15,73]. Under normal conditions, these repetitive elements are tightly silenced. However, when repressive epigenetic marks, such as DNA methylation or H3K9me3, are lost due to pharmacological inhibition or cellular stress, these endogenous retroelements undergo massive bi-directional transcription. The resulting complementary transcripts readily hybridize into long dsRNAs, which are highly prone to forming Z-RNA structures and engaging ZBP1-mediated viral mimicry[16,74].
When spliceosome function is impaired, whether by cellular stress, mutations in core splicing factors such as SF3B1, or pharmacologic inhibition, widespread intron retention ensues[13,18]. If nonsense-mediated decay is unable to efficiently clear these aberrant transcripts, retained introns and mis-spliced RNAs accumulate in the nucleus. Unlike properly processed mRNAs, these stranded transcripts are not fully protected by mature RNA-binding protein complexes. Their internal regions, particularly those enriched in alternating purine-pyrimidine motifs, are structurally prone to adopting stable left-handed conformations.
A related source of endogenous Z-form ligands emerges from R-loop accumulation. R-loops are three-stranded nucleic acid structures composed of an RNA:DNA hybrid and a displaced single-stranded DNA strand[75,76]. Their abundance increases under conditions such as prolonged transcriptional pausing, replication fork stalling, and spliceosome stress[77]. Within transcriptionally active chromatin, local torsional strain can promote a conformational transition of the RNA:DNA hybrid into a left-handed state[77]. This Z-conformation RNA:DNA hybrid can be recognized by the Zα domain of ZBP1, thereby representing an additional class of endogenous ligands[21,78]. In addition, recent evidence has expanded the ligand repertoire of ZBP1 beyond classical Z-form nucleic acids to include G-quadruplexes (G4s). In vitro biophysical studies have demonstrated that both the Zα1 and Zα2 domains of ZBP1 can directly bind to various DNA and RNA G4 structures with nanomolar affinities[79]. Specifically, the Zα2 domain of the truncated ZBP1 isoform acts as a direct sensor for G4 structures formed within guanine-rich telomeric repeat-containing RNA (TERRA). This specific engagement with TERRA G4s promotes higher-order ZBP1 oligomerization. The resulting ZBP1 assembly subsequently activates MAVS-dependent innate immune signaling, robust type I interferon responses, and inflammatory cell death[80].
Taken together, these observations indicate that, even in the absence of infection, defects in RNA processing and genome maintenance can generate Z-conformation nucleic acids capable of engaging innate immune surveillance (Figure 1).
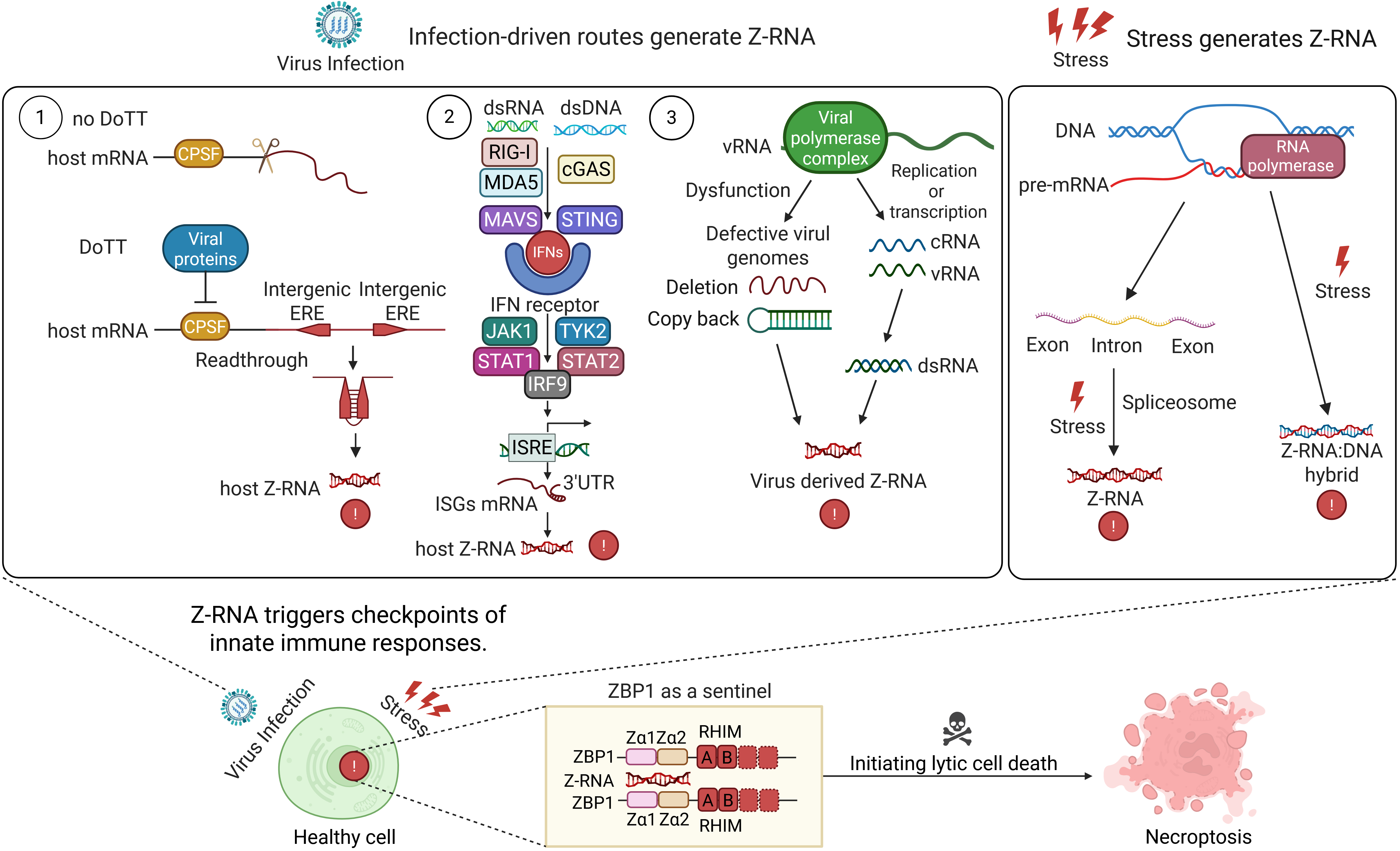{kind=link}
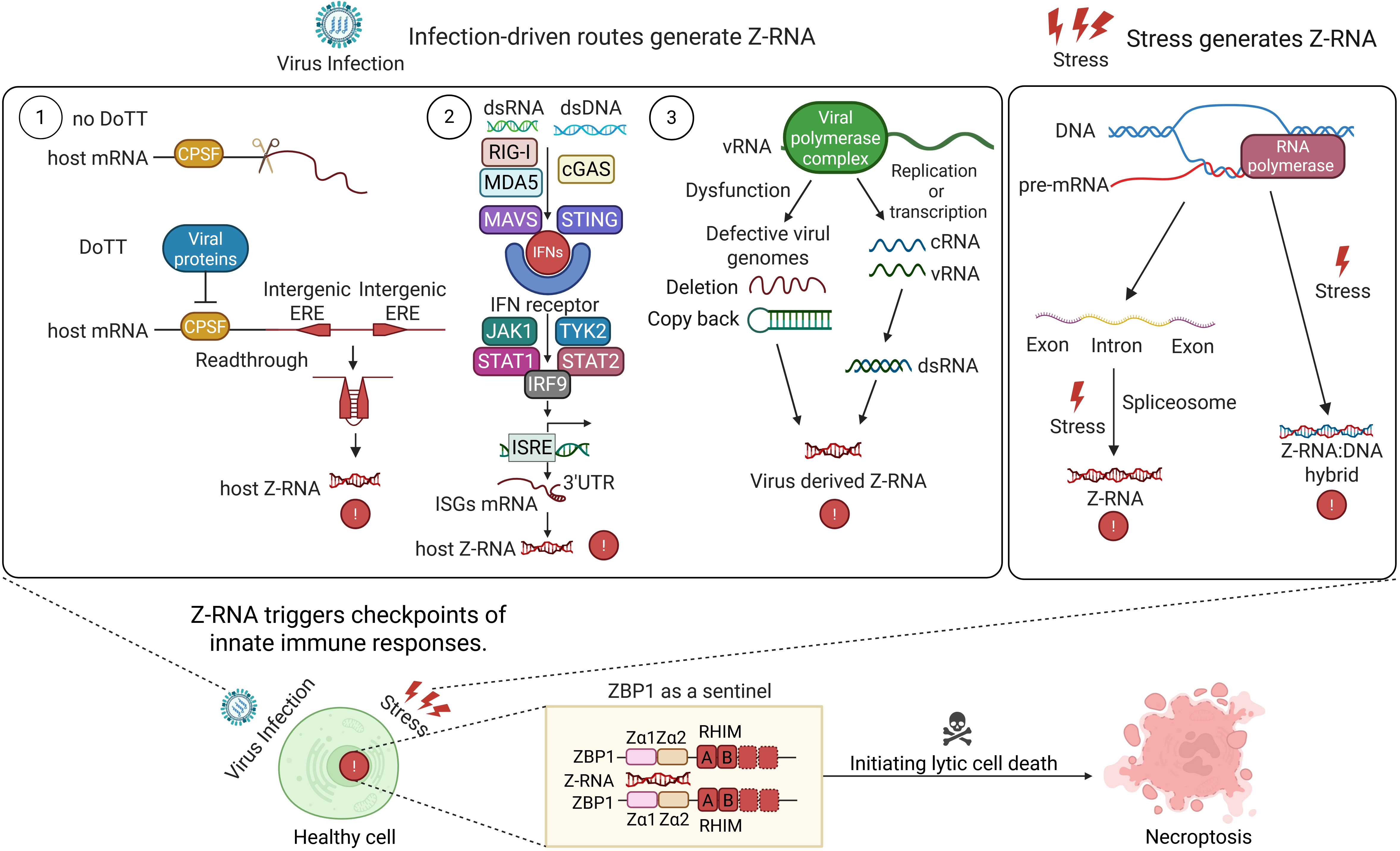
Figure 1. Infection-and stress-driven routes that generate Z-RNA and trigger innate immune checkpoints. Left: Three infection-driven pathways generate Z-RNA. Viral inhibition of the CPSF complex produces readthrough transcripts; interferon signalling induces ISG mRNAs with Z-prone 3’UTRs; and error-prone viral replication yields DVGs. All three sources form Z-RNA. Right: Non-infectious stressors also generate Z-RNA. Spliceosome stress causes intron retention, and R-loop accumulation leads to Z-RNA: DNA hybrids under torsional strain. These endogenous Z-RNAs converge on the sensor ZBP1, triggering necroptosis. Created in BioRender. Zhong, X. (2026) https://biorender.com/dxqiatw. DoTT: differential processing of transcript termini; CPSF: cleavage and polyadenylation specificity factor; ERE: exonic repeat element; dsRNA: double-stranded RNA; dsDNA: double-stranded DNA; RIG-I: retinoic acid-inducible gene I; MDA5: melanoma differentiation-associated protein 5; MAVS: mitochondrial antiviral signaling protein; cGAS: cyclic GMP-AMP synthase; STING: stimulator of interferon genes; IFNs: interferons; JAK1: Janus kinase 1; TYK2: tyrosine kinase 2; STAT: signal transducer and activator of transcription; IRF9: interferon regulatory factor 9; ISRE: interferon-stimulated response element; 3’UTR: three-prime untranslated region; ISGs: interferon-stimulated genes; vRNA: viral RNA; cRNA: complementary RNA; ZBP1: Z-nucleic acid-binding protein 1; RHIM: receptor-interacting protein homotypic interaction motif; DVGs: defective viral genomes.
3. ZBP1 as the Central Sentinel of Z-RNA-Mediated Checkpoints in Innate Immunity
The range of Z-RNA sources is therefore broader than initially appreciated. Viral DoTT, interferon-driven transcriptional amplification, replication-derived viral RNA products, and non-infectious cellular stress can all generate ligands for ZBP1. This expanded landscape places ZBP1 in a role that extends well beyond conventional pathogen sensing. Rather than functioning solely as a detector of pathogen invasion, ZBP1 appears to operate as a broader checkpoint sentinel that monitors genomic instability, transcriptomic stress, and failures in RNA processing.
This function has important implications for cellular quality control. Severe genomic instability is commonly accompanied by widespread transcriptional and splicing defects. If cells bearing such abnormalities evade elimination, malformed transcripts may be translated into dysfunctional proteins, potentially disrupting tumor suppressor pathways or generating oncogenic fusion products. In this setting, ZBP1 provides surveillance at the level of RNA metabolism by initiating non-lytic inflammatory signaling alongside lytic cell death before aberrant proteins accumulate to pathogenic levels. Necroptotic rupture, or even sub-lethal membrane perforation, of these compromised cells also releases nucleic acids and other DAMPs, thereby generating a paracrine alarm signal that recruits immune cells and may facilitate presentation of neoantigens (Figure 2).
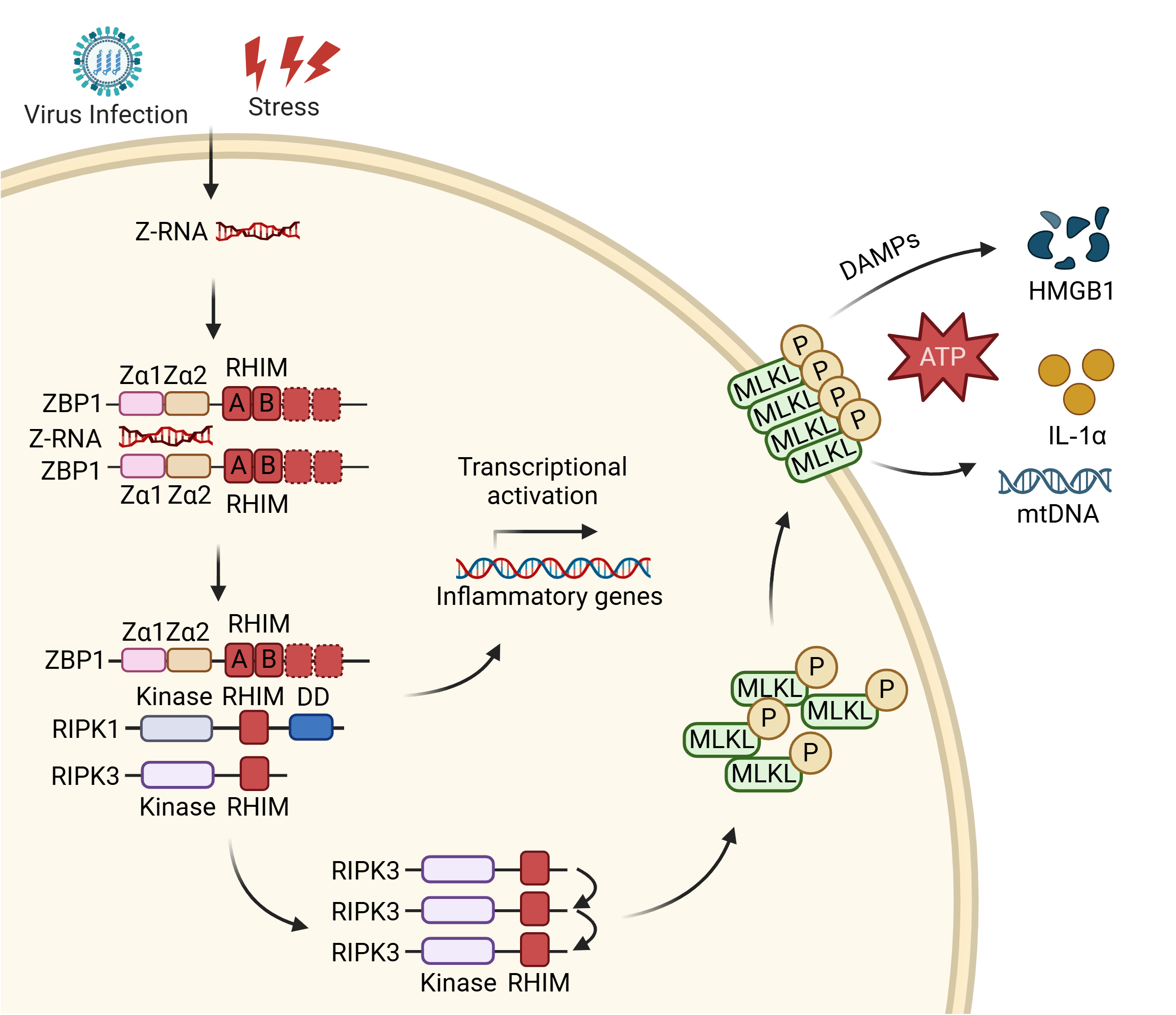{kind=link}
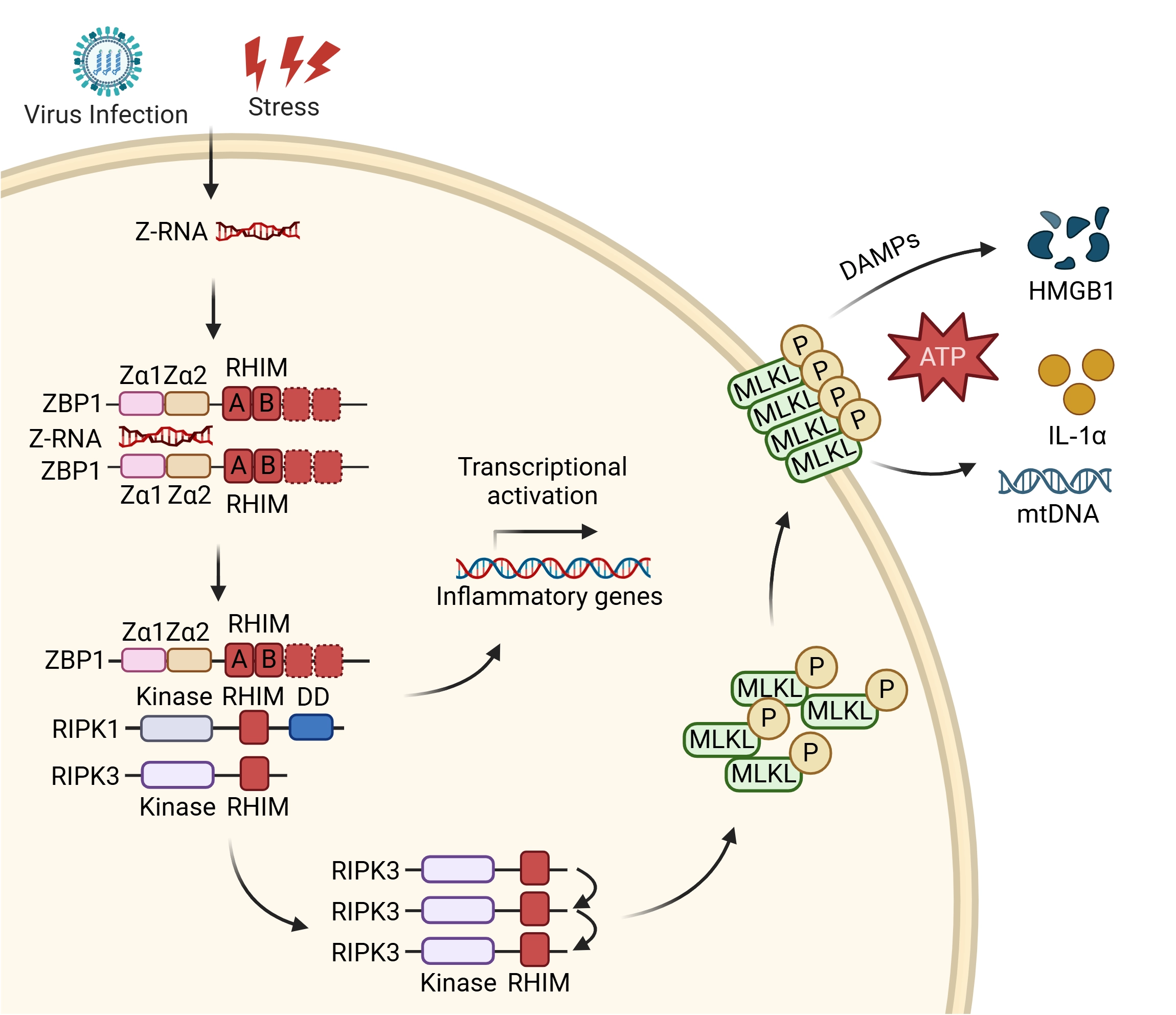
Figure 2. Z-RNA activates ZBP1-mediated necroptosis and inflammatory gene transcription. Z-RNA from various sources binds the Zα domains of ZBP1, inducing its activation. ZBP1 then recruits RIPK1 and RIPK3 via RHIM–RHIM interactions to assemble a signaling scaffold. From this complex, the pathway can bifurcate: it engages non-lytic signaling cascades leading to the transcriptional activation of inflammatory genes, or it proceeds to execute cell death. For necroptosis execution, activated RIPK3 phosphorylates MLKL, which oligomerizes and forms plasma membrane pores, leading to necroptotic lysis (or sublethal membrane perforation) and the release of DAMPs (HMGB1, ATP, IL-1α, mtDNA). Created in BioRender. Zhong, X. (2026) https://BioRender.com/ftogojj. Z-RNA: Z-form double-stranded RNA; ZBP1: Z-nucleic acid-binding protein 1; RIPK: receptor-interacting serine/threonine-protein kinase; RHIM: receptor-interacting protein homotypic interaction motif; MLKL: mixed lineage kinase domain-like protein; P: phosphorylation; DAMPs: damage-associated molecular patterns; HMGB1: high mobility group box 1; ATP: adenosine triphosphate; IL-1α: interleukin-1 alpha; mtDNA: mitochondrial DNA.
3.1 Molecular recognition and activation of ZBP1 by Z-RNA
ZBP1 was originally identified as DLM-1, also termed DAI, and was first described as a cytosolic sensor of double-stranded DNA[3]. Subsequent structural and biochemical work has substantially revised that view, establishing ZBP1 as a principal sensor of left-handed Z-form nucleic acids[8,81]. In contrast to the canonical right-handed helix, Z-conformations are defined by a left-handed helical twist and a characteristic zigzag phosphate backbone[5,82]. ZBP1 detects these structures through its conserved Zα domains, which recognize the distinctive backbone geometry of Z-form nucleic acids. Binding is mediated by a network of hydrogen bonds and contacts with features of the altered minor groove characteristic of the Z-helix[7,8,77,81].
Engagement of Z-RNA by the Zα domain induces a conformational change in ZBP1[83]. This transition is thought to release autoinhibitory constraints and promote oligomerization of the receptor[7]. Biophysical studies further suggest that activated ZBP1 can undergo liquid-liquid phase separation or assemble into higher-order complexes, giving rise to macromolecular signaling condensates in the cytoplasm or nucleus[83,84]. These condensates function as spatially restricted signaling platforms that concentrate pathway components and accelerate the protein-protein interactions required for downstream cell death execution[23,84].
3.2 Execution of ZBP1-mediated necroptosis
The best-characterized consequence of ZBP1 activation is induction of necroptosis, an inflammatory form of programmed lytic cell death[23,24]. Unlike apoptosis, which relies on caspase activation and packages cellular contents into membrane-bound apoptotic bodies for relatively silent clearance, necroptosis is immunologically conspicuous[85]. It not only halts intracellular viral replication but also broadcasts tissue distress to the surrounding immune environment[86,87]. In functional terms, this makes necroptosis a particularly effective response when rapid destruction of the intracellular replication niche is required. By rupturing the membrane rather than preserving it, the pathway limits completion of viral assembly, exposes immature viral components, and reduces viral burst size, thereby constraining spread to neighboring cells.
Signal propagation downstream of ZBP1 depends on the RIP homotypic interaction motif, or receptor-interacting protein homotypic interaction motif (RHIM)[23]. ZBP1 contains two RHIM domains, which mediate association with downstream kinases[88]. Upon activation, ZBP1 recruits receptor-interacting protein kinase 3 through RHIM-RHIM interactions. This interaction seeds formation of an amyloid-like signaling scaffold known as the necrosome[23,89]. Within the assembled complex, RIPK3 undergoes trans-autophosphorylation and activation[90]. Activated RIPK3 then phosphorylates specific serine and threonine residues in MLKL[85]. Phosphorylation relieves the autoinhibitory conformation of MLKL and exposes its N-terminal execution domain[84,85]. MLKL subsequently oligomerizes and translocates to the inner leaflet of the plasma membrane. Once inserted into the lipid bilayer, these oligomers form cation-permeable channels that drive sodium and calcium influx, osmotic swelling, and ultimately plasma membrane rupture[23,85]. In addition to lytic necroptosis, ZBP1 can also engage RIPK1 to drive apoptosis, a parallel death outcome not typically co-observed with necroptosis in the same cell. Furthermore, STING modulates ZBP1 activity at multiple levels, controlling both the generation of its ligand, Z-RNA, and the assembly of the ZBP1–RIPK1–RIPK3 complex. This regulatory axis identifies STING as a previously unrecognized checkpoint in the ZBP1-dependent necroptotic pathway and highlights the close functional crosstalk between two critical nucleic acid-sensing pathways of innate immunity[91].
3.3 Release of DAMPs and immune chemotaxis
The rupture of necroptotic cells has consequences that extend beyond local tissue injury; it directly shapes communication with the immune system[87]. Cell lysis releases intracellular contents into the extracellular space, including multiple classes of DAMPs[87,92]. Among the best-characterized are high mobility group box 1 (HMGB1), which functions extracellularly as a proinflammatory signal despite its nuclear role as a DNA-binding protein; extracellular adenosine triphosphate (ATP), which acts as a chemotactic cue for phagocytes; interleukin-1α; and fragments of unmethylated mitochondrial DNA[87,92]. In many settings, necroptosis is therefore accompanied by abundant extracellular release of ATP, HMGB1, IL-33, and free host or viral nucleic acids, generating a strong inflammatory signal in the surrounding tissue. Notably, membrane rupture is not a prerequisite for DAMP release; necroptotically engaged but still-intact cells can be major producers of DAMPs[93].
These molecules diffuse through the stromal compartment and engage Toll-like receptors and purinergic receptors expressed by tissue-resident dendritic cells and macrophages[93]. Such signaling promotes dendritic cell maturation and upregulation of major histocompatibility complex molecules together with costimulatory receptors[92,94]. At the same time, DAMPs act as chemoattractants and immune activators, recruiting both innate and adaptive immune cells to the site of injury and establishing a local inflammatory barrier for early, nonspecific containment. Mature dendritic cells can then internalize viral antigens released from lysed cells and migrate to draining lymph nodes, where they cross-present these antigens to naive CD8+ T cells[87,92]. The resulting clonal expansion of virus-specific cytotoxic T lymphocytes supports targeted clearance at sites of infection[92,94]. In this way, ZBP1-driven necroptosis functions not simply as a terminal cell-death program but as an adjuvant-like process that links innate sensing to adaptive immunity[87].
3.4 Non-lytic signaling: ZBP1-mediated inflammatory gene transcription
While ZBP1 is classically defined by its execution of lytic necroptosis, it provides broader surveillance by initiating non-lytic inflammatory signaling alongside cell death. Beyond outright lysis, ZBP1 can engage non-lytic programs of inflammatory gene transcription by directly binding RIPK1 to drive NF-κB activation[95,96]. Rather than proceeding directly to complete MLKL-driven membrane rupture, the assembled ZBP1-RIPK1-RIPK3 scaffold can engage alternative signaling cascades. For instance, RIPK3 downstream of ZBP1 can promote type I interferon responses through the adaptor protein TIR-domain-containing adapter-inducing interferon-β[97]. Alternatively, RIPK3 activation can trigger sublethal necroptosis, a state in which restricted MLKL-dependent membrane perforation releases DAMPs without causing terminal cell death[90,95-97].
This non-cytolytic transcriptional capacity and sublethal DAMP release hold profound physiological and pathological significance across different cell types. In neurons, for example, RIPK3-mediated signaling can restrict viral replication independently of cell death by coordinating protective chemokine responses, as observed in West Nile virus encephalitis, or by driving metabolic rewiring, such as itaconate production during Zika virus infection[98,99]. Moreover, RIPK3 performs critical non-lethal functions in the tumor immune microenvironment. In certain myeloid-derived suppressor cell subsets, RIPK3 acts primarily as a transcriptional hub rather than a death executioner, promoting inflammatory cytokine production and supporting tumor-promoting Th17 expansion[100]. By establishing a localized inflammatory or chemotactic state prior to, or completely independently of, outright necroptotic lysis, ZBP1 ensures a rapid and coordinated innate immune response while preserving transient tissue integrity.
4. ROS Regulates ZBP1-Mediated Necroptosis
Recognition of Z-RNA by ZBP1 initiates necroptotic signaling, but the efficiency and extent of pathway execution are shaped just as strongly by the metabolic condition of the cell, particularly its redox state. Necroptosis is not governed solely by a linear sequence of kinase phosphorylation events; it is also embedded within broader metabolic control and redox-dependent regulation[39,101]. In this context, ROS, especially mitochondrial superoxide and hydrogen peroxide, function as both amplifiers and regulatory nodes within the ZBP1-RIPK1-RIPK3 axis[102,103]. Consistent with this, preventing ROS accrual by blocking NF-κB, and consequently limiting its downstream antioxidant target SOD2, sensitizes cells to IFN-driven necroptosis[104].
Rather than acting as passive by-products of stress, ROS, and mitochondrial ROS in particular, serve as active signaling intermediates that influence both the initiation and propagation of ZBP1-dependent necroptosis[105,106]. The relationship between redox homeostasis and the necroptotic machinery is best understood as a feed-forward circuit: oxidative stress promotes early activation of sensors and kinases, while the assembled necrosome perturbs mitochondrial function and further enhances oxidative injury[107] (Figure 3).
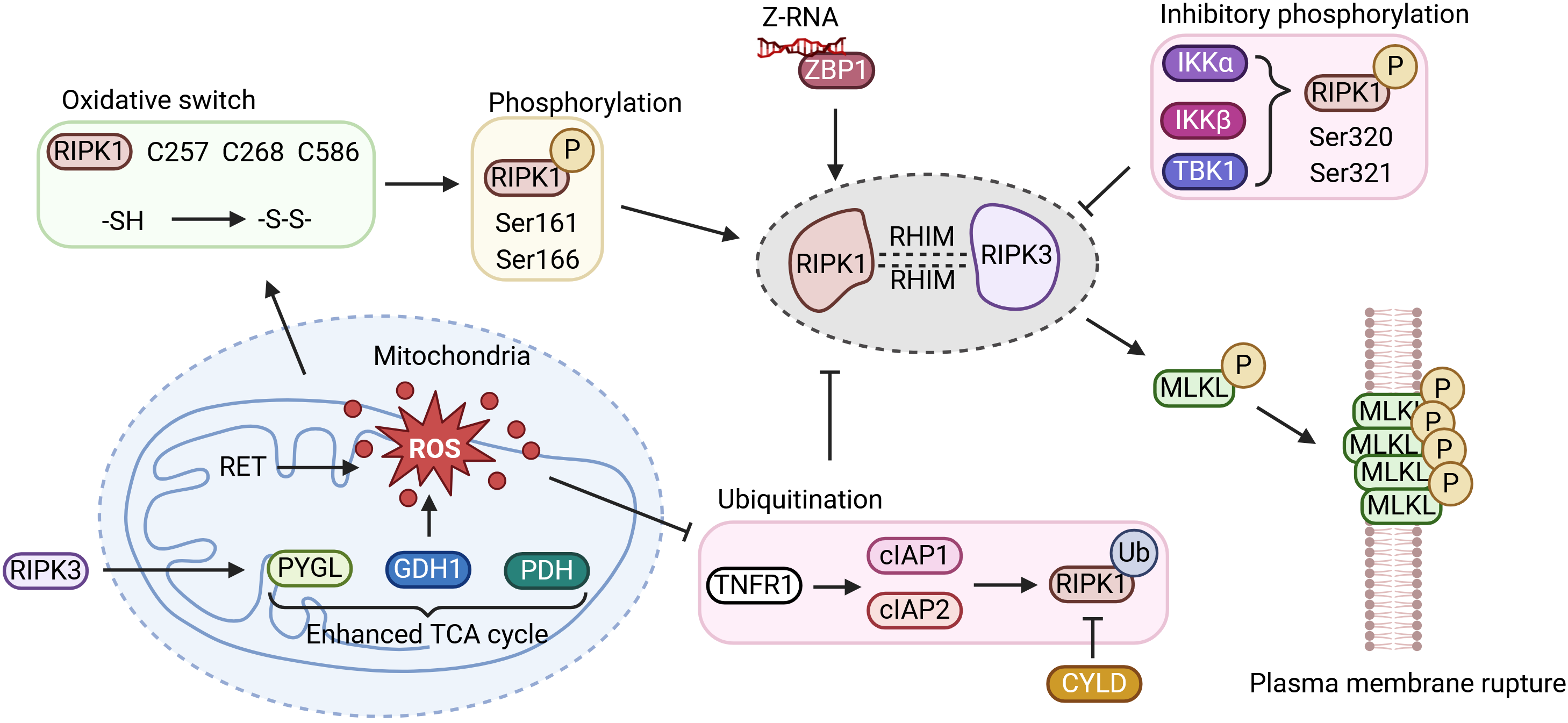{kind=link}
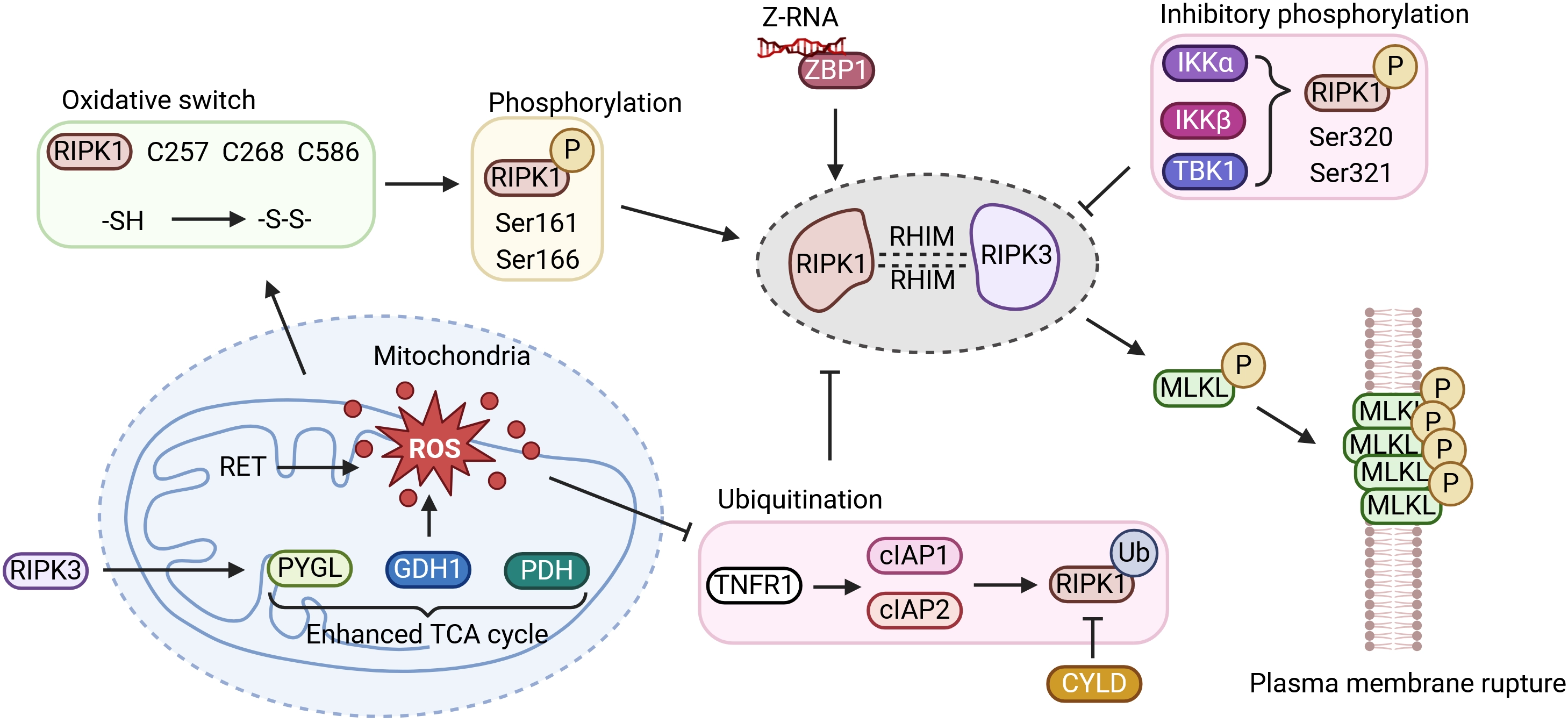
Figure 3. ROS regulates ZBP1-mediated necroptosis. ROS oxidize critical cysteine residues (C257, C268, C586) in RIPK1, promoting its oligomerization and autophosphorylation at S161/S166. Concurrently, oxidative stress triggers c-IAP1 degradation and CYLD-mediated deubiquitination, releasing RIPK1. In human cells, ZBP1 recruits RIPK1 as an essential scaffold; this complex then engages RIPK3 to form the necrosome, which phosphorylates MLKL and drives membrane lysis. Activated RIPK3 also phosphorylates metabolic enzymes (e.g., PYGL, GLUD1, PDH), enhancing glycolytic flux and glutamine-dependent anaplerosis into the TCA cycle, which in turn promotes RET at complex I and generates a second wave of mtROS. This establishes a feed-forward loop that amplifies necroptosis. Created in BioRender. Zhong, X. (2026) https://BioRender.com/6wjsrqn. RIPK: receptor-interacting serine/threonine-protein kinase; P: phosphorylation; Z-RNA: Z-form RNA; ZBP1: Z-nucleic acid-binding protein 1; IKKα: inhibitor of nuclear factor kappa-B kinase subunit alpha; IKKβ: inhibitor of nuclear factor kappa-B kinase subunit beta; TBK1: TANK-binding kinase 1; RET: reverse electron transport; ROS: reactive oxygen species; PYGL: glycogen phosphorylase L; GDH1: glutamate dehydrogenase 1; PDH: pyruvate dehydrogenase; TCA: tricarboxylic acid; TNFR1: tumor necrosis factor receptor 1; cIAP1/2: cellular inhibitor of apoptosis protein 1 and 2; Ub: ubiquitin.
4.1 Oxidative stress and ubiquitination cooperatively determine RIPK1 activation
A key question is how oxidative stress is translated into commitment to necroptosis. In mammalian cells, RIPK1 occupies a central position between upstream sensing and downstream execution pathways. Before full necrosome assembly occurs, RIPK1 remains highly sensitive to the intracellular redox environment. An early burst of ROS acts upstream of necrosome formation, and the kinase domain of RIPK1 is intrinsically responsive to oxidative conditions. Its activation reflects the combined effects of cysteine oxidation and serine autophosphorylation[100,108].
Structurally, RIPK1 contains an N-terminal kinase domain, an intermediate RHIM-containing region, and a C-terminal death domain. Under oxidative stress, mitochondria-derived ROS modify three critical cysteine residues, C257, C268, and C586. Mass spectrometry studies have shown that these thiol groups are reduced under basal conditions but become oxidized when ROS levels rise, forming intra- or intermolecular disulfide bonds[100]. This oxidation favors oligomerization of RIPK1 and promotes a conformational rearrangement in the activation loop of its kinase domain, exposing the catalytic pocket for ATP binding[108]. The resulting open conformation facilitates autophosphorylation at Ser161 and Ser166. Among these sites, phosphorylation at Ser161 is particularly important: it is required for RIPK1 activity and for recruitment of RIPK3, and the S161N mutation abolishes RIPK3 engagement and blocks necroptosis altogether[28,109]. By contrast, Ser166 is widely used as a marker of RIPK1 activation but appears partially redundant with Ser161. Consistent with this distinction, combined mutation of S161 and S166 suppresses necroptosis, whereas the phosphomimetic S161E substitution is sufficient to drive cell death, underscoring the dominant role of Ser161 in specifying necroptotic fate[28,109].
Not all phosphorylation events on RIPK1 promote death signaling. Phosphorylation imposed by upstream pro-survival pathways serves an opposing function. IKKα/IKKβ and TBK1, for example, can phosphorylate Ser320 and Ser321 within the intermediate region of RIPK1, modulating kinase activity and retaining RIPK1 within receptor-associated survival complexes, thereby restraining necroptosis[110]. These observations suggest that RIPK1 integrates multiple site-specific phosphorylation inputs, creating a regulatory code through which cells interpret the intensity and context of oxidative stress.
Ubiquitination introduces an additional checkpoint that strongly influences the behavior of the RIPK1-RIPK3 axis. During pro-survival signaling, such as after TNFR1 activation, c-IAP1 and c-IAP2 function as E3 ubiquitin ligases and decorate RIPK1 with K63-, K48-, K11-, and M1-linked polyubiquitin chains. These modifications serve a dual purpose: they promote NF-κB-dependent survival signaling while constraining RIPK1 in a non-death conformation[90]. Under oxidative stress, however, or following treatment with Smac mimetics, redox imbalance drives auto-ubiquitination and proteasomal degradation of c-IAP1[90]. Once this inhibitory layer is removed, deubiquitinases, most notably CYLD, process the ubiquitin chains on RIPK1. Freed from complex I, RIPK1 relocates to the cytoplasm, where its exposed cysteine residues become more susceptible to ROS-mediated oxidation. This oxidative priming favors autophosphorylation at Ser161 and Ser166 and commits the cell to necroptosis[111].
4.2 Human ZBP1 signals through RIPK1 as a scaffold because of RHIM divergence
Although oxidative regulation of RIPK1 is broadly conserved, the architectural role of RIPK1 within the necrosome differs substantially across species. Much of what is known about ZBP1 signaling has come from rodent studies, in which murine ZBP1 can bind RIPK3 directly through homologous RHIM-RHIM interactions and thereby promote MLKL phosphorylation and membrane rupture[112]. Structural and comparative analyses, however, have shown that this arrangement is not fully preserved in humans. In human cells, endogenous ZBP1 does not assemble a fully functional signaling complex directly with human RIPK3. Instead, RIPK1 serves as an essential scaffold that bridges human ZBP1 to RIPK3[9].
The species-specific dependency of ZBP1 on RIPK1 arises from variation in the RIPK3 RHIM tetrapeptide core (e.g., human VQVG versus mouse). Replacing the human RHIM with the murine sequence enables direct ZBP1–RIPK3 binding and RIPK1-independent necroptosis. More broadly, mutational analysis reveals functional plasticity: human VQVG→AAAA or naturally occurring variants like TQIG and VQSG retain pro-death activity but lose NF-κB activation, suggesting that RHIM diversification tunes the balance between inflammatory signaling and lytic cell death rather than acting as a simple on/off switch[88,113].
Within the human pathway, recruitment of RIPK1 represents a shift in signaling logic rather than a minor structural variation. During HSV-1 infection or under endogenous oxidative stress, complex assembly appears to proceed in a stepwise manner. Available evidence suggests that, after activated human ZBP1 recognizes nucleic acids through its N-terminal Zα domain, it recruits free RIPK1 through its RHIM-A domain[9]. This event occurs before RIPK3 incorporation and can still take place in RIPK3-deficient cells. Once bound, RIPK1 undergoes conformational rearrangement and provides a multimeric platform that enables RIPK3 recruitment, mutual phosphorylation, and formation of amyloid-like fibrillar structures that propagate downstream signaling[114,115].
Functional perturbation studies support this model. When RIPK1 is eliminated, either by PROTAC-mediated degradation with agents such as R1-ICR-3 or by CRISPR-Cas9 knockout, assembly of the human ZBP1-RIPK3 complex is disrupted, and downstream MLKL phosphorylation and plasma membrane rupture no longer occur[9]. Because RIPK1 kinase activity is itself highly responsive to intracellular redox changes, this human-specific reliance on RIPK1 directly couples ZBP1-driven necroptosis to ROS-dependent regulation[116].
4.3 Necroptosis induces a mitochondrial ROS burst and establishes a feed-forward loop
ROS do not act only at the initiation stage. Once upstream oxidative stress has promoted RIPK1 activation, a second wave of ROS frequently emerges from mitochondria during pathway execution. Activated ZBP1-RIPK1-RIPK3 signaling interacts with the mitochondrial electron transport chain, creating a bidirectional positive-feedback system[117].
A common example arises in ischemia-reperfusion injury and bacterial infection, where succinate accumulates within the mitochondrial matrix and alters the behavior of succinate dehydrogenase and the mitochondrial membrane potential[118]. Under these conditions, electrons can flow backward through complex I, a process termed reverse electron transport[119]. RET is a well-established source of mitochondrial superoxide under pathological conditions[120]. The resulting mtROS enters the cytosol, where it can oxidize inactive RIPK1, promote conformational opening and autophosphorylation, and potentially influence the generation of endogenous Z-RNA that recruits and activates ZBP1[120,121]. Experimental support for this model comes from studies using RET inhibitors or metabolic modulators such as dimethyl fumarate, both of which reduce ROS levels and interrupt necrosome assembly and MLKL activation[121,122].
RIPK3 also contributes directly to metabolic rewiring. Once activated, it phosphorylates not only core signaling proteins but also a range of cytoplasmic and mitochondrial metabolic enzymes, thereby influencing substrate availability for oxidative metabolism. Phosphoproteomic analyses have identified liver glycogen phosphorylase, glutamate dehydrogenase 1, glutamine-ammonia ligase, and the pyruvate dehydrogenase complex among RIPK3-regulated targets[123]. Through these modifications, RIPK3 enhances glycolytic flux and glutamine-dependent anaplerosis, channeling pyruvate and related metabolites into the tricarboxylic acid cycle[124,125]. When this increased substrate supply occurs in mitochondria already primed by oxidative modification, the respiratory chain becomes a major generator of ROS[126]. Necroptosis therefore emerges not simply as a terminal signaling event but as a metabolically reinforced program in which mitochondrial dysfunction and ROS production intensify one another.
5. Stress-Driven Innate Immune Checkpoints and Associated ROS Production Robustly Activate Antitumor Immunity
How might this molecular circuitry be exploited therapeutically? The interaction among ZBP1, endogenous Z-form nucleic acids, and ROS provides a framework for addressing one of the most persistent obstacles in cancer therapy: the immunosuppressive, poorly inflamed “cold” tumor. Such tumors are characterized by limited cytotoxic T-cell infiltration and poor responsiveness to immune checkpoint blockade[127,128]. By inducing viral mimicry and engaging stress-responsive innate immune checkpoints, ZBP1-centered signaling offers a plausible route for reshaping these resistant TMEs[14,129].
This strategy, however, cannot be considered solely from the perspective of tumor cells. ZBP1 is also functionally important in multiple immune populations, particularly within the myeloid compartment, and these cellular contexts influence both therapeutic efficacy and toxicity[130].
Macrophage state is one determinant of susceptibility. During polarization toward an M1-like phenotype, macrophages upregulate basal expression of ZBP1, RIPK3, and MLKL[11,130]. In this state, they are better positioned to deploy ZBP1-dependent defense programs when confronted with stress or danger signals[131,132]. Yet the same lytic response may also produce immunopathology, indicating that excessive activation of this pathway could damage tissues and should be evaluated carefully in therapeutic settings[133].
Dendritic cells illustrate a different but equally important balance. Their function depends on maintaining sufficient survival to process and present antigen. In the absence of RIPK1, endogenous nucleic acids can activate ZBP1 and trigger extensive necroptosis in dendritic cells, thereby impairing antigen presentation while releasing self-derived nucleic acid fragments and DAMPs that may promote autoimmune pathology[131,134]. These findings suggest that precise spatiotemporal control of ZBP1 activity in the TME may be required if inducible necroptosis in dendritic cells is to be used as an in situ adjuvant strategy[135]. To address the issue that dendritic cell necroptosis markedly impairs their antigen-presenting function, targeted delivery systems, such as TME-responsive nanocarriers, must be employed. These advanced nanomaterials are engineered to remain stable in circulation but undergo structural transformations (e.g., polymer degradation or swelling) to release ZBP1 agonists and ROS-generating agents exclusively in response to TME-specific stimuli, such as acidic pH, reductive conditions, or overexpressed enzymes. Furthermore, optimizing their physicochemical properties, such as size and surface modifications, ensures precise cellular internalization and activation of regulated cell death[41,42].
Additional myeloid circuits further complicate this landscape. Neutrophil extracellular traps (NETs) generated under stress can amplify ZBP1 signaling[132]. NET formation promotes mitochondrial damage and mtDNA leakage, and leaked mtDNA may adopt Z-form structures that are sensed by ZBP1[82,132]. In this setting, ZBP1 can recruit cGAS into a signaling complex that amplifies STING-IRF3-dependent interferon responses[136,137]. A self-reinforcing loop may therefore arise in which NETs promote mtDNA release, ZBP1-cGAS activation, and necroptosis, which in turn fuels further inflammatory output and NET production. Because this cycle may exacerbate tissue damage, targeting NETs has been proposed as one way to modulate ZBP1 activity[138,139]. This observation reinforces the idea that therapeutic manipulation of the ZBP1-RIPK3 axis must account for context-specific signaling in immune cells[100,140]. Hematopoietic stem and progenitor cells provide yet another layer of concern: their homeostasis depends on suppression of ZBP1 by RIPK1, and inappropriate activation can deplete the stem cell pool through extensive necroptosis[110,141]. Any attempt to induce viral mimicry therapeutically must therefore consider consequences for hematopoietic reserve and systemic immune balance[142,143].
Taken together, these findings highlight the central therapeutic tension. Activation of ZBP1-mediated necroptosis in tumor cells can remodel the TME by releasing antigens and endogenous adjuvants[144,145], but vulnerable immune or stem-cell compartments must be protected to avoid systemic toxicity[146,147]. It is against this backdrop that viral mimicry-based interventions have been developed[148,149].
5.1 Pharmacological and genetic approaches for direct induction of viral mimicry and ZBP1 activation
Although ZBP1 is classically viewed as a viral sensor, pharmacologic approaches can induce formation of its cognate ligands, Z-DNA and Z-RNA, within tumor cells in the absence of infection[32,82]. This has led to the development of compounds that function as non-infectious agonists of ZBP1 signaling[150,151].
One prominent example is the small molecule Curaxin CBL0137, which intercalates into chromatin and promotes conversion of canonical B-form DNA into left-handed Z-DNA[32,82]. Through this alteration in DNA topology, CBL0137 activates ZBP1 directly through Z-DNA generation, thereby inducing ZBP1-dependent necroptosis in tumor cells and cancer-associated fibroblasts[32,152]. CBL0137 was originally designed as an inhibitor of the FACT chromatin chaperone complex[82,153]. Its chromatin-binding properties allow it to act as a DNA intercalator, generating topological strain across the genome and forcing segments of right-handed B-DNA into Z-DNA conformations[32,82]. Accumulation of genomic Z-DNA can in turn activate ZBP1-dependent necroptosis[154,155]. Analyses of clinical samples suggest that tumors retaining an intact necroptotic machinery, such as certain melanomas, often exhibit increased infiltration of monocytes and CD8+ T cells, consistent with the idea that activation of ZBP1-dependent necroptosis can help reprogram the tumor immune microenvironment[32,156]. By contrast, tumors in which the necroptotic pathway is silenced tend to display immune-poor phenotypes. Preclinical studies have shown that CBL0137 can induce necroptosis in malignant cells and in cancer-associated fibroblasts[157-159]. By generating Z-DNA ligands that are not subject to ADAR1-mediated editing, it may enhance the immunogenicity of the TME[32,160]. This translational potential is reflected in its ongoing clinical evaluation in solid tumors, central nervous system tumors, neuroblastoma, and melanoma[82,161].
Another route to ligand generation involves disrupting transcription termination and post-transcriptional RNA processing[150,162]. Inspired by virus-induced DoTT, investigators have examined JTE-607, a small-molecule prodrug originally developed for inflammatory disorders. After cellular uptake, endogenous esterases convert JTE-607 to its active metabolite, Compound 2, which inhibits CPSF73. As described earlier, pharmacologic inhibition of CPSF73 mimics DoTT by causing accumulation of readthrough transcripts capable of forming Z-RNA. The inhibitory effect of JTE-607 on cleavage is influenced by the nucleotide composition surrounding the mRNA cleavage site, with particular affinity for U/A-rich motifs. Beyond pharmacological agents, genetic interventions offer precise strategies to induce viral mimicry. For instance, CRISPR-Cas9-mediated depletion of the RNA editing enzyme ADAR1, or the targeted silencing of core spliceosome components, intentionally forces the accumulation of unedited endogenous Z-RNAs. This genetically driven ligand generation robustly triggers ZBP1 activation and downstream immunogenic cell death[32,58].
Small-cell lung cancer (SCLC) illustrates the relevance of this concept in a clinically difficult setting. SCLC is a prototypical “cold” tumor, marked by neuroendocrine differentiation and poor responsiveness to standard immunotherapy[18,127]. Transcriptomic studies have identified a vulnerability in the splicing machinery, prompting investigation of spliceosome inhibitors such as pladienolide B, which targets the SF3b complex[18]. Consistent with the proposed connection between spliceosome stress and Z-RNA generation, PladB-induced intron retention gives rise to ZBP1 ligands, including both Z-RNA and Z-form RNA:DNA hybrids. In SCLC tumors and associated fibroblasts that retain functional ZBP1-RIPK3 signaling, these inhibitors trigger necroptosis. This approach effectively turns dependence on continuous RNA processing into a therapeutic liability. Importantly, the therapeutic induction of Z-form nucleic acids is not restricted to SCLC. Similar regulatory vulnerabilities have been exploited in other drug-resistant solid malignancies, including breast cancer and melanoma. For instance, in melanoma models, the administration of the small molecule curaxin CBL0137 directly intercalates into chromatin, forcing the topological conversion of canonical B-DNA into immunogenic Z-DNA within the TME[32]. Conversely, in metastatic breast cancer models, ultrasound-targeted nanotherapy can be deployed to generate substantial localized oxidative stress, which serves as a potent, time- and dose-dependent inducer of endogenous Z-RNA formation[145]. In both models, triggering Z-DNA or Z-RNA accumulation effectively breaks the immunosuppressive barrier and drives tumor regression, underscoring the broad universality of this innate immune strategy.
5.2 Localized ROS generation through nanotherapy and physical stimulation
Because ROS act both as upstream priming factors for RIPK1 and as downstream amplifiers of the necroptotic cascade, precise manipulation of tumor redox balance has become an important therapeutic objective[39,101]. To limit systemic toxicity, current nanomedicine approaches aim to confine oxidative bursts to the TME[145,148]. Photodynamic and sonodynamic platforms are particularly attractive because they use externally applied light or ultrasound to trigger localized ROS generation. Two-dimensional Iridium(III)-carbon nitride nanocomplexes, for example, have been engineered to generate ROS under ultrasound while simultaneously inducing endoplasmic reticulum stress, thereby creating oxidative conditions favorable for RIPK1-dependent necroptosis. Similarly, chlorin e6-encapsulated nanobubbles act as sonosensitizers that produce ROS upon ultrasound exposure and induce necroptosis, improving sonodynamic immunotherapy in breast cancer models. In bone metastatic breast cancer, an ultrasound-activated Janus hydrogel has likewise been used to release ROS and cooperate with ZBP1 in activating necroptotic pathways.
Catalytic nanozymes and radiosensitizers provide additional means of amplifying oxidative stress[144]. Platinum-based redox centers loaded onto non-stoichiometric tungsten oxide substrates can mimic NADPH oxidase activity, inducing endoplasmic reticulum stress and immunogenic cell death. In radiotherapy settings, gold single-atom artificial enzymes function as radio-activatable agents[146]. Upon irradiation, they increase intracellular ROS, intensify DNA damage, and initiate necroptotic death. Radiation-triggerable bioreactors extend this concept by delivering inducible nitric oxide synthase gene circuits together with β-lapachone, thereby generating both nitric oxide and ROS to drive cancer stem cells toward immunogenic necroptosis[147].
The necrosome also feeds back on endogenous ROS production through metabolic regulation[123]. Biomimetic nanosystems have begun to exploit this feature by co-delivering metabolic inhibitors and photosensitizers[148]. One such system incorporates the glutamine metabolism inhibitor BPTES, which perturbs tumor metabolism while producing a ROS burst under irradiation, thereby promoting necroptosis[148]. Ion-targeting strategies provide yet another route. Piezocatalytic Mg2+-doped hydroxyapatite nanoparticles, when activated by ultrasound, release ROS, promote intracellular calcium influx, upregulate death receptors, and induce tumor-cell necroptosis[149].
5.3 Synergistic sensitization of immune checkpoint blockade
Induction of necroptosis, particularly when coupled with localized ROS production, can profoundly alter the immunologic state of a tumor[151]. Because necroptosis is lytic, it promotes release of DAMPs and tumor-associated antigens. This form of immunogenic cell death supports dendritic-cell recruitment and maturation and ultimately increases infiltration of cytotoxic CD8+ T cells[152].
Preclinical studies indicate that such changes can sensitize tumors to checkpoint immunotherapy[151,153]. Lipid nanoparticles co-delivering shikonin and manganese ions, for example, induce necroptosis while activating the cGAS-STING pathway. In squamous cell carcinoma models, necroptosis-driven antigen release together with Mn2+-enhanced interferon signaling promoted dendritic-cell maturation and improved responsiveness to anti-PD-1 treatment[151,154].
These strategies are not limited to conventional PD-1 or PD-L1 blockade[152,155]. Photodynamically activated Iridium(III) complexes have been reported to reduce CD47 expression while inducing necroptosis, thereby favoring M1 macrophage polarization[36]. A-D-A-type photovoltaic molecule nanoparticles have also been used to induce immunogenic cell death through combined photothermal and photodynamic activity; when paired with anti-TIGIT antibodies targeting exhausted natural killer cells, they produced an enhanced antitumor immune response[43]. In immune-checkpoint-refractory melanoma models, intratumoral administration of CBL0137 together with systemic anti-PD-1 antibodies increased recruitment of activated CD8+ T cells into tumors and promoted their expansion in draining lymph nodes, resulting in tumor regression that depended on host ZBP1 expression. By converting a poorly inflamed tumor milieu into an inflammatory one, the combined use of targeted ROS generation and programmed necroptosis offers a mechanistically grounded strategy for improving cancer immunotherapy[151,156]. Furthermore, combining ZBP1-driven viral mimicry with mRNA-based cancer vaccines represents a highly promising synergistic frontier. While mRNA vaccines precisely deliver tumor neoantigens to prime specific CD8+ T cells, concurrent ZBP1 activation provides a potent inflammatory adjuvant signal. This dual approach effectively amplifies antigen cross-presentation and maximizes the efficacy of targeted anti-tumor immunity[163,164] (Figure 4).
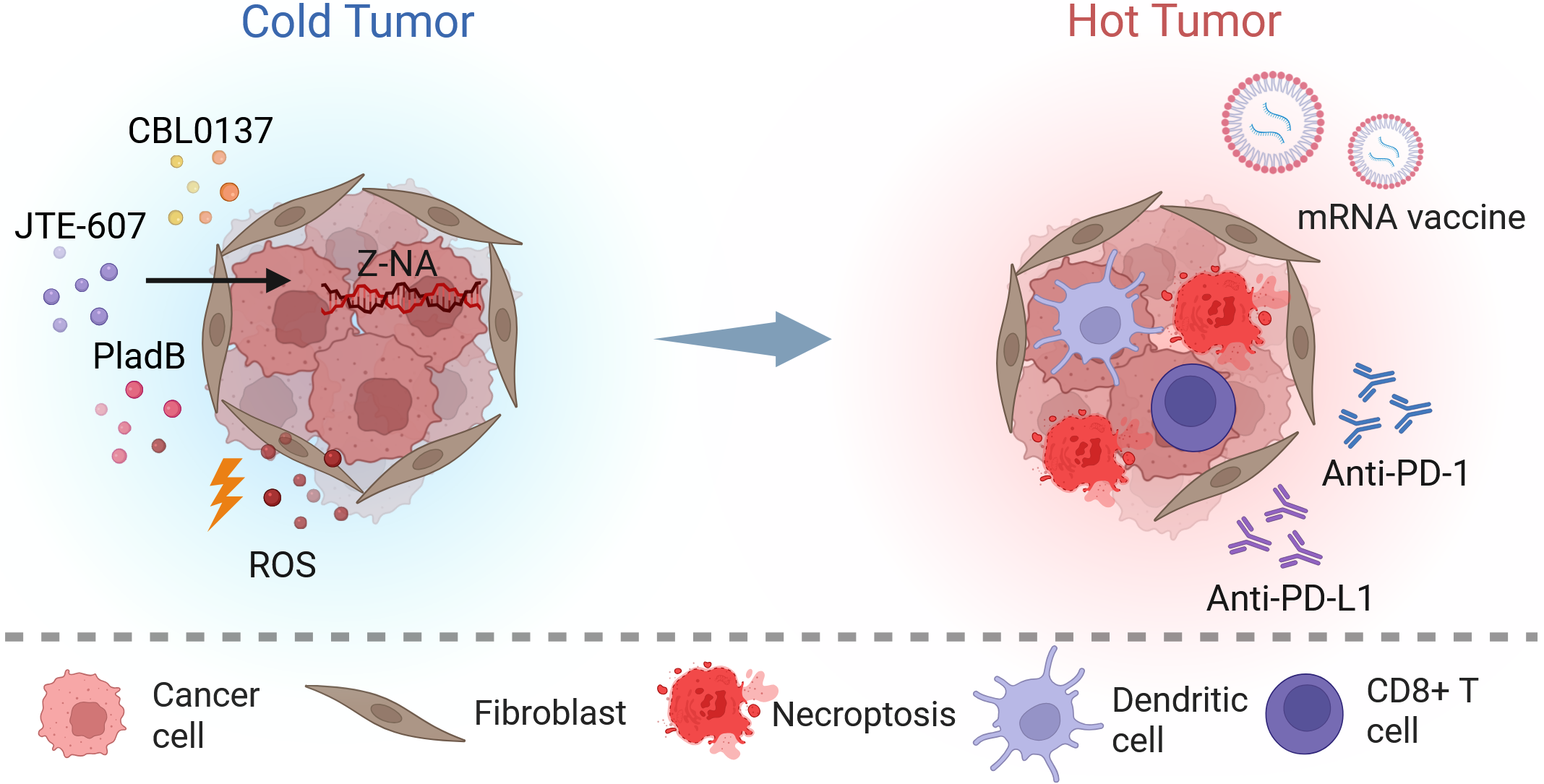{kind=link}
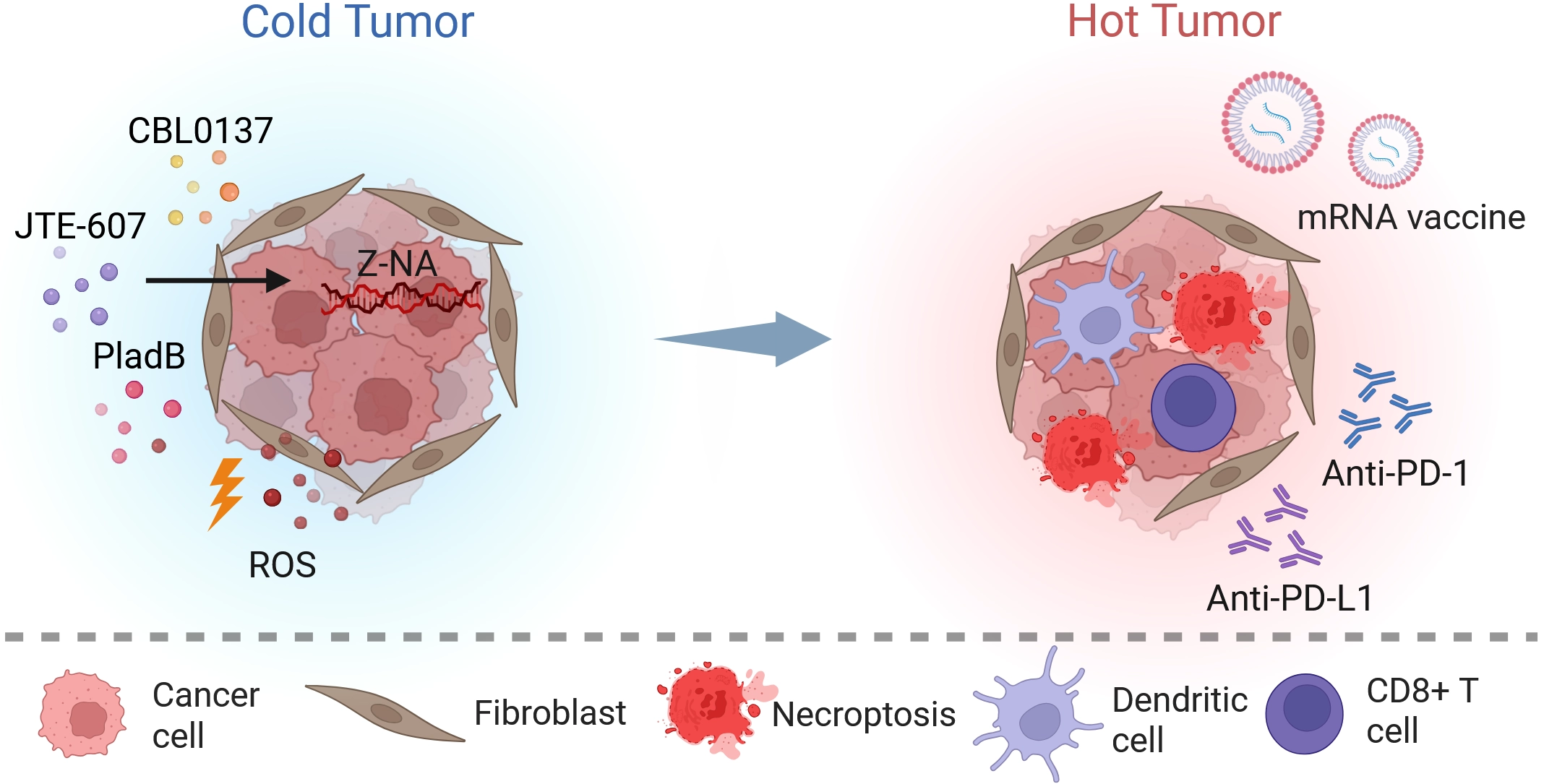
Figure 4. Stress-driven innate immune checkpoints and associated ROS generation potently activate anti-tumor immunity. Pharmacological Z-NA induction (e.g., CBL0137, JTE-607, PladB) and ROS generation cooperatively trigger immunogenic necroptosis, releasing DAMPs and tumor antigens. Dendritic cells capture these antigens and, synergizing with mRNA vaccines, robustly prime CD8+ T cells. Immune checkpoint blockade (anti-PD-1/PD-L1) further potentiates effector T cell function, driving the cold-to-hot transition. Created in BioRender. Zhong, X. (2026) https://BioRender.com/em5hbuc. ROS: reactive oxygen species; Z-NA: Z-form nucleic acid; PladB: pladienolide B; anti-PD-1: anti-programmed cell death protein 1 antibody; anti-PD-L1: anti-programmed death-ligand 1 antibody.
6. Conclusions and Perspectives
Recognition of endogenous Z-form nucleic acids by ZBP1, together with its close interplay with ROS signaling, defines a fundamental mechanism linking innate immunity to tumor biology[4,165]. Taken together, these observations indicate that ZBP1 can no longer be regarded simply as an antiviral receptor. Rather, it functions as a broader sensor of cellular stress, responding not only to infectious challenge but also to disturbances in genomic integrity and RNA transcription or processing[16,20]. Notably, ZBP1-mediated signaling pathways are tightly integrated with the cellular redox environment: ROS provide the oxidative priming required for RIPK1 activation and later amplify necroptosis during the mitochondrial execution phase[28,102,105].
In essence, ZBP1 operates as a central quality-control node that responds both to pathogen invasion and to endogenous nucleic acid instability. During infection, it surveils DVGs and host transcriptional disruption, drives both gene transcription and necroptosis, and promotes immune-cell recruitment. Under sterile conditions, it monitors RNA splicing fidelity, detects aberrant cytoplasmic Z-RNAs, and may eliminate premalignant cells before oncogenic protein accumulation occurs. This coupling of severe nucleic acid dysregulation to inflammatory lytic death represents a core host-defense mechanism and provides a conceptual basis for reprogramming the TME. Pharmacologic or nanomaterial-based activation of this innate checkpoint can trigger immunogenic necroptosis, release DAMPs and tumor antigens, and convert a “cold” tumor into an inflamed state that is receptive to adaptive checkpoint blockade such as anti-PD-1 or anti-PD-L1 therapy. The therapeutic value of this approach lies precisely in the synergy between innate and adaptive immune checkpoints.
To translate this pathway into clinically useful therapies, several challenges, along with equally promising opportunities, merit careful investigation[33,42]. One major direction lies in combining viral-mimicry-inducing strategies with epigenetic modulation[74,166]. Because epigenetic dysregulation is a defining feature of many cancers, agents targeting DNA methyltransferases, histone deacetylases, or Polycomb repressive complex 2 can relieve repression of silenced endogenous retroelements[72,167]. Tumor preconditioning with these epigenetic therapies or, alternatively, pharmacologic activation of p53 may substantially expand the pool of endogenous immunogenic nucleic acids[168,169]. Integrating such epigenetic priming with tumor-targeted ROS-generating nanomedicines could enhance ZBP1-driven necroptosis and further sensitize resistant tumors to immunotherapy[170-172].
Another priority is to define the context dependence of ZBP1-initiated cell death. Although ZBP1 robustly induces RIPK3-MLKL-dependent necroptosis under oxidative stress, it can also trigger caspase-dependent apoptosis depending on cellular context, availability of downstream effectors, and the post-translational state of regulatory proteins[90,173]. Future work should therefore identify the molecular checkpoints that determine death-pathway choice, including quantitative thresholds of mitochondrial ROS, dynamic ubiquitination states of necrosome components, and the differential engagement of RHIM interactions[23,112,174]. A detailed understanding of this bifurcation will be essential for designing therapies that reliably channel tumor cells toward immunogenic necroptosis rather than relatively silent apoptosis[129,152].
Safety remains an equally important consideration. Therapeutic strategies that amplify ROS or activate ZBP1 indiscriminately carry an inherent risk of provoking autoinflammatory damage or collateral tissue injury[175,176]. To reduce this risk, next-generation nanomaterials should be designed to respond selectively to features of the TME, such as hypoxia, mild acidity, or overexpression of tumor-associated proteases. Such designs could spatially and temporally confine photodynamic, sonodynamic, or chemodynamic ROS bursts to tumor tissue and thereby improve precision while minimizing off-target toxicity[153,160,177].
Equally critical is the identification of predictive biomarkers. Baseline expression of key necroptosis components: ZBP1, RIPK3, and MLKL, together with ADAR1 expression and activity, spliceosome mutational status, and the intrinsic oxidative and metabolic profiles of tumors, will likely determine which patients are most likely to benefit[18,32,178]. Establishing such biomarkers will support rational patient stratification and help realize precision medicine approaches for cancer immunotherapy[20,40,179,180].
Throughout this review, the unifying concept has been the nucleic-acid conformational checkpoint surveilled by ZBP1: a rigid innate immune clearance mechanism in which ZBP1 senses aberrant Z-form nucleic acids and initiates necroptosis. Whether considered in the context of the three-stage checkpoints operating during infection, the accumulation of endogenous Z-RNAs during non-infectious stress, or the therapeutic induction of viral mimicry in combination with adaptive checkpoint blockade, ZBP1 remains positioned at the central point of sensing and decision-making. Clarifying the intrinsic relationship between this ZBP1-centered innate immune checkpoint and adaptive immune checkpoints, including PD-1 and PD-L1, should provide a conceptual foundation for the development of more comprehensive and effective combination immunotherapy strategies.
Acknowledgements
The authors declare that AI tools (ChatGPT-5.5 and Claude-4.5) were used solely for language polishing during the manuscript preparation process. The authors take full responsibility for the integrity, originality, and accuracy of the work.
Authors contribution
Zhang T: Conceptualization, methodology, supervision, writing-review & editing.
Zhong X: Investigation, data curation, writing-original draft.
Balachandran S: Supervision, writing-review & editing.
Conflicts of interest
The authors declare no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
This work was supported by the National Natural Science Foundation of China (Grant No. 32370157), 1.3.5 project for disciplines of excellence from West China Hospital of Sichuan University (Grant No. ZYYC24008), and Fundamental Research Funds for the Central Universities (Grant No. 20822041J4064).
Copyright
© The Author(s) 2026.
References
-
1. Kassiotis G. The immunological conundrum of endogenous retroelements. Annu Rev Immunol. 2023;41:99-125.[DOI]
-
2. Song Z, Xie X, Chen Y, Zhang B, Li X, Yang Y, et al. Innate immune sensing of Z-nucleic acids by ZBP1-RIPK1 axis drives neuroinflammation in Alzheimer’s disease. Immunity. 2025;58(10):2574-2592.[DOI]
-
3. Jiao H, Wachsmuth L, Kumari S, Schwarzer R, Lin J, Eren RO, et al. Z-nucleic-acid sensing triggers ZBP1-dependent necroptosis and inflammation. Nature. 2020;580(7803):391-395.[DOI]
-
4. Maelfait J, Rehwinkel J. The Z-nucleic acid sensor ZBP1 in health and disease. J Exp Med. 2023;220(8):e20221156.[DOI]
-
5. Herbert A. The ancient Z-DNA and Z-RNA specific Zα fold has evolved modern roles in immunity and transcription through the natural selection of flipons. R Soc Open Sci. 2024;11(6):240080.[DOI]
-
6. Diallo MA, Pirotte S, Hu Y, Morvan L, Rakus K, Suárez NM, et al. A fish herpesvirus highlights functional diversities among Zα domains related to phase separation induction and A-to-Z conversion. Nucleic Acids Res. 2023;51(2):806-830.[DOI]
-
7. Gabriel L, Srinivasan B, Kuś K, Mata JF, João Amorim M, Jansen LET, et al. Enrichment of Zα domains at cytoplasmic stress granules is due to their innate ability to bind to nucleic acids. J Cell Sci. 2021;134(10):jcs258446.[DOI]
-
8. Romero MF, Krall JB, Nichols PJ, Vantreeck J, Henen MA, Dejardin E, et al. Novel Z-DNA binding domains in giant viruses. J Biol Chem. 2024;300(8):107504.[DOI]
-
9. Amusan OT, Wang S, Yin C, Koehler HS, Li Y, Tenev T, et al. RIPK1 is required for ZBP1-driven necroptosis in human cells. PLoS Biol. 2025;23(2):e3002845.[DOI]
-
10. Balachandran S, Mocarski ES. Viral Z-RNA triggers ZBP1-dependent cell death. Curr Opin Virol. 2021;51:134-140.[DOI]
-
11. Li S, Zhang Y, Guan Z, Ye M, Li H, You M, et al. SARS-CoV-2 Z-RNA activates the ZBP1-RIPK3 pathway to promote virus-induced inflammatory responses. Cell Res. 2023;33(3):201-214.[DOI]
-
12. Zhang T, Yin C, Boyd DF, Quarato G, Ingram JP, Shubina M, et al. Influenza virus Z-RNAs induce ZBP1-mediated necroptosis. Cell. 2020;180(6):1115-1129.[DOI]
-
13. Yang ZH, Wu P, Zhang BX, Yang CR, Huang J, Wu L, et al. ZBP1 senses splicing aberration through Z-RNA to promote cell death. Mol Cell. 2025;85(9):1775-1789.[DOI]
-
14. Chour M, Porteu F, Depil S, Alcazer V. Endogenous retroelements in hematological malignancies: From epigenetic dysregulation to therapeutic targeting. Am J Hematol. 2025;100(1):116-130.[DOI]
-
15. Chen C, Cui Y, Wang S, Yang Y, Liu Z, Jin S, et al. Human endogenous retroviruses and diseases. MedComm. 2025;6(11):e70452.[DOI]
-
16. Chen R, Ishak CA, De Carvalho DD. Endogenous retroelements and the viral mimicry response in cancer therapy and cellular homeostasis. Cancer Discov. 2021;11(11):2707-2725.[DOI]
-
17. Yang T, Wang G, Zhang M, Hu X, Li Q, Yun F, et al. Triggering endogenous Z-RNA sensing for anti-tumor therapy through ZBP1-dependent necroptosis. Cell Rep. 2023;42(11):113377.[DOI]
-
18. Jiang X, Ma X, Zhou Y, Liu X, Zhang T, Kim W, et al. Spliceosome inhibition induces Z-RNA and ZBP1-driven cell death in small cell lung cancer. Cell Rep. 2025;44(10):116384.[DOI]
-
19. Seetharam D, Chandar J, Ramsoomair CK, Desgraves JF, Medina AA, Hudson AJ, et al. Activating antiviral immune responses potentiates immune checkpoint inhibition in glioblastoma models. J Clin Invest. 2025;135(6):e183745.[DOI]
-
20. Rosenberg L, Vabret N. Viral mimicry in cancer therapy. Trends Cancer. 2025;11(12):1185-1202.[DOI]
-
21. He J, Zhu Y, Tian Z, Liu M, Gao A, Fu W, et al. ZBP1 senses spliceosome stress through Z-RNA: DNA hybrid recognition. Mol Cell. 2025;85(9):1790-1805.[DOI]
-
22. Nagata M, Carvalho Schäfer Y, Wachsmuth L, Pasparakis M. A shorter splicing isoform antagonizes ZBP1 to modulate cell death and inflammatory responses. EMBO J. 2024;43(21):5037-5056.[DOI]
-
23. Riebeling T, Kunzendorf U, Krautwald S. The role of RHIM in necroptosis. Biochem Soc Trans. 2022;50(4):1197-1205.[DOI]
-
24. Molnár T, Mázló A, Tslaf V, Szöllősi AG, Emri G, Koncz G. Current translational potential and underlying molecular mechanisms of necroptosis. Cell Death Dis. 2019;10(11):860.[DOI]
-
25. Liu Y, Liu T, Lei T, Zhang D, Du S, Girani L, et al. RIP1/RIP3-regulated necroptosis as a target for multifaceted disease therapy (Review). Int J Mol Med. 2019;44:771-786.[DOI]
-
26. Zhou Y, Wu R, Wang X, Jiang Y, Xu W, Shao Y, et al. Activation of UQCRC2-dependent mitophagy by tetramethylpyrazine inhibits MLKL-mediated hepatocyte necroptosis in alcoholic liver disease. Free Radic Biol Med. 2022;179:301-316.[DOI]
-
27. Rius-Pérez S, Pérez S, Toledano MB, Sastre J. Mitochondrial reactive oxygen species and lytic programmed cell death in acute inflammation. Antioxid Redox Signal. 2023;39:708-727.[DOI]
-
28. Zhang Y, Su SS, Zhao S, Yang Z, Zhong CQ, Chen X, et al. RIP1 autophosphorylation is promoted by mitochondrial ROS and is essential for RIP3 recruitment into necrosome. Nat Commun. 2017;8:14329.[DOI]
-
29. Wang P, Zheng SY, Jiang RL, Wu HD, Li YA, Lu JL, et al. Necroptosis signaling and mitochondrial dysfunction cross-talking facilitate cell death mediated by chelerythrine in glioma. Free Radic Biol Med. 2023;202:76-96.[DOI]
-
30. Lei K, Chen J, Deng Y, Peng Y, Zhai X, Ren X, et al. Cracking the code of cancer immunotherapy resistance: Emerging roles of pyroptosis and necroptosis. J Exp Clin Cancer Res. 2025;44:308.[DOI]
-
31. Wang X, Hua P, He C, Chen M. Non-apoptotic cell death-based cancer therapy: Molecular mechanism, pharmacological modulators, and nanomedicine. Acta Pharm Sin B. 2022;12(9):3567-3593.[DOI]
-
32. Zhang T, Yin C, Fedorov A, Qiao L, Bao H, Beknazarov N, et al. ADAR1 masks the cancer immunotherapeutic promise of ZBP1-driven necroptosis. Nature. 2022;606(7914):594-602.[DOI]
-
33. Lu J, He R, Liu Y, Zhang J, Xu H, Zhang T, et al. Exploiting cell death and tumor immunity in cancer therapy: Challenges and future directions. Front Cell Dev Biol. 2024;12:1416115.[DOI]
-
34. Um W, Ko H, You DG, Lim S, Kwak G, Shim MK, et al. Necroptosis-inducible polymeric nanobubbles for enhanced cancer sonoimmunotherapy. Adv Mater. 2020;32(16):1907953.[DOI]
-
35. Liu Z, Zhang J, Liu H, Shen H, Meng N, Qi X, et al. BSA-AIE nanoparticles with boosted ROS generation for immunogenic cell death immunotherapy of multiple myeloma. Adv Mater. 2023;35(7):2208692.[DOI]
-
36. Yu LB, Wang P, Shen QH, Guan QX, Li ZY, Han YY, et al. Ir(III) complexes convert cold to hot tumors via ferroptosis/necroptosis-driven immunogenic cell death and photosensitized CD47 downregulation. Adv Sci. 2026;13(8):e14256.[DOI]
-
37. Yatim N, Jusforgues-Saklani H, Orozco S, Schulz O, Barreira da Silva R, Reis e Sousa C, et al. RIPK1 and NF-κB signaling in dying cells determines cross-priming of CD8+T cells. Science. 2015;350(6258):328-334.[DOI]
-
38. Chen XY, Dai YH, Wan XX, Hu XM, Zhao WJ, Ban XX, et al. ZBP1-mediated necroptosis: Mechanisms and therapeutic implications. Molecules. 2023;28(1):52.[DOI]
-
39. Chen YS, Tian HX, Rong DC, Wang L, Chen S, Zeng J, et al. ROS homeostasis in cell fate, pathophysiology, and therapeutic interventions. Mol Biomed. 2025;6:89.[DOI]
-
40. Wang R, Dong X, Zhang X, Liao J, Cui W, Li W. Exploring viral mimicry combined with epigenetics and tumor immunity: New perspectives in cancer therapy. Int J Biol Sci. 2025;21(3):958-973.[DOI]
-
41. Linderman SW, DeRidder L, Sanjurjo L, Foote MB, Alonso MJ, Kirtane AR, et al. Enhancing immunotherapy with tumour-responsive nanomaterials. Nat Rev Clin Oncol. 2025;22(4):262-282.[DOI]
-
42. Luo Y, Lin X, Su C, Zheng Z, Li Y, Guo W, et al. Nanomaterial-driven regulated cell death: Mechanistic insights and novel strategies for tumor therapy. Int J Nanomed. 2025;20:13183-13208.[DOI]
-
43. Chen L, Huang J, Liu L, Tong M, He X, Zhao S, et al. Multimodal cascade-amplified phototheranostics for enhanced anti-tumor immunity. Biomaterials. 2026;325:123634.[DOI]
-
44. Nichols PJ, Krall JB, Henen MA, Vögeli B, Vicens Q. Z-RNA biology: A central role in the innate immune response? RNA. 2023;29(3):273-281.[DOI]
-
45. Chiang DC, Li Y, Ng SK. The role of the Z-DNA binding domain in innate immunity and stress granules. Front Immunol. 2021;11:625504.[DOI]
-
46. Mandel CR, Kaneko S, Zhang H, Gebauer D, Vethantham V, Manley JL, et al. Polyadenylation factor CPSF-73 is the pre-mRNA 3’-end-processing endonuclease. Nature. 2006;444(7121):953-956.[DOI]
-
47. Marc D. How influenza A virus NS1 impedes transcription of the host cell’s messenger RNAs. npj Viruses. 2025;3:55.[DOI]
-
48. Wang X, Hennig T, Whisnant AW, Erhard F, Prusty BK, Friedel CC, et al. Herpes simplex virus blocks host transcription termination via the bimodal activities of ICP27. Nat Commun. 2020;11:293.[DOI]
-
49. Chen R, Zou J, Chen J, Zhong X, Kang R, Tang D. Pattern recognition receptors: Function, regulation and therapeutic potential. Sig Transduct Target Ther. 2025;10:216.[DOI]
-
50. Onomoto K, Onoguchi K, Yoneyama M. Regulation of RIG-I-like receptor-mediated signaling: Interaction between host and viral factors. Cell Mol Immunol. 2021;18(3):539-555.[DOI]
-
51. Trishna S, Lavon A, Shteinfer-Kuzmine A, Dafa-Berger A, Shoshan-Barmatz V. Overexpression of the mitochondrial anti-viral signaling protein, MAVS, in cancers is associated with cell survival and inflammation. Mol Ther Nucleic Acids. 2023;33:713-732.[DOI]
-
52. Dong L, Hou YR, Xu N, Gao XQ, Sun Z, Yang QK, et al. Cyclic GMP-AMP synthase recognizes the physical features of DNA. Acta Pharmacol Sin. 2025;46(2):264-270.[DOI]
-
53. Liu S, Cai X, Wu J, Cong Q, Chen X, Li T, et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science. 2015;347(6227):aaa2630.[DOI]
-
54. Manes NP, Nita-Lazar A. Molecular mechanisms of the toll-like receptor, STING, MAVS, inflammasome, and interferon pathways. mSystems. 2021;6(3):e00336-e00321.[DOI]
-
55. de Weerd NA, Kurowska AK, Mendoza JL, Schreiber G. Structure–function of type I and III interferons. Curr Opin Immunol. 2024;86:102413.[DOI]
-
56. Rengachari S, Groiss S, Devos JM, Caron E, Grandvaux N, Panne D. Structural basis of STAT2 recognition by IRF9 reveals molecular insights into ISGF3 function. Proc Natl Acad Sci U S A. 2018;115(4):E601-E609.[DOI]
-
57. Platanitis E, Demiroz D, Schneller A, Fischer K, Capelle C, Hartl M, et al. A molecular switch from STAT2-IRF9 to ISGF3 underlies interferon-induced gene transcription. Nat Commun. 2019;10:2921.[DOI]
-
58. Jiao H, Wachsmuth L, Wolf S, Lohmann J, Nagata M, Kaya GG, et al. ADAR1 averts fatal type I interferon induction by ZBP1. Nature. 2022;607(7920):776-783.[DOI]
-
59. Hubbard NW, Ames JM, Maurano M, Chu LH, Somfleth KY, Gokhale NS, et al. ADAR1 mutation causes ZBP1-dependent immunopathology. Nature. 2022;607(7920):769-775.[DOI]
-
60. de Reuver R, Verdonck S, Dierick E, Nemegeer J, Hessmann E, Ahmad S, et al. ADAR1 prevents autoinflammation by suppressing spontaneous ZBP1 activation. Nature. 2022;607(7920):784-789.[DOI]
-
61. de Reuver R, Dierick E, Wiernicki B, Staes K, Seys L, De Meester E, et al. ADAR1 interaction with Z-RNA promotes editing of endogenous double-stranded RNA and prevents MDA5-dependent immune activation. Cell Rep. 2021;36(6):109500.[DOI]
-
62. Nakahama T, Kawahara Y. The RNA-editing enzyme ADAR1: A regulatory hub that tunes multiple dsRNA-sensing pathways. Int Immunol. 2023;35(3):123-133.[DOI]
-
63. Vignuzzi M, López CB. Defective viral genomes are key drivers of the virus–host interaction. Nat Microbiol. 2019;4(7):1075-1087.[DOI]
-
64. French H, Pitré E, Oade MS, Elshina E, Bisht K, King A, et al. Transient RNA structures cause aberrant influenza virus replication and innate immune activation. Sci Adv. 2022;8(36):eabp8655.[DOI]
-
65. Moeller NH, Shi K, Demir Ö, Belica C, Banerjee S, Yin L, et al. Structure and dynamics of SARS-CoV-2 proofreading exoribonuclease ExoN. Proc Natl Acad Sci U S A. 2022;119(9):e2106379119.[DOI]
-
66. Zhou T, Gilliam NJ, Li S, Spandau S, Osborn RM, Connor S, et al. Generation and functional analysis of defective viral genomes during SARS-CoV-2 infection. mBio. 2023;14(3):e00250-e00223.[DOI]
-
67. Grimm C, Bartuli J, Fischer U. Cytoplasmic gene expression: Lessons from poxviruses. Trends Biochem Sci. 2022;47(10):892-902.[DOI]
-
68. Koehler H, Cotsmire S, Zhang T, Balachandran S, Upton JW, Langland J, et al. Vaccinia virus E3 prevents sensing of Z-RNA to block ZBP1-dependent necroptosis. Cell Host Microbe. 2021;29(8):1266-1276.[DOI]
-
69. DeAntoneo C, Herbert A, Balachandran S. Z-form nucleic acid-binding protein 1 (ZBP1) as a sensor of viral and cellular Z-RNAs: Walking the razor’s edge. Curr Opin Immunol. 2023;83:102347.[DOI]
-
70. Herbert A, Cherednichenko O, Lybrand TP, Egli M, Poptsova M. Zα and zβ localize ADAR1 to flipons that modulate innate immunity, alternative splicing, and nonsynonymous RNA editing. Int J Mol Sci. 2025;26(6):2422.[DOI]
-
71. Li S, Deng X, Pathak D, Basavaraj R, Sun L, Cheng Y, et al. Deficiency of m6a RNA methylation promotes ZBP1-mediated cell death. bioRxiv [Preprint]. 2024.[DOI]
-
72. Liu Y, Hu L, Wu Z, Yuan K, Hong G, Lian Z, et al. Loss of PHF8 induces a viral mimicry response by activating endogenous retrotransposons. Nat Commun. 2023;14:4225.[DOI]
-
73. Tang Y, Tang S, Yang W, Zhang Z, Wang T, Wu Y, et al. MED12 loss activates endogenous retroelements to sensitise immunotherapy in pancreatic cancer. Gut. 2024;73(12):1999-2011.[DOI]
-
74. Guil S, Esteller M. PRC2 loss and DNMT inhibition boost viral mimicry in cancer. Cancer Discov. 2022;12(9):2020-2022.[DOI]
-
75. Shi X, Teng H, Sun Z. An updated overview of experimental and computational approaches to identify non-canonical DNA/RNA structures with emphasis on G-quadruplexes and R-loops. Brief Bioinform. 2022;23(6):bbac441.[DOI]
-
76. Pan Z, Zeng Z, Xiong W, Fan C. The roles and mechanisms of non-canonical RNA secondary structures in tumors. Biochim Biophys Acta Rev Cancer. 2025;1880(4):189363.[DOI]
-
77. Wang S, Xu Y. Z-form DNA-RNA hybrid blocks DNA replication. Nucleic Acids Res. 2025;53(5):gkaf135.[DOI]
-
78. Fischer H, Tschachler E, Eckhart L. Pangolins lack IFIH1/MDA5, a cytoplasmic RNA sensor that initiates innate immune defense upon coronavirus infection. Front Immunol. 2020;11:939.[DOI]
-
79. Kroft CW, Krall JB, Warchol M, Welty R, Herbert A, Henen MA, et al. Zα and Zβ domains of ADAR1 and ZBP1 bind G-quadruplexes with nanomolar affinities, establishing Zβ as a G-quadruplex-specific domain. Nucleic Acids Res. 2026;54(8):gkag419.[DOI]
-
80. Qin G, Zhao C, Gao C, Hu W, Zhang N, Ren J, et al. Telomeric repeat-containing RNA G-quadruplexes trigger ZBP1-mediated cell death. Nat Commun. 2026;17:3076.[DOI]
-
81. Srinivasan B, Kuś K, Athanasiadis A. Thermodynamic analysis of Zα domain-nucleic acid interactions. Biochem J. 2022;479(16):1727-1741.[DOI]
-
82. Liu F, Wang S, Xu Y. Solution structure of Z-form DNA bound to a curaxin ligand CBL0137. Nucleic Acids Res. 2026;54(4):gkag104.[DOI]
-
83. Xie F, Wu D, Huang J, Liu X, Shen Y, Huang J, et al. ZBP1 condensate formation synergizes Z-NAs recognition and signal transduction. Cell Death Dis. 2024;15(7):487.[DOI]
-
84. Li S, Li H, Zhou Z, Ye M, Wang Y, Li W, et al. A viral necrosome mediates direct RIPK3 activation to promote inflammatory necroptosis. Proc Natl Acad Sci U S A. 2025;122(22):e2420245122.[DOI]
-
85. Ekhlak M, Kulkarni PP, Singh V, Chaurasia SN, Mohapatra SK, Chaurasia RN, et al. Necroptosis executioner MLKL plays pivotal roles in agonist-induced platelet prothrombotic responses and lytic cell death in a temporal order. Cell Death Differ. 2023;30(8):1886-1899.[DOI]
-
86. Yin C, Fedorov A, Guo H, Crawford JC, Rousseau C, Zhong X, et al. Host cell Z-RNAs activate ZBP1 during virus infections. Nature. 2025;648(8094):707-716.[DOI]
-
87. Mishchenko T, Mitroshina E, Balalaeva I, Krysko O, Vedunova M, Krysko DV. An emerging role for nanomaterials in increasing immunogenicity of cancer cell death. Biochim Biophys Acta Rev Cancer. 2019;1871(1):99-108.[DOI]
-
89. Baker MODG, Shanmugam N, Pham CLL, Ball SR, Sierecki E, Gambin Y, et al. The RHIM of the immune adaptor protein TRIF forms hybrid amyloids with other necroptosis-associated proteins. Molecules. 2022;27(11):3382.[DOI]
-
90. Koerner L, Wachsmuth L, Kumari S, Schwarzer R, Wagner T, Jiao H, et al. ZBP1 causes inflammation by inducing RIPK3-mediated necroptosis and RIPK1 kinase activity-independent apoptosis. Cell Death Differ. 2024;31(7):938-953.[DOI]
-
91. Kelepouras K, Saggau J, Bonasera D, Kiefer C, Locci F, Rakhsh-Khorshid H, et al. STING induces ZBP1-mediated necroptosis independently of TNFR1 and FADD. Nature. 2025;647(8090):735-746.[DOI]
-
92. Li Z, Chu Z, Yang J, Qian H, Xu J, Chen B, et al. Immunogenic cell death augmented by manganese zinc sulfide nanoparticles for metastatic melanoma immunotherapy. ACS Nano. 2022;16(9):15471-15483.[DOI]
-
93. Tait SWG, Oberst A. Cellular memory of sub-lethal stress. Immunity. 2026;59(4):801-812.[DOI]
-
94. Mishra S, Jain D, Dey AA, Nagaraja S, Srivastava M, Khatun O, et al. Bat RNA viruses employ viral RHIMs orchestrating species-specific cell death programs linked to Z-RNA sensing and ZBP1-RIPK3 signaling. iScience. 2024;27(12):111444.[DOI]
-
95. Kaiser WJ, Upton JW, Mocarski ES. Receptor-interacting protein homotypic interaction motif-dependent control of NF-κB activation via the DNA-dependent activator of IFN regulatory factors. J Immunol. 2008;181(9):6427-6434.[DOI]
-
96. Rebsamen M, Heinz LX, Meylan E, Michallet MC, Schroder K, Hofmann K, et al. DAI/ZBP1 recruits RIP1 and RIP3 through RIP homotypic interaction motifs to activate NF-κB. EMBO Rep. 2009;10(8):916-922.[DOI]
-
97. Kofman SB, Chu LH, Ames JM, Chavarria SD, Lichauco K, Daniels BP, et al. RIPK3 coordinates RHIM domain–dependent antiviral inflammatory transcription in neurons. Sci Signal. 2025;18(880):eado9745.[DOI]
-
98. Daniels BP, Kofman SB, Smith JR, Norris GT, Snyder AG, Kolb JP, et al. The nucleotide sensor ZBP1 and kinase RIPK3 induce the enzyme IRG1 to promote an antiviral metabolic state in neurons. Immunity. 2019;50(1):64-76.[DOI]
-
99. Daniels BP, Snyder AG, Olsen TM, Orozco S, Oguin TH, Tait SWG, et al. RIPK3 restricts viral pathogenesis via cell death-independent neuroinflammation. Cell. 2017;169(2):301-313.[DOI]
-
100. Jayakumar A, Bothwell ALM. RIPK3-induced inflammation by I-MDSCs promotes intestinal tumors. Cancer Res. 2019;79(7):1587-1599.[DOI]
-
101. Park WH. The mitochondrial nexus: Targeting metabolic vulnerabilities, oxidative stress, and immunomodulation to induce cancer cell death. Biochim Biophys Acta Rev Cancer. 2025;1880(6):189491.[DOI]
-
102. Rius-Pérez S. p53 at the crossroad between mitochondrial reactive oxygen species and necroptosis. Free Radic Biol Med. 2023;207:183-193.[DOI]
-
103. Weindel CG, Martinez EL, Zhao X, Mabry CJ, Bell SL, Vail KJ, et al. Mitochondrial ROS promotes susceptibility to infection via gasdermin D-mediated necroptosis. Cell. 2022;185(17):3214-3231.[DOI]
-
104. Thapa RJ, Basagoudanavar SH, Nogusa S, Irrinki K, Mallilankaraman K, Slifker MJ, et al. NF-κB protects cells from gamma interferon-induced RIP1-dependent necroptosis. Mol Cell Biol. 2011;31(14):2934-2946.[DOI]
-
105. An X, Yu W, Liu J, Tang D, Yang L, Chen X. Oxidative cell death in cancer: Mechanisms and therapeutic opportunities. Cell Death Dis. 2024;15(8):556.[DOI]
-
106. Diaconescu IB, Dumitru AV, Tataru CP, Toader C, Șerban M, Covache-Busuioc RA, et al. From electron imbalance to network collapse: Decoding the redox code of ischemic stroke for biomarker-guided precision neuroprotection. Int J Mol Sci. 2025;26(22):10835.[DOI]
-
107. Du J, Wang Z. Regulation of RIPK1 phosphorylation: Implications for inflammation, cell death, and therapeutic interventions. Biomedicines. 2024;12(7):1525.[DOI]
-
108. Srivastava A, Tomar B, Sharma P, Kumari S, Prakash S, Rath SK, et al. RIPK3-MLKL signaling activates mitochondrial CaMKII and drives intrarenal extracellular matrix production during CKD. Matrix Biol. 2022;112:72-89.[DOI]
-
109. Mohammed S, Nicklas EH, Thadathil N, Selvarani R, Royce GH, Kinter M, et al. Role of necroptosis in chronic hepatic inflammation and fibrosis in a mouse model of increased oxidative stress. Free Radic Biol Med. 2021;164:315-328.[DOI]
-
110. Roderick-Richardson JE, Lim SE, Suzuki S, Ahmad MH, Selway J, Suleiman R, et al. ZBP1 activation triggers hematopoietic stem and progenitor cell death resulting in bone marrow failure in mice. Proc Natl Acad Sci U S A. 2024;121(4):e2309628121.[DOI]
-
111. López-Pérez W, González-Calderón RE, Sai K, Rai P, MacStudy JM, Sakamachi Y, et al. TAK1 inhibition activates pore-forming proteins to block intracellular bacterial growth through modulating mitochondria. Cell Death Dis. 2025;16:456.[DOI]
-
112. Ju E, Park KA, Shen HM, Hur GM. The resurrection of RIP kinase 1 as an early cell death checkpoint regulator: A potential target for therapy in the necroptosis era. Exp Mol Med. 2022;54(9):1401-1411.[DOI]
-
113. Xie X, Liu J, Liang W, Zhang Y, Gong X, Yuan S, et al. Repression of RIPK1 kinase by INPP5D inhibits expression of diverse proinflammatory mediators and late-onset Alzheimer’s disease risk factors. Immunity. 2026;59(2):419-437.[DOI]
-
114. Zhang JB, Zhang QR, Jin Q, Yang J, Lin SZ, Fan JG. Sestrin2 maintains hepatic immune homeostasis and redox balance partially via inhibiting RIPK3-mediated necroptosis in metabolic dysfunction-associated steatohepatitis. Mol Metab. 2024;80:101865.[DOI]
-
115. Kang JS, Cho NJ, Lee SW, Lee JG, Lee JH, Yi J, et al. RIPK3 causes mitochondrial dysfunction and albuminuria in diabetic podocytopathy through PGAM5-Drp1 signaling. Metabolism. 2024;159:155982.[DOI]
-
116. Wu L, Zhang X, Zheng L, Zhao H, Yan G, Zhang Q, et al. RIPK3 orchestrates fatty acid metabolism in tumor-associated macrophages and hepatocarcinogenesis. Cancer Immunol Res. 2020;8(5):710-721.[DOI]
-
117. Li G, Wu M, Chen K, Xu Y, Zhang X, Chen Y, et al. ROS-mediated M1 polarization-necroptosis crosstalk involved in Di-(2-ethylhexyl) phthalate-induced chicken liver injury. Poult Sci. 2025;104(1):104558.[DOI]
-
119. Wang L, Wang L, Shi X, Xu S. Chlorpyrifos induces the apoptosis and necroptosis of L8824 cells through the ROS/PTEN/PI3K/AKT axis. J Hazard Mater. 2020;398:122905.[DOI]
-
120. Lei Y, Xu T, Sun W, Wang X, Gao M, Lin H. Evodiamine alleviates DEHP-induced hepatocyte pyroptosis, necroptosis and immunosuppression in grass carp through ROS-regulated TLR4/MyD88/NF-κB pathway. Fish Shellfish Immunol. 2023;140:108995.[DOI]
-
121. Kong Y, Tian X, Zhang R, Deng H, Wang G, Wu D, et al. F-53B exposure induced testicular premature aging through ZBP1-mediated programmed necrosis. J Hazard Mater. 2025;500:140351.[DOI]
-
122. Lian CY, Xia WH, Sun MC, Wan XM, Zhou XL, Wang L. Cadmium targeting MLKL-Drp1 axis to trigger mitochondrial oxidative stress contributes to necroinflammation in rat kidney. J Adv Res. 2026;81:1079-1093.[DOI]
-
123. Xu T, Liu Q, Chen D, Liu Y. Atrazine exposure induces necroptosis through the P450/ROS pathway and causes inflammation in the gill of common carp (Cyprinus carpio L.). Fish Shellfish Immunol. 2022;131:809-816.[DOI]
-
124. Guan S, Lu S, Zhang R, Wang Y, Yao X, Deng X, et al. Lactoferrin alleviates LPS-induced oxidative stress and necroptosis in liver by promoting mitophagy. J Agric Food Chem. 2025;73(19):11948-11959.[DOI]
-
125. Chen J, Jiang WD, Feng L, Wu P, Liu Y, Jin XW, et al. Myo-inositol: A potential game-changer in preventing gill cell death and alleviating “gill rot” in grass carp (Ctenopharyngodon idellus). Fish Shellfish Immunol. 2024;153:109850.[DOI]
-
126. Guo J, Hu JP, Liu M, Chen Y, Zhang S, Guan S. Apigenin-mediated ESCRT-III activation and mitophagy alleviate LPS-induced necroptosis in renal cells. J Agric Food Chem. 2025;73(16):9906-9919.[DOI]
-
127. Tran TH, Tran TTP. Current status of nanoparticle-mediated immunogenic cell death in cancer immunotherapy. Int Immunopharmacol. 2024;142:113085.[DOI]
-
128. Lee AV, Nestler KA, Chiappinelli KB. Therapeutic targeting of DNA methylation alterations in cancer. Pharmacol Ther. 2024;258:108640.[DOI]
-
129. Sepand MR, Ranjbar S, Kempson IM, Akbariani M, Muganda WCA, Müller M, et al. Targeting non-apoptotic cell death in cancer treatment by nanomaterials: Recent advances and future outlook. Nanomed Nanotechnol Biol Med. 2020;29:102243.[DOI]
-
130. Hao Q, Idell S, Tang H. M1 macrophages are more susceptible to necroptosis. J Cell Immunol. 2021;3(2):97.[DOI]
-
131. O’Donnell JA, Lehman J, Roderick JE, Martinez-Marin D, Zelic M, Doran C, et al. Dendritic cell RIPK1 maintains immune homeostasis by preventing inflammation and autoimmunity. J Immunol. 2018;200(2):737-748.[DOI]
-
132. Zhang H, Wang Z, Lu J, Li J, Jia Y, Xie X, et al. Neutrophil extracellular traps prime the ZBP1-cGAS sensor complex, triggering necroptosis and inflammatory injury in acute pancreatitis. Int J Biol Sci. 2026;22(5):2398-2417.[DOI]
-
133. Gong D, Sun K, Yin K, Wang X. Selenium mitigates the inhibitory effect of TBBPA on NETs release by regulating ROS/MAPK pathways-induced carp neutrophil apoptosis and necroptosis. Fish Shellfish Immunol. 2023;132:108501.[DOI]
-
134. Sun B, Wang JD, Wu MY, Chen HY, Yang SQ, Chen YM, et al. Sestrin2 alleviates sepsis-induced immunosuppression of dendritic cells by regulating mitochondrial dynamics. Free Radic Biol Med. 2025;240:583-596.[DOI]
-
135. Tan X, He Y, Yu P, Deng Y, Xie Z, Guo J, et al. The dual role of FSP1 in programmed cell death: Resisting ferroptosis in the cell membrane and promoting necroptosis in the nucleus of THP-1 cells. Mol Med. 2024;30:102.[DOI]
-
136. Liu Y, Li H, Dai Y, Niu J, Jiang L, Tang J, et al. Activation of ZBP1/RIPK3/MLKL-dependent necroptosis by pseudorabies virus restricts viral infection in BV2 microglia cells. Transbound Emerg Dis. 2025;2025:8510846.[DOI]
-
137. Xiao C, Wang X, Li S, Zhang Z, Li J, Deng Q, et al. A cuproptosis-based nanomedicine suppresses triple negative breast cancers by regulating tumor microenvironment and eliminating cancer stem cells. Biomaterials. 2025;313:122763.[DOI]
-
138. Sun D, Zou Y, Song L, Han S, Yang H, Chu D, et al. A cyclodextrin-based nanoformulation achieves co-delivery of ginsenoside Rg3 and quercetin for chemo-immunotherapy in colorectal cancer. Acta Pharm Sin B. 2022;12(1):378-393.[DOI]
-
139. Wang X, Bian L, Feng Z, O’Shaughnessy K, Kwong AC, Ha EH, et al. A PNPLA3–NACC1–RIPK3 pathway mediates macrophage necroptosis and inflammation in MASLD. Hepatol Commun. 2026;10(2):e0890.[DOI]
-
140. Khuanjing T, Ongnok B, Maneechote C, Siri-Angkul N, Prathumsap N, Arinno A, et al. Acetylcholinesterase inhibitor ameliorates doxorubicin-induced cardiotoxicity through reducing RIP1-mediated necroptosis. Pharmacol Res. 2021;173:105882.[DOI]
-
141. Chen L, Zhang X, Ou Y, Liu M, Yu D, Song Z, et al. Advances in RIPK1 kinase inhibitors. Front Pharmacol. 2022;13:976435.[DOI]
-
142. Wu X, Li Z, Yin T, Shu J, Xu YS, Liang J, et al. Two-dimensional iridium(III)–carbon nitride nano complexes induce simultaneous oncosis and necroptosis for synergistic chemo- and sono-immunotherapy in hypoxic melanoma. J Am Chem Soc. 2026;148(9):10105-10123.[DOI]
-
143. Tian S, Liu Y, Tan Y, Cui X, Liu R, Liu C, et al. Necroptosis-inducing nanobubbles for effective oxygen delivery and enhanced sonodynamic immunotherapy of breast cancer via UTND. Eur J Pharm Biopharm. 2025;210:114675.[DOI]
-
144. Han C, Xiao S, Xing Z, Xu X, Wang M, Han X, et al. NADPH oxidases-inspired reactive oxygen biocatalysts with electron-rich Pt sites to potently amplify immune checkpoint blockade therapy. Adv Mater. 2025;37(3):2407644.[DOI]
-
145. Li Y, Liu Y, Lou C, Chen X, Zhang Y, Shi H, et al. Sono-controllable Janus hydrogel platform for sequential tumor eradication and bone regeneration in metastatic breast cancer. Adv Sci. 2025;12(43):e06386.[DOI]
-
146. Zhu Y, Min Y, Geng W, Wei Z, Xiao S, Peng X, et al. Radio-activable gold-single-atom-based artificial enzymes with cascade biocatalysis and enhanced radiosensitization to prevent metastasis and recurrence in malignancies. J Am Chem Soc. 2026;148(7):7515-7531.[DOI]
-
147. Yao X, Yang H, Guo S, Liu Y, Zhang Q, Zhou Z, et al. Radiation-triggerable bioreactors enable bioenergetic reprograming of cancer stem cell plasticity via targeted arginine metabolism disruption for augmented radio-immunotherapy. Biomaterials. 2025;322:123391.[DOI]
-
148. Zheng Q, Zou T, Wang W, Zhang C, Hu S, Cheng X, et al. Necroptosis-mediated synergistic photodynamic and glutamine-metabolic therapy enabled by a biomimetic targeting nanosystem for cholangiocarcinoma. Adv Sci. 2024;11(29):2309203.[DOI]
-
149. Yang J, Du Y, Yao Y, Liao Y, Wang B, Yu X, et al. Employing piezoelectric Mg2+-doped hydroxyapatite to target death receptor-mediated necroptosis: A strategy for amplifying immune activation. Adv Sci. 2024;11(13):2307130.[DOI]
-
150. Liu L, Yu AM, Wang X, Soles LV, Teng X, Chen Y, et al. The anticancer compound JTE-607 reveals hidden sequence specificity of the mRNA 3’ processing machinery. Nat Struct Mol Biol. 2023;30(12):1947-1957.[DOI]
-
151. Yu S, Li J, Zhang J, Zeng G, Zeng B, Song S, et al. Nanosized shikonin disrupts tumor-cell mismatch repair and synergizes with manganese to sensitize squamous carcinoma to immunotherapy. ACS Nano. 2025;19(14):13889-13905.[DOI]
-
152. Yu L, Huang K, Liao Y, Wang L, Sethi G, Ma Z. Targeting novel regulated cell death: Ferroptosis, pyroptosis and necroptosis in anti-PD-1/PD-L1 cancer immunotherapy. Cell Prolif. 2024;57(8):e13644.[DOI]
-
153. Li Z, Ren G, Wang X, Li X, Ding L, Zhu J, et al. Tumor microenvironment responsive nano-PROTAC for BRD4 degradation enhanced cancer photo-immunotherapy. Biomaterials. 2025;322:123387.[DOI]
-
154. Zhang W, Yang LM, Zhao Y, Li MY, Shi YQ, Lu Y, et al. Stable cyclometalated gold(III) complex engaging isoquinoline derivative and disulfur ligand elicits necroptosis-dependent immunogenic cell death. J Med Chem. 2026;69(4):4932-4944.[DOI]
-
155. Jiang Y, Zhang Y, Hou J, Liu H, Dai X, Hou Y. Single-cell sequencing-guided design of synergistic chemo-immunotherapy nanodrugs for cGAS-STING activation in prostate cancer therapy. J Nanobiotechnol. 2026;24:198.[DOI]
-
156. Egbulefu C, Black K, Su X, Karmakar P, Habimana-Griffin L, Sudlow G, et al. Radionuclide-stimulated dynamic therapy induces complementary immunogenic necroptosis and apoptosis cancer cell death pathways. Commun Biol. 2026;9:275.[DOI]
-
157. Liu H, He Q, Wang DX, Liu J, Qu Y, Zhang D, et al. Delivery of a ZBP1 agonist enhances radiotherapy-induced antitumour immunity in hepatocellular carcinoma. J Control Release. 2026;395:114991.[DOI]
-
158. Chen Z, Sang L, Bian D, Liu Y, Bai Z. Piezo-catalytic immunotherapy: Mechanisms and feasibility in cancer treatment. Theranostics. 2025;15(13):6236-6252.[DOI]
-
159. Li Y, Chen J, Xia Q, Shang J, He Y, Li Z, et al. Photothermal Fe3O4 nanoparticles induced immunogenic ferroptosis for synergistic colorectal cancer therapy. J Nanobiotechnol. 2024;22:630.[DOI]
-
160. Cai Y, Chai T, Nguyen W, Liu J, Xiao E, Ran X, et al. Phototherapy in cancer treatment: Strategies and challenges. Sig Transduct Target Ther. 2025;10:115.[DOI]
-
161. Lee SH, Cho Y, Bang S, Sung D, Koo J, Kim S, et al. Nanoshield-assisted viral gene therapy with induction of non-apoptotic cell death and durable antitumor immunity. Adv Sci. 2025;12(41):e07550.[DOI]
-
162. Chu T, Maksoudian C, Pedrotti S, Izci M, Perez Gilabert I, Koutsoumpou X, et al. Nanomaterial-mediated delivery of MLKL plasmids sensitizes tumors to immunotherapy and reduces metastases. Adv Healthc Mater. 2024;13(27):2401306.[DOI]
-
163. Sahin U, Derhovanessian E, Miller M, Kloke BP, Simon P, Löwer M, et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature. 2017;547(7662):222-226.[DOI]
-
164. Miao L, Zhang Y, Huang L. mRNA vaccine for cancer immunotherapy. Mol Cancer. 2021;20:41.[DOI]
-
165. Li Y, Guo Y, Zhang K, Zhu R, Chen X, Zhang Z, et al. Cell death pathway regulation by functional nanomedicines for robust antitumor immunity. Adv Sci. 2024;11(3):2306580.[DOI]
-
166. Goyal A, Bauer J, Hey J, Papageorgiou DN, Stepanova E, Daskalakis M, et al. DNMT and HDAC inhibition induces immunogenic neoantigens from human endogenous retroviral element-derived transcripts. Nat Commun. 2023;14(1):6731.[DOI]
-
167. Huang C, Gao Y, Chen J, Hong JH, Jiang Y, Chai KXY, et al. Priming with DNMT inhibitors potentiates PD-1 immunotherapy by triggering viral mimicry in relapsed/refractory NK/T-cell lymphoma. Cancer Discov. 2025;15(12):2450-2467.[DOI]
-
168. Zhou X, Singh M, Sanz Santos G, Guerlavais V, Carvajal LA, Aivado M, et al. Pharmacologic activation of p53 triggers viral mimicry response thereby abolishing tumor immune evasion and promoting antitumor immunity. Cancer Discov. 2021;11(12):3090-3105.[DOI]
-
169. Liu Y, Stockwell BR, Jiang X, Gu W. p53-regulated non-apoptotic cell death pathways and their relevance in cancer and other diseases. Nat Rev Mol Cell Biol. 2025;26(8):600-614.[DOI]
-
170. Casagrande Raffi G, Chen J, Feng X, Chen Z, Lieftink C, Deng S, et al. An antibiotic that mediates immune destruction of senescent cancer cells. Proc Natl Acad Sci U S A. 2024;121(52):e2417724121.[DOI]
-
171. Catanzaro E, Feron O, Skirtach AG, Krysko DV. Immunogenic cell death and role of nanomaterials serving as therapeutic vaccine for personalized cancer immunotherapy. Front Immunol. 2022;13:925290.[DOI]
-
172. Ma S, Zhu W, Ji X, Liu C, Chen N, Guo D, et al. Multiplex methionine modulating hydrogel for cancer metabolic therapy. Adv Mater. 2025;37(30):2420445.[DOI]
-
173. Liu R, Cao H, Zhang S, Cai M, Zou T, Wang G, et al. ZBP1-mediated apoptosis and inflammation exacerbate steatotic liver ischemia/reperfusion injury. J Clin Invest. 2024;134(13):e180451.[DOI]
-
174. Rojas-Rivera D, Beltrán S, Muñoz-Carvajal F, Ahumada-Montalva P, Abarzúa L, Gomez L, et al. The autophagy protein RUBCNL/PACER represses RIPK1 kinase-dependent apoptosis and necroptosis. Autophagy. 2024;20(11):2444-2459.[DOI]
-
175. Devos M, Tanghe G, Gilbert B, Dierick E, Verheirstraeten M, Nemegeer J, et al. Sensing of endogenous nucleic acids by ZBP1 induces keratinocyte necroptosis and skin inflammation. J Exp Med. 2020;217(7):e20191913.[DOI]
-
176. Shen T, Wang Y, Cheng L, Bode AM, Gao Y, Zhang S, et al. Oxidative complexity: The role of ROS in the tumor environment and therapeutic implications. Bioorg Med Chem. 2025;127:118241.[DOI]
-
177. Xin L, Ning S, Wang H, Shi R. Tumor microenvironment responsive and platelet membrane coated polydopamine nanoparticles for cancer radiosensitization by inducing cuproptosis. Int J Nanomed. 2025;20:3643-3652.[DOI]
-
178. Mani S, Ralph SJ, Swargiary G, Rani M, Wasnik S, Singh SP, et al. Therapeutic targeting of mitochondrial plasticity and redox control to overcome cancer chemoresistance. Antioxid Redox Signal. 2023;39:591-619.[DOI]
-
179. Sun X, Cai X, Li S, Pi R, Guo Z, Jiang J, et al. High-dose ascorbic acid selectively induces pyroptosis in LKB1-deficient lung cancer and sensitizes immunotherapy. Cell Rep Med. 2025;6(9):102291.[DOI]
-
180. Hsu SK, Li CY, Lin IL, Syue WJ, Chen YF, Cheng KC, et al. Inflammation-related pyroptosis, a novel programmed cell death pathway, and its crosstalk with immune therapy in cancer treatment. Theranostics. 2021;11(18):8813-8835.[DOI]
Copyright
© The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Publisher’s Note
Science Exploration remains a neutral stance on jurisdictional claims in published
maps
and institutional affiliations. The views expressed in this article are solely those
of
the author(s) and do not reflect the opinions of the Editors or the publisher.
Share And Cite



Science Exploration Style
Zhong X, Balachandran S, Zhang T. ZBP1-mediated sensing of genomic stress in cancer therapy. Ferroptosis Oxid Stress. 2026;2:202621. https://doi.org/10.70401/fos.2026.0035
Tips
Copy completed.
Submit a Manuscript
Author Instructions
Cite this Article
Article Metrics
0
View
0
Download
Cited
Article Updates

- Abstract
- Keywords
- 1. Introduction
- 2. Z-RNA Triggers Checkpoints of Innate Immune Responses in Pathogenesis
- 3. ZBP1 as the Central Sentinel of Z-RNA-Mediated Checkpoints in Innate Immunity
- 4. ROS Regulates ZBP1-Mediated Necroptosis
- 5. Stress-Driven Innate Immune Checkpoints and Associated ROS Production Robustly Activate Antitumor Immunity
- 6. Conclusions and Perspectives
- Acknowledgements
- Authors contribution
- Conflicts of interest
- Ethical approval
- Consent to participate
- Consent for publication
- Availability of data and materials
- Funding
- References
- Copyright
Science Exploration Style
Zhong X, Balachandran S, Zhang T. ZBP1-mediated sensing of genomic stress in cancer therapy. Ferroptosis Oxid Stress. 2026;2:202621. https://doi.org/10.70401/fos.2026.0035
copy
Share Link
copy