Chromatin fatigue: An epigenetic legacy of DNA repair
Lingjiang Chen
Zhiyong Mao
*

,
Yu Chen
*
*Correspondence to:
Zhiyong Mao, Shanghai Key Laboratory of Maternal Fetal Medicine, Clinical and Translational Research Center of Shanghai First Maternity and Infant Hospital, Frontier Science Center for Stem Cell Research, School of Life Sciences and Technology, Tongji University, Shanghai 200092, China.
E-mail: zhiyong_mao@tongji.edu.cn
Yu Chen, Shanghai Key Laboratory of Maternal Fetal Medicine, Clinical and Translational Research Center of Shanghai First Maternity and Infant Hospital, Frontier Science Center for Stem Cell Research, School of Life Sciences and Technology, Tongji University, Shanghai 200092, China. E-mail: y_chen@tongji.edu.cn
Yu Chen, Shanghai Key Laboratory of Maternal Fetal Medicine, Clinical and Translational Research Center of Shanghai First Maternity and Infant Hospital, Frontier Science Center for Stem Cell Research, School of Life Sciences and Technology, Tongji University, Shanghai 200092, China. E-mail: y_chen@tongji.edu.cn
Ageing Cancer Res Treat. 2026;3:202610. 10.70401/acrt.2026.0020
Received: March 16, 2026Accepted: May 11, 2026Published: May 13, 2026
Abstract
While genomic instability is a hallmark of aging, and unrepaired or mutagenic double-strand breaks (DSBs) are established drivers, recent evidence suggests that even accurately repaired DSBs contribute to aging. Here, we focus on an intriguing study by Bantele et al. published in Science, which demonstrates that Cas9-induced DSB repair can induce persistent, heritable alterations in higher-order chromatin structure and function, termed "chromatin fatigue". These alterations, characterized by changes in chromatin topology and gene expression, persist long after DNA sequence restoration and are inherited through cell divisions. Crucially, they impair transcriptional responsiveness to physiological stimuli. This finding provides a novel mechanism for DNA damage-driven aging independent of mutations, potentially explaining age-related epigenetic dysfunction. The commentary also highlights key unresolved questions regarding the permanence, locus-specificity, and physiological impact of chromatin fatigue, and explores its interaction with age-related DNA repair decline. This striking molecular phenomenon challenges the notion that faithful repair ensures full functional restoration and opens avenues for future research into interventions against aging and other age-related diseases, such as cancer.
Keywords
Chromatin fatigue, DNA repair, double-strand break, aging, epigenetics
The three-dimensional (3D) architecture of the eukaryotic genome provides a foundational scaffold for the precise regulation of gene expression. Key structural elements, such as topologically associating domains (TADs) and chromatin compartments, maintain genomic functional integrity by coordinating interactions between regulatory elements and their target genes[1,2]. Genomic instability is an established hallmark of aging, and DNA damage accumulation is a major driver of this instability[3]. The intricate relationship between the repair of DNA double-strand breaks (DSBs), the most severe form of DNA damage, and the aging process has long been a research focus[4]. Conventionally, the primary threat posed by DSBs has been considered their potential to cause sequence alterations (such as mutations) or trigger inflammatory responses, thereby driving aging phenotypes[5,6]. However, emerging evidence indicates that even accurately repaired DSBs contribute to aging[7], although the precise underlying mechanisms remain unclear.
A recent Science study by Bantele et al. reports that DSB repair can induce a heritable form of genome functional impairment termed “chromatin fatigue”[8] (Figure 1). This finding provides a possible mechanism for the long-term consequences of DNA damage independent of mutations and a potential contributor to biological aging and cancer. Focusing on the c-MYC locus, the study employed a specifically designed Cas9-induced DSB system targeting 12 sites across its 2.8 Mb TAD region. Results demonstrate that persistent alterations remain long after DNA sequence integrity is restored, including chromatin topological rearrangement, increased local compaction, reduced c-MYC expression, and decreased RNA retention within the c-MYC TAD[8]. This chromatin dysfunction exhibits significant distance-dependent effects on transcription, where DSB sites closer to the c-MYC gene induce more robust transcriptional repression. Notably, this aberrant chromatin state is stably heritable through cell division. The authors demonstrated that daughter cells inherit both the chromatin topological perturbations and the repressed gene expression phenotype from parent cells.
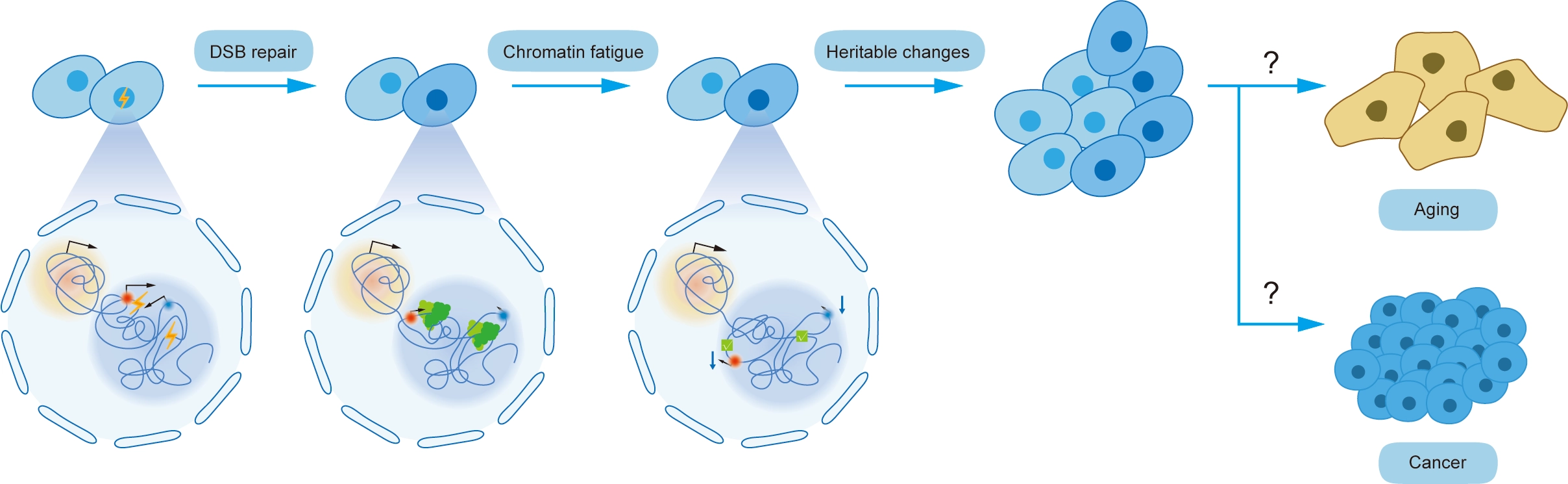
Figure 1. Repair of DNA double-strand breaks causes chromatin fatigue. DSB: double-strand break.
This study raises many interesting questions for future research. First, it is crucial to investigate if the DNA repair initiated by endogenous DSBs results in changes similar to those caused by Cas9-induced repair. DSBs can be repaired via distinct subpathways. The choice of pathway is influenced by multiple factors, including the chromatin context where the damage occurs and the nature of the DNA ends[9]. Consequently, the physiological relevance of Cas9-induced DSBs as a model for the diverse spectrum of endogenous lesions must be further evaluated. Second, histone modifications play important roles in regulating gene expression. A key question is whether successful DSB repair, beyond altering topology, also influences the histone modifications surrounding the damage sites. Moreover, in their study, Bantele et al. induced a single DSB and observed significant consequent changes. Given that an estimated 10-50 DSBs occur per cell per day in mammals[10,11], a key question arises: how does the simultaneous occurrence of DSBs at multiple genomic loci impact chromatin topology and histone modifications? This warrants further investigation. Additionally, although the authors confirmed that these persistent chromatin alterations can be passed through cell division, a key unresolved question is their duration. Are these changes truly permanent, or can they be gradually restored over extended cell divisions? Future work involving extended lineage tracing is needed to answer this question.
While DNA sequence integrity can be restored upon faithful DNA repair, whether higher-order chromatin architecture and its functional state are fully restored post-repair remains unclear. This study provides evidence that damage-induced changes in higher-order chromatin architecture may not be repaired. Determining the long-term consequences of these changes is an intriguing future direction. Epigenetic dysregulation has been proposed as another primary hallmark of aging. Based on unique age-related alterations in DNA methylation patterns, multiple DNA methylation-based epigenetic clocks have been established in recent years to predict biological age[12-15]. While these clocks are widely used in aging research, the upstream drivers of this epigenetic drift remain obscure. Given the prevalence of endogenous DNA damage, Bantele et al. provide a potential mechanistic explanation for age-associated epigenetic changes. In addition, responsiveness to regulatory cues is essential for tissue homeostasis. The authors showed that persistent chromatin alterations blunt this responsiveness, as cells exhibit reduced c-MYC responsiveness to upstream signaling even when DSBs occur outside its coding region. This impairment may hinder cellular adaptability to changing environments or responsiveness to stimuli, potentially contributing to age-related functional decline. Previous work, including our own, has demonstrated an age-related decline in DNA repair capacity[16]; for example, we have shown that XRCC4 transcription declines with age[17,18]. An interesting question is how impaired DNA repair processes influence the chromatin fatigue phenotype. Could decreased repair capacity increase the likelihood and severity of these alterations? Conversely, efficient DNA repair is associated with longevity, and activation of DNA repair pathways contributes to delayed aging and reduced age-related degenerative decline[19-23]. Could we potentially ameliorate these chromatin alterations by actively enhancing DNA repair? These questions warrant further exploration. But first, it must be established whether the chromatin fatigue phenotype is present in naturally aged cells or organisms.
Is chromatin fatigue involved in oncogenesis and tumor development? Advances in research techniques have revealed alterations in the 3D genome in cancer[24]. Given the rapid proliferation and high metabolic activity of cancer cells, a large number of DSBs are likely to occur due to replication stress and reactive oxygen species. However, it remains unclear whether a causal link exists between such DNA damage and the observed spatial genome reorganization in cancer. Furthermore, mutations in DNA repair factors are known to facilitate tumorigenesis[25,26]. Similar to the discussion on aging, a key question is how such DNA repair deficiencies impact chromatin fatigue and, consequently, modulate cancer progression. Notably, studies have also indicated that epigenetic alterations compromise DNA damage recognition and repair in cancer[27]. This raises an additional question: could chromatin fatigue, in turn, impair DNA repair processes, thereby establishing a vicious cycle that exacerbates both repair deficiency and chromatin disorganization? Finally, a key future goal should be to explore whether targeting chromatin fatigue can lead to new interventions for cancer.
In summary, this study establishes persistent chromatin alterations as a key mechanistic link between DNA damage repair and epigenetic dysfunction, challenging the traditional paradigm that efficient DNA repair ensures full functional restoration. Future research addressing unresolved questions regarding the reversibility, locus specificity, and physiological relevance of these alterations will further solidify the role of chromatin fatigue in aging and cancer and could potentially inform new therapeutic strategies for healthy longevity.
Acknowledgements
The authors declare that the Deepseek-V3 was used solely for language polishing during the manuscript preparation process. All research content, including study design, interpretations, and figures was not generated using AI tools.
Authors contribution
Chen Y: Conceptualization, writing-review & editing, funding acquisition.
Chen L: Writing-original draft, writing-review & editing.
Mao Z: Funding acquisition.
Conflicts of interest
Zhiyong Mao is an Associate Editor, and Yu Chen is a Youth Editorial Board Member of Ageing and Cancer Research & Treatment. The other author declares no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
This work was funded by National Natural Science Foundation of China (NSFC) (Grant Nos. 82522035 and 32470796 to Yu Chen), National Key R&D Program of China (Grant No. 2025YFA1805500 to Yu Chen), the Fundamental Research Funds for the Central Universities (Grant No. 22120250374), and Peak Disciplines (Type IV) of Institutions of Higher Learning in Shanghai (This program does not have a grant number).
Copyright
© The Author(s) 2026.
References
-
2. Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326(5950):289-293.[DOI]
-
4. White RR, Vijg J. Do DNA double-strand breaks drive aging? Mol Cell. 2016;63(5):729-738.[DOI]
-
5. Chen Y, Geng A, Zhang W, Qian Z, Wan X, Jiang Y, et al. Fight to the bitter end: DNA repair and aging. Ageing Res Rev. 2020;64:101154.[DOI]
-
6. Xiang W, Wang S, Pan Y, Cong X, Lu J, Zhang Y. Cytoplasmic chromatin fragments: Divergent roles in senescence and cancer. Ageing Cancer Res Treat. 2026;3(2):202603.[DOI]
-
7. Yang JH, Hayano M, Griffin PT, Amorim JA, Bonkowski MS, Apostolides JK, et al. Loss of epigenetic information as a cause of mammalian aging. Cell. 2023;186(2):305-326.[DOI]
-
12. Li YY, Tay FR. The epigenetic rejuvenation promise: Partial reprogramming as a therapeutic strategy for aging and disease. Ageing Res Rev. 2026;115:103009.[DOI]
-
14. Hao Y, Han K, Wang T, Yu J, Ding H, Dao F. Exploring the potential of epigenetic clocks in aging research. Methods. 2024;231:37-44.[DOI]
-
15. Zheng Z, Li J, Liu T, Fan Y, Zhai QC, Xiong M, et al. DNA methylation clocks for estimating biological age in Chinese cohorts. Protein Cell. 2024;15(8):575-593.[DOI]
-
16. Tang Y, Zhang D, Wang K, Mao Z, Chen Y. Decoding DNA repair regulation across human lifespan variability. Ageing Res Rev. 2025;111:102833.[DOI]
-
17. Li Z, Zhang W, Chen Y, Guo W, Zhang J, Tang H, et al. Impaired DNA double-strand break repair contributes to the age-associated rise of genomic instability in humans. Cell Death Differ. 2016;23(11):1765-1777.[DOI]
-
18. Chen Y, Zhen Z, Chen L, Wang H, Wang X, Sun X, et al. Androgen signaling stabilizes genomes to counteract senescence by promoting XRCC4 transcription. EMBO Rep. 2023;24(12):e56984.[DOI]
-
19. Chen Y, Chen Z, Wang H, Cui Z, Li KL, Song Z, et al. A cGAS-mediated mechanism in naked mole-rats potentiates DNA repair and delays aging. Science. 2025;390(6769):eadp5056.[DOI]
-
20. Jiang R, Wang H, Zhang W, Li J, Huang S, Chen L, et al. DHT ameliorates cardiac aging in progeroid mice by XRCC4-mediated genome stabilization. Mech Ageing Dev. 2026;229:112141.[DOI]
-
21. Zhang W, Yang J, Chen Y, Xue R, Mao Z, Lu W, et al. Lycorine hydrochloride suppresses stress-induced premature cellular senescence by stabilizing the genome of human cells. Aging Cell. 2021;20(2):e13307.[DOI]
-
22. Tian X, Firsanov D, Zhang Z, Cheng Y, Luo L, Tombline G, et al. SIRT6 is responsible for more efficient DNA double-strand break repair in long-lived species. Cell. 2019;177(3):622-638.[DOI]
-
23. Firsanov D, Zacher M, Tian X, Sformo TL, Zhao Y, Tombline G, et al. Evidence for improved DNA repair in the long-lived bowhead whale. Nature. 2025;648(8094):717-725.[DOI]
-
26. Jiang M, Jia K, Wang L, Li W, Chen B, Liu Y, et al. Alterations of DNA damage repair in cancer: From mechanisms to applications. Ann Transl Med. 2020;8(24):1685.[DOI]
-
27. Rembiałkowska N, Rekiel K, Urbanowicz P, Mamala M, Marczuk K, Wojtaszek M, et al. Epigenetic dysregulation in cancer: Implications for gene expression and DNA repair-associated pathways. Int J Mol Sci. 2025;26(13):6531.[DOI]
Copyright
© The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Publisher’s Note
Science Exploration remains a neutral stance on jurisdictional claims in published
maps
and institutional affiliations. The views expressed in this article are solely those
of
the author(s) and do not reflect the opinions of the Editors or the publisher.
Share And Cite



Science Exploration Style
Chen L, Mao Z, Chen Y. Chromatin fatigue: An epigenetic legacy of DNA repair. Ageing Cancer Res Treat. 2026;3:202610. https://doi.org/10.70401/acrt.2026.0020
Tips
Copy completed.
Submit a Manuscript
Author Instructions
Cite this Article
Article Metrics
0
View
0
Download
Cited
Article Updates

Science Exploration Style
Chen L, Mao Z, Chen Y. Chromatin fatigue: An epigenetic legacy of DNA repair. Ageing Cancer Res Treat. 2026;3:202610. https://doi.org/10.70401/acrt.2026.0020
copy
Share Link
copy