cGAS-STING pathway drives cellular senescence and inflammaging
Yali Chen

Kun Chen
*
*Correspondence to:
Kun Chen, State Key Laboratory of Cardiovascular Diseases and Medical Innovation Center, Shanghai East Hospital, School of Life Sciences and Technology, Tongji University, Shanghai 200127, China.
E-mail: chenk@tongji.edu.cn
Ageing Cancer Res Treat. 2026;3:202615. 10.70401/acrt.2026.0026
Received: March 31, 2026Accepted: June 12, 2026Published: June 15, 2026
This article belongs to the Special lssue Inflammation in Aging and Tumorigenesis
Abstract
Cellular senescence is a cell fate triggered by diverse endogenous and exogenous stresses, including DNA damage, telomere dysfunction, and metabolic dysregulation. It is characterized by irreversible cell cycle arrest and a hypersecretory state known as the
Keywords
Cellular senescence, inflammaging, cGAS, STING, DNA damage
1. Introduction
Organisms experience a progressive decline in the function of multiple organs and an increased susceptibility to various diseases throughout their lifespan. This functional deterioration is referred to as aging, a common, complex, and natural biological process. Aging is increasingly characterized by systemic chronic inflammation, coupled with cellular senescence, inflammaging[1]. Inflammaging refers to a state of mild, sterile, chronic damage driven by elevated levels of pro-inflammatory mediators[2]. In parallel, cellular senescence is characterized by cell cycle arrest accompanied by distinct molecular alterations in damaged or aged cells[3,4]. Currently, senescence, described as a relatively quiescent, non-proliferative, and irreversible cellular state, is recognized as a core hallmark of aging[4]. Senescent cells generally increase with the aging process, which is closely linked to the decline of physiological functions and the pathogenesis of age-associated disorders.
As a hallmark of aging, cellular senescence, its progression and accumulation significantly contributes to organ damage, tumor suppression and inflammatory diseases. Multiple factors have been identified as triggers for cellular senescence, including DNA damage, telomere shortening, oncogene activation, oxidative stress, and chromatin rearrangement[3-6]. Among these, DNA damage serves as a pivotal driver of cellular senescence and inflammation, plays a critical role in age-related diseases. It is well established that DNA damage induces the expression of type I interferon and inflammatory factors. Recent mechanistic insights further reveal the connection between DNA damage and either senescence or inflammation is mediated by cytoplasmic DNA sensing pathways[7,8].
DNA sensing pathways constitute a critical host defense mechanism responsible for detecting aberrant nucleic acids and initiating type I interferon and inflammatory responses[8-10]. Cyclic guanosine monophosphate (GMP)-adenosine monophosphate (AMP) synthase (cGAS), serves as a core intracellular DNA sensor for detecting foreign and aberrant self-DNA, thereby triggering powerful innate immune responses. Upon activation after binding to DNA, cGAS sythethesizes the second messenger cyclic GMP-AMP (cGAMP), which activates the adaptor protein stimulator of interferon genes (STING). STING further triggers a signaling cascade, leading to the production of immune and inflammatory mediators, including type I and III interferons[8,11,12]. Given its capacity to sense cytoplasmic DNA arising from genomic instability or mitochondrial damage, the cGAS-STING pathway is closely related to cellular senescence. The activated cGAS-STING pathway in senescent cells can also induce the senescence-associated secretory phenotype (SASP), leading to chronic inflammation and subsequent cellular senescence[9,13]. In this review, we primarily reviewed the regulatory mechanisms of cGAS-STING in cellular senescence and inflammaging. We further emphasize its potential as a therapeutic target for age-related human diseases, providing a comprehensive summary of cGAS-STING inhibitors currently under investigation for conditions, including neurodegenerative and cardiovascular diseases.
2. Characteristics of Cellular Senescence
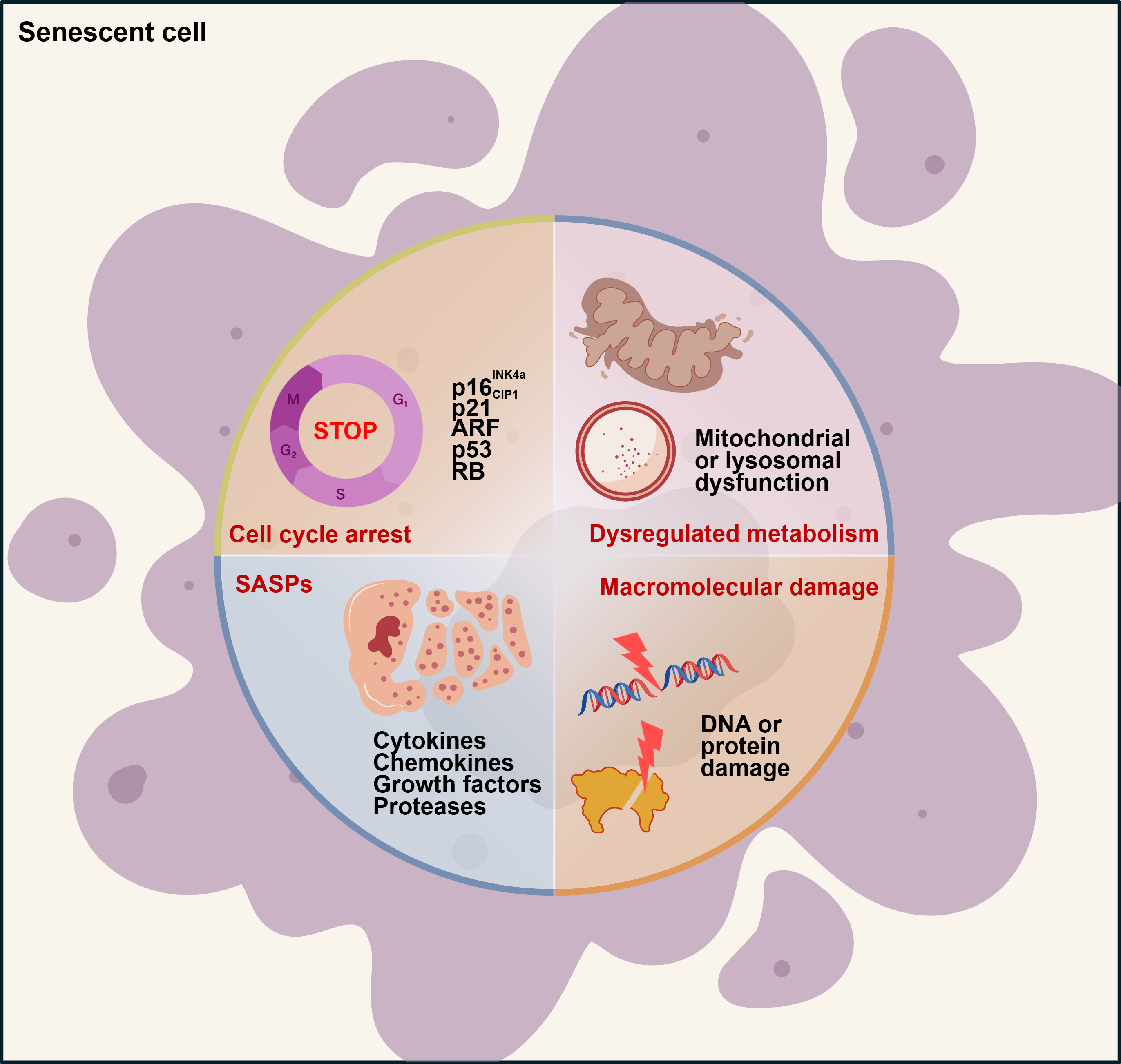
Cellular senescence is a nondividing state caused by various stress stimuli and certain physiological processes, exhibiting distinct phenotypes and hallmarks. The hallmarks of cellular senescence encompass irreversible cell cycle arrest, the SASP, metabolic disorders, and macromolecular damage[4,14,15]. Although these characteristics are often interconnected, no single marker is sufficient to definitively identify the specific state of cellular senescence. Therefore, a combinatorial approach utilizing multiple assays and markers is essential to accurately characterize specific senescence phenotypes (Figure 1).
{kind=link}
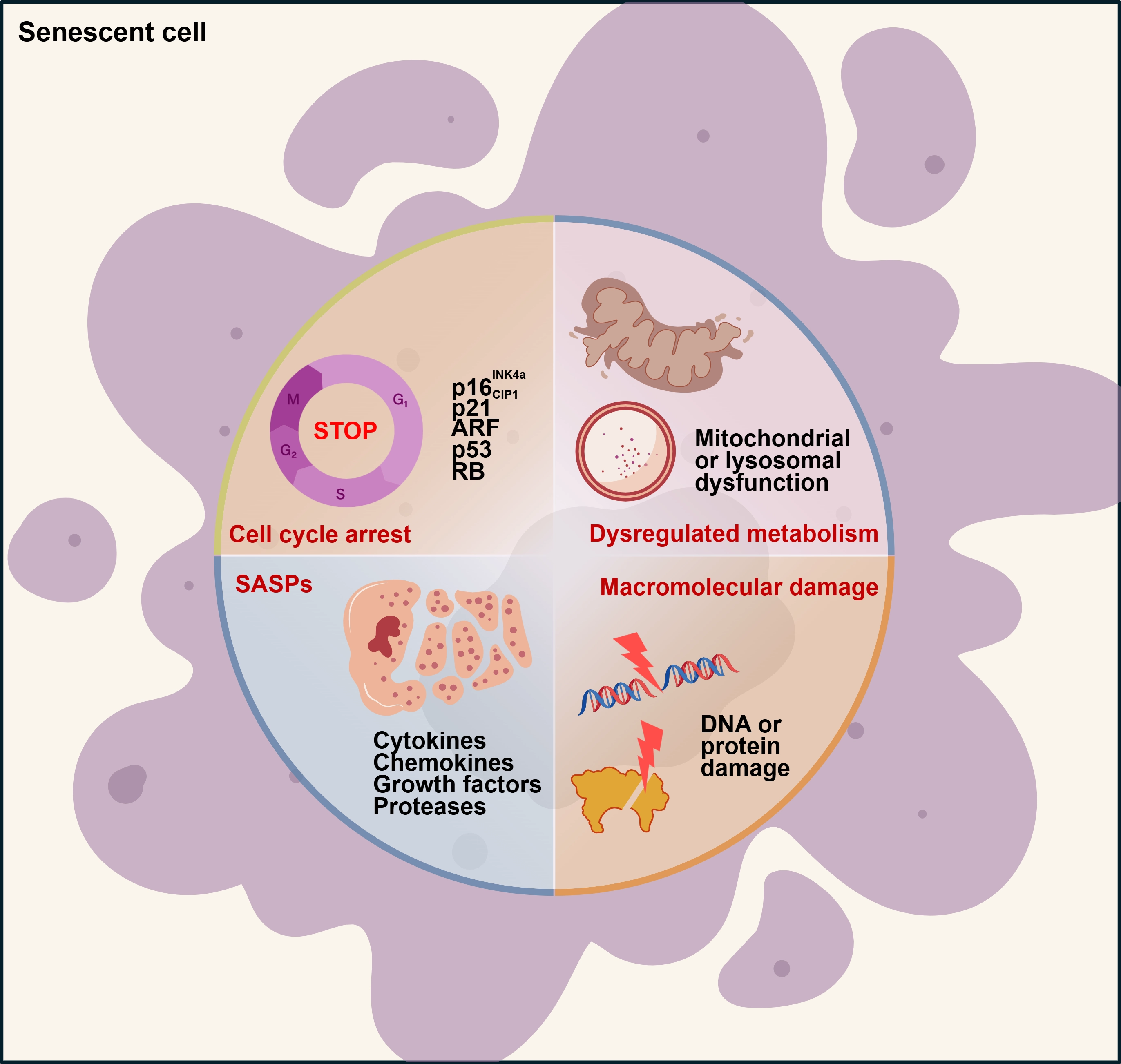
Figure 1. The characteristics of cellular senescence. Senescent cells exhibit four interdependent characteristics: irreversible cell cycle arrest, the SASP, macromolecular damage, and metabolic dysregulation. ARF: alternate reading frame; RB: retinoblastoma; SASP: senescence-associated secretory phenotype.
2.1 Irreversible cell cycle arrest
Senescence-associated cell cycle arrest is defined as a durable and irreversible withdrawal from the cell cycle, typically triggered by harmful stimuli or aberrant proliferation. While cyclin-dependent kinases (CDKs) phosphorylation participates in cell cycle progression, cell cycle arrest is accompanied by the upregulation of CDK inhibitors. CDKN2A (p16INK4a, hereafter p16) and CDKN1A
2.2 Senescence-associated secretory phenotype
During cellular senescence, cells secrete a variety of factors, mainly categorized into pro-inflammatory cytokines (e.g., interleukins), chemokines (e.g., CXCL chemokines), growth factors (e.g., VEGF, TGF-β), proteases (such as matrix metalloproteinases and plasminogen activators), receptors, and other mediators. This characteristic is termed the SASP[4]. The SASP can exacerbate cellular senescence through autocrine and paracrine pathways. Senescent cells can secrete SASP cytokines, and cytokine signaling in turn induces the expression of cell cycle inhibitors, including p16, thereby reinforcing cell cycle arrest and senescence in an autocrine manner. Simultaneously, secreted SASP factors can also trigger neighboring normal cells senescence in a paracrine manner, which is termed the bystander effect[9]. Thus, the SASP accounts for certain harmful and pro-aging effects of senescent cells. However, senescence inducers, cell types, or the duration of stimuli (acute or chronic) can endow SASP components with distinct biological activities, such as wound healing and regeneration, tumor development, and inflammaging[5].
2.3 Macromolecular damage
Senescence can be induced by diverse stressors, including radiation, chemotherapeutic agents, oxidative stress, and telomerase dysfunction. These stimuli lead to abnormal accumulation of damaged macromolecules, such as DNA damage, protein damage, and lipid damage[16]. During cellular senescence, DNA damage occurs alongside telomere shortening and cell cycle arrest. Additionally, senescent cells also exhibit significant damage to proteins and lipids. Protein damage is primarily induced by reactive oxygen species (ROS), which oxidize methionine and cysteine residues, thereby altering protein folding and function[17]. ROS can induce lipid damage and promote lipid deposition during cellular senescence[4]. Among these various forms of macromolecular damage, DNA damage is recognized as a defining phenotype of cellular senescence.
2.4 Metabolic alterations
Metabolic alterations during cellular senescence manifest as disruptions in molecular and protein homeostasis, accompanied by mitochondrial or lysosomal dysfunction. Senescent cells exhibit significant perturbations in mitochondrial function, dynamics, and morphology. Specifically, mitochondrial dysfunction in these cells includes defects in electron transfer chain (ETC) activity, complex I assembly, mitochondrial dynamics (fission and fusion), and impairment of the tricarboxylic acid (TCA) cycle[18,19]. For instance, ETC inhibtion induces senescence in an IL-1β-dependent manner[19]. Additionally, in senescent cells, metabolic changes also include the accumulation of senescence-associated β-galactosidase (SA-β-gal), a marker of increased lysosomal mass[14]. Dysregulation of lysosomal function results in increased production of ROS in mitochondria, which in turn further compromises lysosomal integrity. This vicious cycle between lysosomes and mitochondria remarkably exacerbates the senescence phenotype[20].
3. Mechanisms Involved in Cellular Senescence
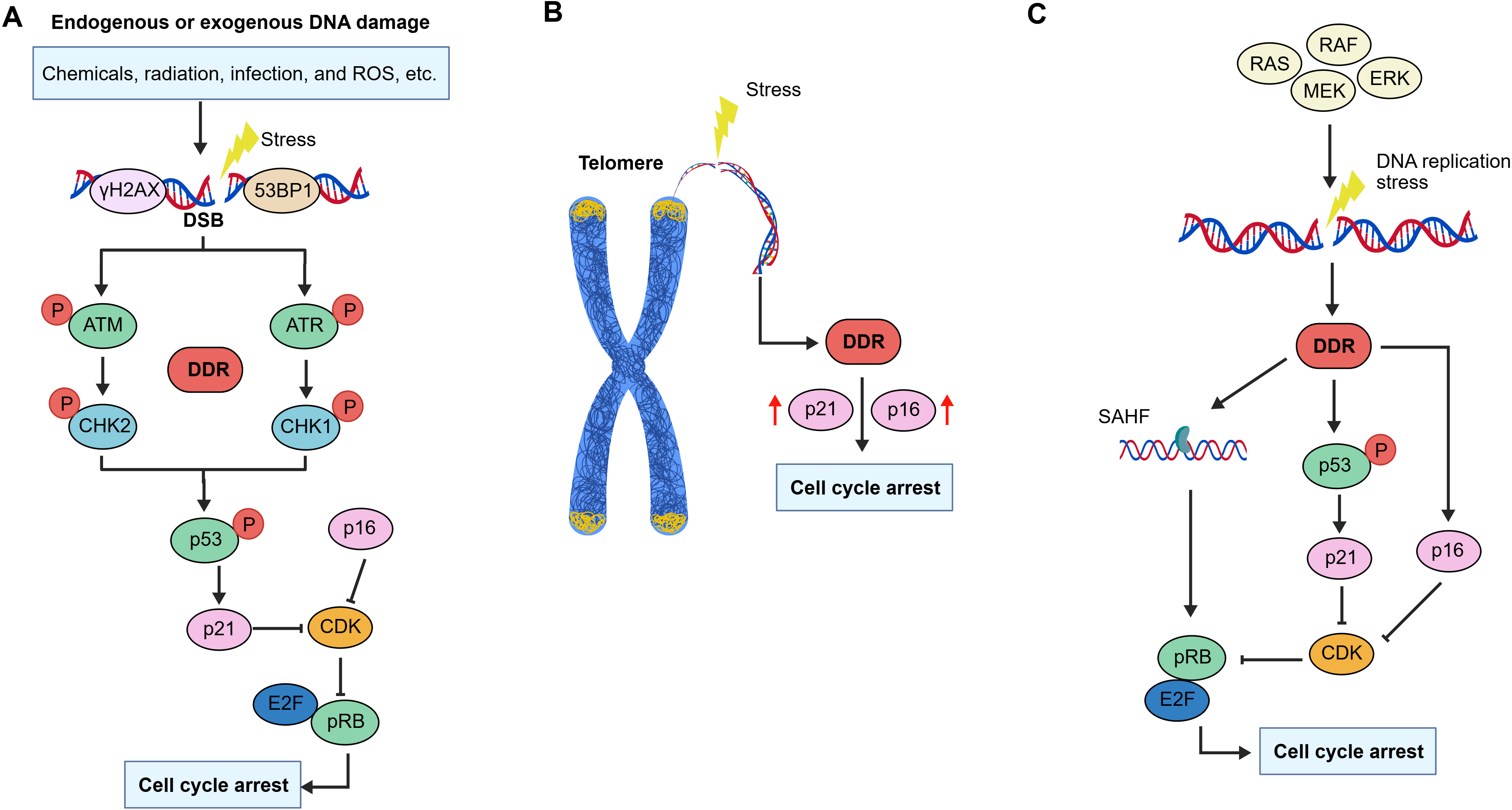
Cellular senescence is driven by a series of interdependent mechanisms, including DNA damage, stress signaling, organelle dysfunction, and chromatin rearrangement (Figure 2).
{kind=link}
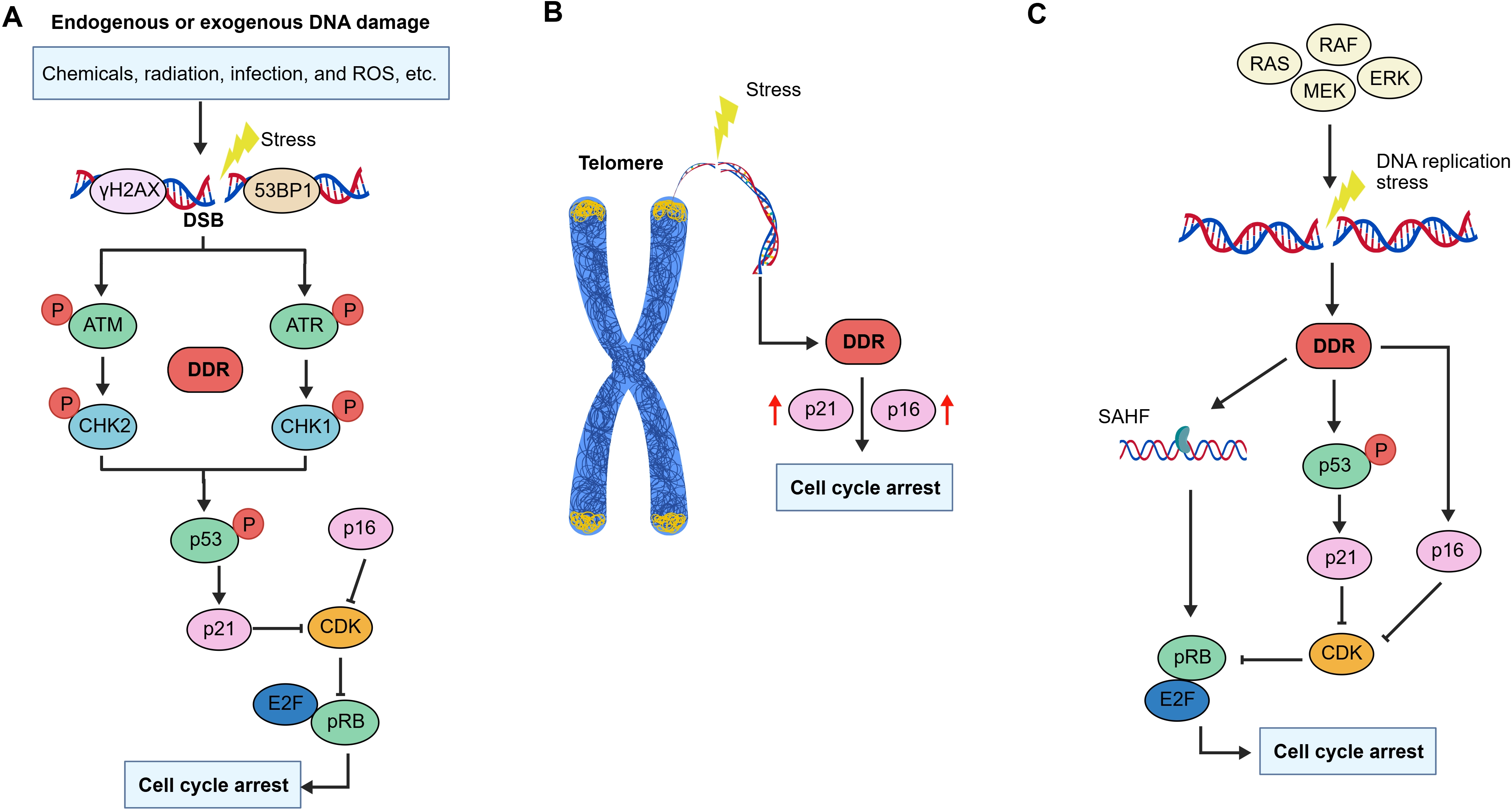
Figure 2. Triggers of cellular senescence. (A) Endogenous or exogenous stress-induced damage causes DSBs and activates the DDR, leading to the phosphorylation of histone H2AX (γ-H2AX) and the recruitment of 53BP1. The recruitment of γ-H2AX and 53BP1 at DSB sites activates the kinases ATM and ATR. ATM and ATR undergo phosphorylation and subsequently activate CHK2 and CHK1, respectively, further activating p53 and its effector molecules to induce cell cycle arrest and cellular senescence; (B) Telomere dysfunction caused by shortened or damaged telomere promote DDR activation, leading to the upregulation of p21 and p16. This ultimately results in cell cycle arrest and cellular senescence; (C) Excessive activation of oncogenes induces cellular senescence and phenotypic changes. Activation of the RAS/MAPK pathway, which includes effector molecules such as RAS, RAF, MEK, and ERK, induces DNA damage and activate DDR. Subsequent activation of p53 or formation of SAHF further induces cell cycle arrest, triggering OIS. DSBs: DNA double-strand breaks; DDR: DNA damage response; 53BP1: p53-binding protein 1; ATM: ataxia-telangiectasia mutated;
3.1 DNA damage-induced senescence
DNA damage, a hallmark of genomic instability, can result in nuclear DNA breaks and activate the DNA damage response (DDR) pathway, thereby promoting cellular senescence. In senescent cells, the DDR halts cell cycle progression, preventing the inheritance of aberrant genetic information by offspring. Key DDR proteins and chromatin modifiers accumulate at sites of DNA damage, these include phosphorylated histone H2AX (γ-H2AX), p53-binding protein 1 (53BP1), and the kinases ataxia-telangiectasia mutated (ATM) and ataxia-telangiectasia and RAD3-related (ATR)[6]. Upon activation, ATM and ATR phosphorylate various substrates, notably the tumor suppressor protein p53. This activation coordinates the expression of genes essential for cell cycle arrest, such as p16 and
In addition, DNA damage can cause telomere shortening in senescent cells. Telomeres are cap-like structures located at the ends of linear chromosomes, composed of repetitive DNA sequences (such as TTAGGG in mammals) and the protective shelterin protein complexes. These structures prevent chromosome ends from being recognized and suppressing the activation of DDR[22,23]. Telomeric damage can induce single-stranded DNA breaks, internal DNA loop structures, and the dissociation of shelterin protein complexes, ultimately leading to cellular senescence[24]. Moreover, DNA replication causes telomeres to gradually shorten. Consequently, telomere dysfunction triggers replicative senescence when telomeres reach a critical length[25]. At this threshold, telomeres fail to recruit sufficient shelterin protein complexes and are misrecognized as exposed DNA ends. This exposure thereby activates the DDR pathway and upregulates the expression of senescence-related proteins such as p21 and p16, initiating cellular senescence[26,27].
Compared with nuclear DNA, mitochondrial DNA (mtDNA) is more susceptible to damage, and such damage tends to persist for a longer period[28]. Mitochondrial ROS oxidizes mtDNA to form oxidized mtDNA (Ox-mtDNA). Ox-mtDNA can release from mitochondria through mitochondrial permeability transition pores (mPTP) and voltage-dependent anion channels (VDAC)-dependent channels, subsequently triggering NLRP3 inflammasome activation[29]. Furthermore, improper modifications of mtDNA also contribute to senescence. Recent studies indicate that dysregulated m6A modifcation of mtDNA inappropriately alters mtDNA copy number and transcription levels, impairs oxidative phosphorylation (OXPHOS), increases oxidative stress, and shortens the lifespan of Candida albicans[30].
3.2 Mitochondrial dysfunction and cellular senescence
The accumulation of dysfunctional mitochondria is a key contributor to the elevated oxidative stress observed in senescent cells. Consequently, these cells exhibit significant alterations in mitochondrial mass, membrane potential, and overall morphology[31]. During cellular senescence, a process called minority mitochondrial outer membrane permeabilization (miMOMP) occurs in a subset of mitochondria. Inhibiting MOMP can suppress the expression of inflammatory factors and prolong the lifespan of aged mice[32]. Autophagy serves as a critical clearance pathway for organelles, specifically damaged mitochondria via mitophagy[33]. Mitophagy can maintain muscle function during aging by preventing the senescence of satellite cells[34]. Impaired mitophagy may result in the accumulation of dysfunctional mitochondria and trigger senescence in fibroblasts[18]. Moreover, senescent cells exhibit reduced mitophagy levels, which leads to defects in the mitochondrial network and contributes to metabolic dysfunction during aging[31].
3.3 Chromatin reorganization
Cellular senescence is related to large-scale chromatin rearrangement, with the most prominent changes being the loss of heterochromatin. In normal cells, heterochromatin is enriched with repressive histone modifications, such as dimethylated and trimethylated histone H3 Lys9 (H3K9me2 and H3K9me3) or Polycomb-mediated H3K27me3[35]. These modificatons, along with high levels of DNA methylation, maintain genomic stability by mediating the transcriptional repression of retrotransposons, such as long interspersed element (LINE-1) and endogenous retroviruses (ERVs). However, in senescent cells, the erosion of heterochromatin leads to the derepression of retrotransposons and the reverse transcription of complementary deoxyribonucleic acid (cDNA), and its translocation into the cytoplasm, ultimately triggering cellular senescence[35].
Notably, the loss of heterochromatin results in the loss of gene silencing and drives the senescence process. In Saccharomyces cerevisiae, heterochromatin loss is associated with interrupted transcriptional gene silencing and replicative senescence[36]. Similar observations of heterochromatin loss were documented in Cockayne syndrome. Deficiency in Cockayne syndrome group B reduces the level of the SETDB1, the methyltransferase required for catalyzing H3K9me3 modification, which induces cellular senescence[37]. Increasing evidence suggests that aging is associated with disruption of heterochromatin homeostasis rather than a simple reduction or increase in heterochromatin content. For instance, KDM4B, a H3K9me3 demethylase, epigenetically coordinates β-catenin/
3.4 Oncogene activation-induced senescence
In normal cells, the hyperactivation of oncogenes induces DNA replication stress, which triggers the DDR pathway, leading to cell cycle arrest and oncogene-induced senescence (OIS). Recent studies have demonstrated that over 50 oncogenes and tumor suppressor genes can induce senescence, including Ras and its downstream effectors Raf and MEK, p53, MYC, and loss of PTEN. Numerous evidence indicates that chromatin reorganization plays a pivotal role in OIS, notably through the formation of
4. The cGAS-STING Pathway in Cellular Senescence
The presence of nuclear DNA damage or the leakage of chromatin fragments into the cytoplasm is a well-established driver of senescence, mainly manifesting as DNA double-strand breaks (DSBs) or cytosolic chromatin fragments (CCFs). Under physiological conditions, genomic DNA is compartmentalized within the nucleus and mitochondria. However, in response to stress signals or infections, nuclear or mitochondrial DNA can leak into the cytoplasm, where it is sensed by specific DNA sensors, initiating downstream signalling cascades. cGAS, a pattern recognition receptor (PRR) of the innate immune system, specifically recognizes dsDNA. In addition to sensing microbial DNA during infection, cGAS is also activated by endogenous DNA, including aberrant genomic nucleic acid products, extranuclear chromatin, and mtDNA released into the cytosol[8,41,42]. In resting cells, cGAS normally exists in an inactive form. Upon binding to DNA, it undergoes conformational changes to become active, catalyzing the synthesis of the second messenger cGAMP from adenosine triphosphate (ATP) and guanosine triphosphate (GTP). This cGAMP is subsequently detected by the cyclic-dinucleotide sensor STING, a dimeric transmembrane protein located on the endoplasmic reticulum (ER). The binding of cGAMP activates STING, which then translocates to the Golgi apparatus. At the Golgi, STING recruits and activates TANK-binding kinase 1 (TBK1) which phosphorylates interferon regulatory factor 3 (IRF3), thereby initiating the type I interferon response. In addition to type I interferon signaling, STING can also activate nuclear factor kappa B (NF-κB) signaling, triggering inflammatory gene expression[8,12].
Recent studies have highlighted the critical role of the cGAS-STING axis in regulating autoimmunity, inflammatory disease,
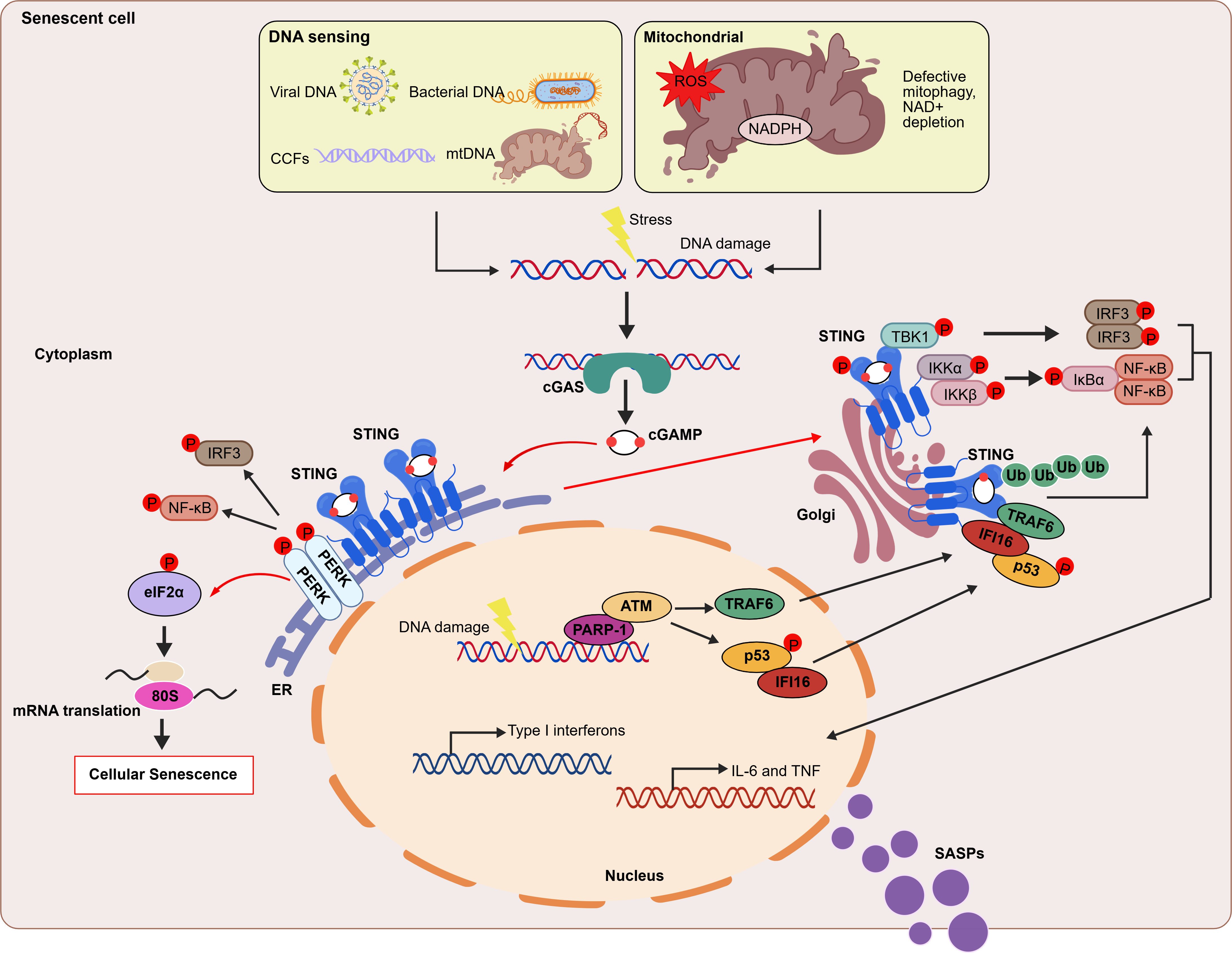{kind=link}
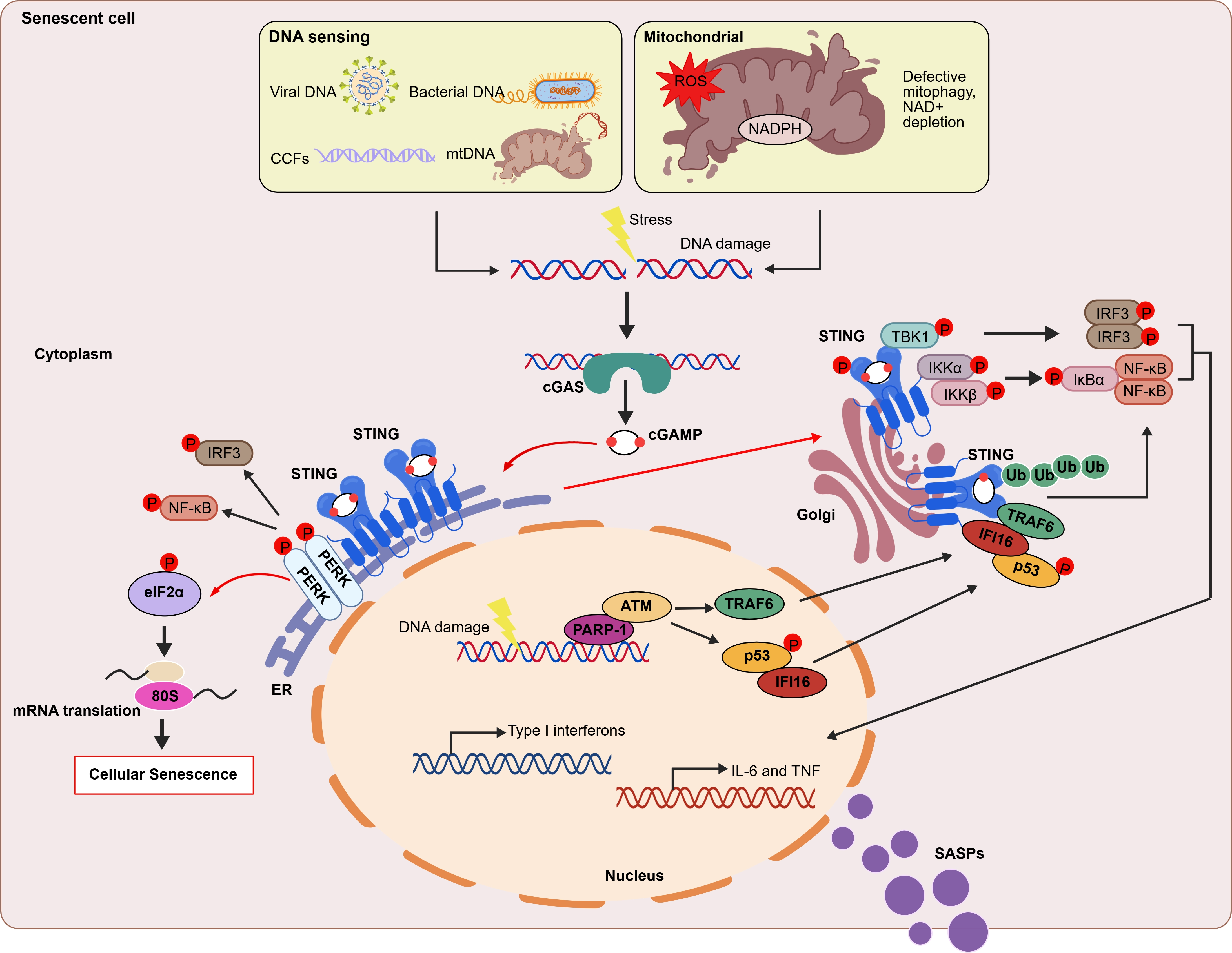
Figure 3. Regulatory mechanisms of the cGAS-STING pathway in cellular senescence. In senescent cell, cGAS senses exogenous DNA (such as bacteria or viruses) and cytosolic self-DNA (DNA damage causing genomic disorder or mitochondrial dysfunction). Upon binding DNA, cGAS synthesizes the second messenger cGAMP. This molecule binds to and activates STING, which is located on the ER membrane, triggering STING translocation to the Golgi apparatus. At the Golgi apparatus, STING recruits and activates TBK1 which in turn phosphorylates IRF3, initiating type I interferon responses. Additionally, STING recruits the IKK kinase and activates NF-κB signaling, triggering the inflammatory response[7]. In the non-canonical STING pathway, ATM, PARP1, p53, and the E3 ubiquitin ligase TRAF6 mediate STING activation. TRAF6 catalyzes the formation of K63-linked ubiquitin chains on STING, thereby activating both the NF-κB pathway and the STING-dependent gene expression program[44]. Moreover, upon cGAMP binding, STING at the ER binds to and directly activates the ER-resident kinase PERK. This triggers either the PERK-eukaryotic initiation factor 2α (eIF2α) signaling axis or the IRF3 and NF-κB pathways[45]. cGAMP: 2′,3′ cyclic GMP-AMP; ER: endoplasmic reticulum; PARP1: poly(ADP-ribose) polymerase 1; PERK: PKR-like ER kinase; NF-κB: nuclear factor kappa B; cGAS: cyclic GMP–AMP synthase; STING: stimulator of interferon genes; TBK1: TANK-binding kinase 1; IRF3: interferon regulatory factor 3;
4.1 DNA damage triggers cGAS-STING activation to drive senescence
Genomic instability is a significant cause of age-related diseases and cancer, severely impacting human health and lifespan. DDR primarily functions to maintain genomic integrity and determine cell fate. A crucial activator of DDR is DSBs. Homologous recombination (HR) repair of DNA is one of the DSB repair pathways and is closely associated with the alleviation of aging in both mice and humans. In a recent study published in Science, Mao’s group reported that cGAS from the naked mole-rat (NMR) promotes DNA repair, thereby stabilizing the genome and potentially extending lifespan. The authors identified that four amino acids within the C-terminal domain of NMR cGAS can facilitate HR repair by mediating the FANCI-dependent recruitment of RAD50. Additionally, NMR cGAS not only significantly reduces cellular senescence but also enhances climbing performance in aged Drosophila. Notably, engineering these four specific amino acid residues of NMR cGAS into human or Drosophila, cGAS eliminates the inhibition of HR repair, resulting in prolonged lifespan of Drosophila or human cells[46]. Moreover, DNA damage can also induce nuclear envelope rupture, potentially leading to the localization of cGAS in micronuclei. Under normal conditions, the nuclear envelope is typically maintained and repaired by the endosomal sorting complexes required for transport (ESCRT-III) machinery[47,48]. Recent studies have shown that DSBs during the cell cycle can lead to the formation of micronuclei during mitosis. Prior to the activation of inflammatory signals, micronuclei serve as a reservoir for cGAS. As mitosis progresses, the relocation of cGAS to micronuclei initiates interferon signaling[49]. In mouse stromal cells, Hippo signalling co-activators YAP and TAZ maintain nuclear envelope integrity by transcriptional regulation of lamin B1 and ACTR2. The deletion of YAP/TAZ triggers cGAS activation on genomic DNA, thereby promoting SASP production and driving cellular senescence[50].
Increasing evidence indicates that CCFs activate the cGAS-STING pathway to produce SASP, leading to senescence in primary IMR90 and BJ fibroblasts, without significantly inducing type I IFN expression[13]. The failure of interferon induction may be due to the activation of p38 MAP-kinase (MAPK) by senescent human fibroblasts, which can inhibit STING mediated interferon induction[51]. However, other studies in MEFs demonstrate that CCFs can induce the production of type I IFN via mediating cGAS-STING signaling during senescence[9]. This discrepancy is likely due to differences in STING activation and function. Generally, upon binding to DNA, cGAS synthesizes the second messenger cGAMP, which binds to and activates STING. Activated STING proteins oligomerize and recruit their downstream effector TBK1 via a conserved PxPLRT/SD motif (where x denotes any amino acid) located in the C-terminal tail (CTT) of STING, thereby promoting trans phosphorylation of TBK1. Phosphorylated TBK1 then phosphorylates the STING CTT at Ser358 and Ser366. This CTT phosphorylation creates a docking site for IRF3, which is subsequently phosphorylated by TBK1. Phosphorylated IRF3 undergoes oligomerization and translocates to the nucleus, where it drives the transcription of type I interferons and interferon-stimulated genes[12,52]. Therefore, the IFN signaling activated by cGAS-STING depends on STING CTT function. However, some studies have found that cGAS-STING-dependent NF-κB activation is independent of STING CTT function[53,54], and may instead be governed by a signal from within the C-terminal ligand-binding domain (LBD) of oligomerized STING. In addition, NF-κB activation depends on TBK1 and IKKε[55]. Consistent with these findings, MEFs deficient in cGAS exhibit immortality, as cGAS deficiency can abrogate the SASP[56]. Overall, these findings suggest that cGAS acts as a key regulator of lifespan by modulating the DNA damage response pathways and chromosomal instability. However, the precise role of nuclear cGAS remains to be fully elucidated.
4.2 Mitochondria dysfunction drives cGAS-STING activation in senescence
Mitochondria are unique organelles with their own genome, the integrity of which is essential for mitochondrial function. Mitochondrial damage or oxidative stress can lead to the release of mtDNA into the cytoplasm[57]. It is well-established that the accumulation of mtDNA in the cytosol triggers cGAS-STING, acting as a key driver for cellular senescence. Aberrant activation of the cGAS-STING pathway by mtDNA is implicated in various senescence-related diseases, including neurodegenerative diseases, kidney inflammation, and cardiovascular diseases[58-60]. During cellular senescence, the activity of the ribonucleotide reductase (RNR) significantly declines. This enzyme is responsible for converting ribonucleotide triphosphates (rNTPs) into deoxyribonucleotide triphosphates (dNTPs). This enzymatic deficiency leads to a nucleotide imbalance and increased ribonucleotide incorporation into mtDNA by DNA polymerase. Such genomic instability in mitochondria further activates cGAS-STING pathway to initiate downstream expression of pro-inflammatory cytokines[59]. However, in prostate cancer, senescent cells release mtDNA into both the cytosolic and extracellular spaces through VDACs, which further triggers the cGAS-STING-NF-κB pathway and enhances the immunosuppressive function of myeloid-derived suppressor cells (MDSCs), contributing to tumor progression[60]. In addition, during apoptosis, the activation of pro-apoptotic proteins BAX and BAK induce mitochondrial outer membrane permeabilization, which leads to the exposure of mtDNA to cGAS[32. Furthermore, under conditions of oxidative stress, mtDNA escapes into cytosol through mitochondrial pores formed by VDAC oligomerization, subsequently triggering an IFN-I response and systemic inflammation[61]. Iron overload induces mitochondrial dysfunction, causing mtDNA release from damaged mitochondria into the cytoplasm. This cytosolic mtDNA activates the cGAS-STING pathway, promoting SASP and cellular senescence[62].
Additionally, defects in mitophagy can induce senescence. Preclinical and clinical studies have demonstrated that impaired mitophagy contributes to the pathogenesis of various age-related diseases[4,63]. Recent insights indicate that mitophagy dysfunction results in the accumulation of damaged mitochondria. This, in turn, activates the cGAS-STING pathway, leading to the production of pro-inflammatory cytokines and exacerbating neuroinflammation[7]. For instance, the primary cause of Parkinson’s disease is frequently due to the functional loss or mutation of PINK1 or Parkin, which are typically required for clearing damaged mitochondria via mitophagy. The human T-cell leukemia virus type 1 (HTLV-1) Tax regulatory protein is recruited to damaged mitochondria through interaction with the IKK regulatory subunit NEMO. This interaction induces PINK1-Parkin-dependent mitophagy, thereby alleviating the activation of cGAS-STING pathway and inhibiting type I interferon production[64,65].
Moreover, mitochondrial dysfunction in senescent cells is accompanied by reduced levels of nicotinamide adenine dinucleotide (NAD+) and ATP[66,67]. In models of doxorubicin-induced cardiotoxicity, it has been found that doxorubicin activated the cGAS-STING pathway and its downstream transcription factor, IRF3, which directly induced CD38 expression. In cardiac endothelial cells, the cGAS-STING-CD38 axis enhances intracellular NAD glycohydrolase (NADase) activity, resulting in reduced NAD levels and subsequent mitochondrial dysfunction[68]. NAD+ supplementation has been shown to inhibit the cGAS-STING/NF-κB signaling pathway, reduce oxidative stress and mitochondrial damage in microglia, and ultimately alleviate proinflammation responses induced by chronic sleep restriction[69]. In summary, these findings underscore that the leakage of mtDNA is a critical cytosolic DNA source that can be sensed by cGAS, linking mitochondrial homeostasis with the onset of cellular senescence.
4.3 Heterochromatin loss triggers cGAS-STING activation in senescence
During cellular senescence, chromatin undergoes rearrangement and degeneration[70]. In aging cells, heterochromatin loss leads to the derepression of retrotransposons and activates the cGAS-STING pathway. For example, SIRT7 deficiency reduces H3K9me3 levels at LINE-1 loci, leading to increased chromatin accessibility and LINE-1 activation in human MSCs. This triggers the phosphorylation of TBK1 and IRF3, as well as the activation of NF-κB signaling, resulting in interferon production, SASP, and premature aging[71]. Additionally, the activity of ERVs plays a regulatory role in senescence. The loss of H3K9me3 and DNA methylation increases the expression of ERV cDNA, which further enhances cGAS-STING activity, inflammation, and senescence in human fibroblasts and neurons[72]. CRISPR-mediated activation of ERVs in proliferating cells is sufficient to induce various senescence hallmarks, including increased SA-β-gal activity, impaired clonal expansion, p16 upregulation, lamin B1 downregulation, increased levels of phosphorylated TBK1, IRF3, and NF-κB, and enhanced SASP[73].
However, in certain contexts, cellular senescence is associated with elevated levels of heterochromatin. OIS results in the appearance of irregular heterochromatin clusters known as SAHFs within the nuclei of senescent cells[74]. During OIS, DNA methyltransferase 1 (DNMT1)-mediated DNA methylation induces SAHFs formation by promoting HMGA2 expression. Additionally, in senescent mouse MSCs lacking KDM4B, H3K9me3 levels increase concomitantly with SAHF formation and the repression of stem cell signature genes[38]. However, it is currently unclear whether SAHF can activate cGAS-STING signaling.
4.4 Non-canonical STING signaling in cellular senescence
Typically, STING activation triggers IRF3 and NF-κB signaling, contributing to IFN production and inflammation, respectively. However, emerging evidence has found that cGAS-STING pathway can also trigger cellular senescence independent of the classical STING-TBK1-IRF3 axis or the STING-NF-κB axis. For instance, in etoposide-treated keratinocytes, DNA damage repair proteins, including ATM, poly-ADP ribose polymerase 1 (PARP1), and the DNA-binding protein IFI16, activate non-canonical STING signaling and assemble an alternative STING signaling complex that includes p53 and the E3 ubiquitin ligase TRAF6.TRAF6 further catalyzes the formation of K63-linked ubiquitin chains on STING proteins, thereby activating the transcription factor NF-κB and inducing
5. Therapeutic Potential of cGAS-STING Inhibition in Age-Related Diseases
Multiple intervention strategies have been developed to address the overactivation of the cGAS-STING pathway in age-related diseases, including small-molecule inhibitors that target the cGAS and STING proteins. cGAS inhibitors or STING inhibitors can effectively alleviate various age-related diseases, such as neurodegenerative and cardiovascular disorders. This section briefly outlines the properties of small-molecule inhibitors targeting the cGAS-STING pathway and their potential applications in neurodegenerative and cardiovascular diseases (Table 1).
Table 1. cGAS-STING pathway inhibitors in neurodegenerative and cardiovascular diseases.
| Diseases | Target | Inhibitor(s) | Mode of Action | Model | Reference |
| Neurodegenerative | cGAS | RU.521 | Inhibit cGAS activity and interferon expression | In macrophages | [79] |
| Suramin | Displace the bound DNA from cGAS and downregulate IFNβ production | In THP1 cells | [80] | ||
| PF-06928215 | Bind to the nucleotide binding site of cGAS and inhibit cGAS activity by preventing cGAMP binding | In vitro screening | [81] | ||
| TDI6570, also known as G097 | Occupy the ATP- and GTP-binding active site and reduce cGAMP accumulation | In primary mouse microglia | [82,89] | ||
| Quinacrine | A DNA intercalator and preventing cGAS from binding to DNA | In L929 cells. | [90] | ||
| STING | C-176 and C-178 | Compound covalently targets the transmembrane cysteine residue 91, and blocking STING activation induced palmitoylation | THP-1 cells, BMDMs and Trex1-/- mice | [92] | |
| Nitro-fatty acids (NO2-FAs) | Inhibit STING palmitoylation and decrease type I IFN production | In THP-1 cells, BMDMs and HEK293T cells | [96] | ||
| Astin C | Bind to STING and reduces the binding affinity of cGAMP to STING. | In Trex1-/- BMDM cells and in Trex1-/- mouse | [97] | ||
| Tetrahydroisoquinolone analogs | Stabilize the open, inactive conformation of STING | In THP-1 cells | [98] | ||
| H-151 | Covalent inhibitors of STING | In fibroblasts | [93,94] | ||
| Cardiovascular aging | cGAS | Antimalarial drugs (Quinacrine, 9-amino-6-chloro-2-methxyacridine, Hydroxychloroquine, Chloroquine, Primaquine, Quinine) | Disrupt cGAS/DNA binding complex interface by interacting with DNA binding sites A and B | In THP1 cells | [91] |
| RU.521 | Inhibit cGAS activity | In human aortic endothelial cells | [85] | ||
| Suramin | Replace dsDAN in cGAS to prevent cGAS activation and I-IFN transcription | In vivo experiments | [87,88] | ||
| PF-06928215 | Inhibit cGAS activity | In vitro screening | [86] | ||
| STING | H-151 | Covalent inhibitors of STING | In BMDMs | [95] | |
| C-176 | Compound covalently targets the transmembrane cysteine residue 91, and blocking STING activation induced palmitoylation | In diabetic cardiomyopathy mice model and H9C2 cells | [102] |
cGAS: cyclic GMP–AMP synthase; cGAMP: cyclic GMP-AMP; ATP: adenosine triphosphate; GTP: guanosine triphosphate; BMDMs: bone marrow-derived macrophages;
5.1 cGAS inhibitor
In recent years, numerous cGAS inhibitors have been developed. Most are synthetic compounds, although some natural products also exhibit cGAS-inhibitory activity. Currently, the primary mechanism of cGAS inhibitors is to inhibit the active site of cGAS or to disrupt cGAS-dsDNA interactions, either by directly binding to the enzyme or to dsDNA[79-82]. In addition, some cGAS inhibitors suppress cGAS activity by blocking its dimerization or interfering with post-translational modifications of the N-terminal dsDNA-binding
In the field of cGAS inhibitors, small-molecule compounds such as RU.521, RU.365, and PF-06928215 have established comprehensive preclinical research systems. RU.521 and RU.365 primarily inhibit cGAS catalytic activity and suppress interferon expression in BMDMs or human aortic endothelial cells[79,85]. Another cGAS competitive inhibitor, PF-06928215, was first identified by Pfizer in 2017 using a novel fluorescence polarization assay. It primarily binds to the nucleotide-binding site of cGAS and inhibits its activity, thereby suppressing cGAMP binding. PF-06928215 is also employed as a preventive strategy against neurodegenerative and cardiovascular diseases[81,86]. Unlike other reported cGAS inhibitors that bind to the ATP/GTP-binding site, suramin inhibits cGAS activation by displacing DNA bound to cGAS, thereby downregulating IFNβ production[80,87,88]. TDI-6570, one of the most potent inhibitors of mouse cGAS, binds to the ATP- and GTP-binding active site and reduces cGAMP accumulation. It exhibits good brain permeability and has been used to evaluate the effects of cGAS inhibition in a mouse model of neurodegenerative disease[82,89]. In addition to some synthetic compound cGAS inhibitors, some natural products also inhibit cGAS activity. The antimalarial drug quinacrine, a DNA intercalator, can prevent the binding of cGAS to DNA[90,91].
5.2 STING inhibitor
Compared with cGAS inhibitors, STING inhibitors have broader therapeutic potential. STING antagonists or inhibitors can block STING activation by targeting either the ligand-binding domain of the STING protein or its post-translational modifications. For example, nitrofuran derivatives (such as C-178 and C-176), indole ureas (such as compound H-151), and nitro-fatty acids (NO2-FAs) can inhibit STING palmitoylation by mediating nitroalkylation of the Cys88 and Cys91 residues, thereby suppressing STING activity and reducing type I interferon production[92-96]. In addition, some STING inhibitors targeting the ligand-binding domain. The cyclopeptide natural product Astin C can bind to STING and reduce the binding affinity of cGAMP to STING[97]. Tetrahydroisoquinoline derivatives also stabilize the open, inactive conformation of STING and bind to the cGAMP-binding site[98]. However, to date, no cGAS or STING inhibitors have entered clinical trials to evaluate their effects on neurodegenerative or cardiovascular diseases. Their safety and efficacy remain to be established.
5.3 Combination and senolytic approaches
More and more research has found that eliminating senescent cells is largely beneficial for anti-aging and appears to have no
6. Conclusions and Perspectives
In this review, we have summarized recent advances in delineating the hallmarks and triggers of cellular senescence, with a specific focus on the mechanisms by which cGAS-STING signaling drives this process. Elucidating the key regulators of the cGAS-STING pathway activation has significantly improved our understanding of the pathogenesis of inflammaging and aging-related diseases, and has identified new avenues for potential therapeutic intervention. Despite the substantial evidence demonstrating the pivotal role of cGAS-STING signaling in cellular senescence and inflammaging, several critical questions remain unclear.
First, the diversity of upstream activators and downstream effectors of the cGAS-STING pathway in senescence requires further characterization. It is unclear how many distinct stimuli can trigger the cGAS-STING pathway across different tissue contexts and whether the resulting SASP exhibits a unique composition or highly context-dependent heterogeneity. Does the cGAS-STING pathway induce type I interferon or primarily SASP factors? Previous research has shown that OIS in human fibroblasts fails to induce IFN gene expression, whereas radiation-induced DNA damage can trigger IFN expression[9,13]. These discrepancies are primarily due to differences in the duration of senescence, the type of stress-induced senescence model, the biological context, and cell-type specificity. Additionally, the functional implications of the sub-cellular localization of cGAS and STING during senescence need further investigation. Specifically, does nuclear cGAS exert distinct roles compared to its cytoplasmic counterpart?
Second, given that cellular senescence intersects with multiple biological processes, including tumorigenesis, age-related diseases, and inflammation, it is crucial to dissect whether inflammation is a primary driver of senescence or a consequential output of senescent phenotype.
Third, significant challenges remain regarding therapeutic strategies. Key questions include how to specifically eliminate senescent cells to prolong lifespan, and how to effectively inhibit SASP production to alleviate age-related inflammatory diseases. At present, small molecule senolytic drugs, which selectively clear senescent cells, primarily function by targeting senescent cell anti-apoptotic pathways (SCAPs), modulating lysosomal activity and mass in senescent cells, or enhancing immune cell-mediated clearance of senescent cells. However, the efficacy and safety of senolytic drugs in both disease models or human clinic trials require further validation. An alternative strategy involves the use of senomorphic agents, such as SASP inhibitors. These inhibitors can directly or indirectly alleviate the SASP by inhibiting key signaling pathways, including NF-κB pathway, JAK-STAT pathway, mTOR pathway, which are crucial for SASP induction and maintenance. A limitation of this approach is the necessity for continuous treatment to maintain SASP suppression. The chronic administration can increase the risk of side effects and off-target effects by suppressing cytokines secretion in non-senescent cells, such as immune cells. Furthermore, in certain contexts, the complete elimination of all senescent cells or the comprehensive inhibition of SASP factors may be harmful. In addition, senescence-targeted therapies often face challenges such as low drug efficacy and unfavorable pharmacokinetics, which hinder their clinical translation. This raises a critical question: is there a specific scavenger capable of eliminating senescent cells with minimal off-target effects and optimal drug penetration? Whether targeting the cGAS-STING pathway holds such potential remains to be further explored.
Finally, the development of selective and potent modulators targeting the cGAS-STING pathway holds considerable promise for therapeutic interventions. In this review, we have summarized emerging small molecule-based strategies to therapeutically target cGAS-STING signalling in neurodegenerative and cardiovascular diseases. However, the potential challenges associated with such interventions, such as side effects, off-target effects, biomarker limitations, and the heterogeneity of senescent cells, necessitate careful consideration. Additionally, cellular senescence acts as a double-edged sword in cancer biology, exerting both
In conclusion, addressing these unresolved questions will not only broaden our understanding of the intricate interplay between inflammation and senescence but also accelerate the development of next-generation therapeutics for age-related pathologies and disease.
Authors contribution
Chen Y, Chen K: Conceptualization, writing-review & editing.
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
This work was supported by the grants from the National Natural Science Foundation of China (Grant No. 32500791), the Fundamental Research Funds for the Central Universities (Grant Nos. 22120250158, 22120250374, and 22120260165).
Copyright
© The Author(s) 2026.
References
-
1. Campisi J, Kapahi P, Lithgow GJ, Melov S, Newman JC, Verdin E. From discoveries in ageing research to therapeutics for healthy ageing. Nature. 2019;571(7764):183-192.[DOI]
-
4. Gorgoulis V, Adams PD, Alimonti A, Bennett DC, Bischof O, Bishop C, et al. Cellular senescence: Defining a path forward. Cell. 2019;179(4):813-827.[DOI]
-
5. Di Micco R, Krizhanovsky V, Baker D, d’Adda di Fagagna F. Cellular senescence in ageing: From mechanisms to therapeutic opportunities. Nat Rev Mol Cell Biol. 2021;22(2):75-95.[DOI]
-
6. Zhao Y, Simon M, Seluanov A, Gorbunova V. DNA damage and repair in age-related inflammation. Nat Rev Immunol. 2023;23(2):75-89.[DOI]
-
7. Decout A, Katz JD, Venkatraman S, Ablasser A. The cGAS–STING pathway as a therapeutic target in inflammatory diseases. Nat Rev Immunol. 2021;21(9):548-569.[DOI]
-
8. Ablasser A, Chen ZJ. cGAS in action: Expanding roles in immunity and inflammation. Science. 2019;363(6431):eaat8657.[DOI]
-
10. Motwani M, Pesiridis S, Fitzgerald KA. DNA sensing by the cGAS–STING pathway in health and disease. Nat Rev Genet. 2019;20(11):657-674.[DOI]
-
11. Li T, Chen ZJ. The cGAS–cGAMP–STING pathway connects DNA damage to inflammation, senescence, and cancer. J Exp Med. 2018;215(5):1287-1299.[DOI]
-
12. Zhang B, Xu P, Ablasser A. Regulation of the cGAS-STING pathway. Annu Rev Immunol. 2025;43:667-692.[DOI]
-
15. Ogrodnik M, Carlos Acosta J, Adams PD, d’Adda di Fagagna F, Baker DJ, Bishop CL, et al. Guidelines for minimal information on cellular senescence experimentation in vivo. Cell. 2024;187(16):4150-4175.[DOI]
-
16. Richardson AG, Schadt EE. The role of macromolecular damage in aging and age-related disease. J Gerontol Ser A Biol Sci Med Sci. 2014;69(Suppl 1):S28-S32.[DOI]
-
18. Correia‐Melo C, Marques FD, Anderson R, Hewitt G, Hewitt R, Cole J, et al. Mitochondria are required for pro‐ageing features of the senescent phenotype. EMBO J. 2016;35(7):724-742.[DOI]
-
19. Wiley CD, Velarde MC, Lecot P, Liu S, Sarnoski EA, Freund A, et al. Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metab. 2016;23(2):303-314.[DOI]
-
22. Shay JW, Wright WE. Telomeres and telomerase: Three decades of progress. Nat Rev Genet. 2019;20(5):299-309.[DOI]
-
23. Chakravarti D, LaBella KA, DePinho RA. Telomeres: History, health, and hallmarks of aging. Cell. 2021;184(2):306-322.[DOI]
-
25. De Rosa M, Barnes RP, Detwiler AC, Nyalapatla PR, Wipf P, Opresko PL. OGG1 and MUTYH repair activities promote telomeric 8-oxoguanine induced senescence in human fibroblasts. Nat Commun. 2025;16:893.[DOI]
-
26. Rossiello F, Jurk D, Passos JF, d’Adda di Fagagna F. Telomere dysfunction in ageing and age-related diseases. Nat Cell Biol. 2022;24(2):135-147.[DOI]
-
27. Bloom SI, Liu Y, Tucker JR, Islam MT, Machin DR, Abdeahad H, et al. Endothelial cell telomere dysfunction induces senescence and results in vascular and metabolic impairments. Aging Cell. 2023;22(8):e13875.[DOI]
-
28. Vodicka P, Vodenkova S, Danesova N, Vodickova L, Zobalova R, Tomasova K, et al. Mitochondrial DNA damage, repair, and replacement in cancer. Trends Cancer. 2025;11(1):62-73.[DOI]
-
30. Hahn A, Hung GCC, Ahier A, Dai CY, Kirmes I, Forde BM, et al. Misregulation of mitochondrial 6mA promotes the propagation of mutant mtDNA and causes aging in C. elegans. Cell Metab. 2024;36(12):2528-2541.e11.[DOI]
-
31. Sun N, Youle RJ, Finkel T. The mitochondrial basis of aging. Mol Cell. 2016;61(5):654-666.[DOI]
-
33. Vargas JNS, Hamasaki M, Kawabata T, Youle RJ, Yoshimori T. The mechanisms and roles of selective autophagy in mammals. Nat Rev Mol Cell Biol. 2023;24(3):167-185.[DOI]
-
34. García-Prat L, Martínez-Vicente M, Perdiguero E, Ortet L, Rodríguez-Ubreva J, Rebollo E, et al. Autophagy maintains stemness by preventing senescence. Nature. 2016;529(7584):37-42.[DOI]
-
36. Lee JH, Kim EW, Croteau DL, Bohr VA. Heterochromatin: An epigenetic point of view in aging. Exp Mol Med. 2020;52(9):1466-1474.[DOI]
-
37. Lee JH, Demarest TG, Babbar M, Kim EW, Okur MN, De S, et al. Cockayne syndrome group B deficiency reduces H3K9me3 chromatin remodeler SETDB1 and exacerbates cellular aging. Nucleic Acids Res. 2019;47(16):8548-8562.[DOI]
-
39. Zhu H, Blake S, Kusuma FK, Pearson RB, Kang J, Chan KT. Oncogene-induced senescence: From biology to therapy. Mech Ageing Dev. 2020;187:111229.[DOI]
-
40. Suram A, Kaplunov J, Patel PL, Ruan H, Cerutti A, Boccardi V, et al. Oncogene-induced telomere dysfunction enforces cellular senescence in human cancer precursor lesions. EMBO J. 2012;31(13):2839-2851.[DOI]
-
41. Wu J, Sun L, Chen X, Du F, Shi H, Chen C, et al. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science. 2013;339(6121):826-830.[DOI]
-
47. Raab M, Gentili M, de Belly H, Thiam HR, Vargas P, Jimenez AJ, et al. ESCRT III repairs nuclear envelope ruptures during cell migration to limit DNA damage and cell death. Science. 2016;352(6283):359-362.[DOI]
-
52. Hopfner KP, Hornung V. Molecular mechanisms and cellular functions of cGAS–STING signalling. Nat Rev Mol Cell Biol. 2020;21(9):501-521.[DOI]
-
56. Yang H, Wang H, Ren J, Chen Q, Chen ZJ. cGAS is essential for cellular senescence. Proc Natl Acad Sci U S A. 2017;114(23):E4612-E4620.[DOI]
-
63. Picca A, Faitg J, Auwerx J, Ferrucci L, D’Amico D. Mitophagy in human health, ageing and disease. Nat Metab. 2023;5(12):2047-2061.[DOI]
-
64. Li J, Yang D, Li Z, Zhao M, Wang D, Sun Z, et al. PINK1/Parkin-mediated mitophagy in neurodegenerative diseases. Ageing Res Rev. 2023;84:101817.[DOI]
-
65. Mohanty S, Suklabaidya S, Mnatsakanyan N, Jacobson S, Harhaj EW. HTLV-1 Tax induces PINK1-Parkin-dependent mitophagy to mitigate activation of the cGAS-STING pathway. bioRxiv [Preprint]. 2025.[DOI]
-
66. Miwa S, Kashyap S, Chini E, von Zglinicki T. Mitochondrial dysfunction in cell senescence and aging. J Clin Investig. 2022;132(13):e158447.[DOI]
-
67. Martini H, Passos JF. Cellular senescence: All roads lead to mitochondria. FEBS J. 2023;290(5):1186-1202.[DOI]
-
68. Luo W, Zou X, Wang Y, Dong Z, Weng X, Pei Z, et al. Critical role of the cGAS-STING pathway in doxorubicin-induced cardiotoxicity. Circ Res. 2023;132(11):e223-e242.[DOI]
-
70. Sen P, Shah PP, Nativio R, Berger SL. Epigenetic mechanisms of longevity and aging. Cell. 2016;166(4):822-839.[DOI]
-
76. Cancado de Faria R, Silva LND, Teodoro-Castro B, McCommis KS, Shashkova EV, Gonzalo S. A noncanonical cGAS–STING pathway drives cellular and organismal aging. Proc Natl Acad Sci U S A. 2025;122(28):e2424666122.[DOI]
-
83. Liu ZS, Cai H, Xue W, Wang M, Xia T, Li WJ, et al. G3BP1 promotes DNA binding and activation of cGAS. Nat Immunol. 2019;20(1):18-28.[DOI]
-
85. Yu H, Liao K, Hu Y, Lv D, Luo M, Liu Q, et al. Role of the cGAS-STING pathway in aging-related endothelial dysfunction. Aging Dis. 2022;13(6):1901.[DOI]
-
87. Dhingra R, Vasan RS. Age as a risk factor. Med Clin N Am. 2012;96(1):87-91.[DOI]
-
88. Ribeiro-Filho HV, de Souza Silva CM, de Siqueira RJ, Lahlou S, dos Santos AA, Magalhães PJC. Biphasic cardiovascular and respiratory effects induced by β-citronellol. Eur J Pharmacol. 2016;775:96-105.[DOI]
-
96. Hansen AL, Buchan GJ, Rühl M, Mukai K, Salvatore SR, Ogawa E, et al. Nitro-fatty acids are formed in response to virus infection and are potent inhibitors of STING palmitoylation and signaling. Proc Natl Acad Sci U S A. 2018;115(33):E7768-E7775.[DOI]
-
98. Siu T, Altman MD, Baltus GA, Childers M, Ellis JM, Gunaydin H, et al. Discovery of a novel cGAMP competitive ligand of the inactive form of STING. ACS Med Chem Lett. 2019;10(1):92-97.[DOI]
-
100. Myakala K, Wang XX, Shults NV, Krawczyk E, Jones BA, Yang X, et al. NAD metabolism modulates inflammation and mitochondria function in diabetic kidney disease. J Biol Chem. 2023;299(8):104975.[DOI]
-
101. Wei P, Zhang X, Yan C, Sun S, Chen Z, Lin F. Mitochondrial dysfunction and aging: Multidimensional mechanisms and therapeutic strategies. Biogerontology. 2025;26(4):142.[DOI]
Copyright
© The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Publisher’s Note
Science Exploration remains a neutral stance on jurisdictional claims in published
maps
and institutional affiliations. The views expressed in this article are solely those
of
the author(s) and do not reflect the opinions of the Editors or the publisher.
Share And Cite



Science Exploration Style
Chen Y, Chen K. cGAS-STING pathway drives cellular senescence and inflammaging. Ageing Cancer Res Treat. 2026;3:202615. https://doi.org/10.70401/acrt.2026.0026
Tips
Copy completed.
Submit a Manuscript
Author Instructions
Cite this Article
Article Metrics
0
View
0
Download
Cited
Article Updates

- Abstract
- Keywords
- 1. Introduction
- 2. Characteristics of Cellular Senescence
- 3. Mechanisms Involved in Cellular Senescence
- 4. The cGAS-STING Pathway in Cellular Senescence
- 5. Therapeutic Potential of cGAS-STING Inhibition in Age-Related Diseases
- 6. Conclusions and Perspectives
- Authors contribution
- Conflict of interest
- Ethical approval
- Consent to participate
- Consent for publication
- Availability of data and materials
- Funding
- References
- Copyright
Science Exploration Style
Chen Y, Chen K. cGAS-STING pathway drives cellular senescence and inflammaging. Ageing Cancer Res Treat. 2026;3:202615. https://doi.org/10.70401/acrt.2026.0026
copy
Share Link
copy