Catalytic asymmetric dearomative formal [4+2] annulation of indoles with O-Silylated hemiaminals as dienes: The dual role of chiral phosphoric acid
*Correspondence to:
Zheng-Hui Guan, Key Laboratory of Synthetic and Nature Molecule of Ministry of Education, Department of Chemistry & Materials Science, Northwest University, Xi’an 710127, Shaanxi, China.
E-mail: guanzhh@nwu.edu.cn
Qingyang Zhao, School of Pharmaceutical Sciences (Shenzhen), Shenzhen Campus of Sun Yat-sen University, Shenzhen 518107, Guangdong, China. E-mail: zhaoqy26@mail.sysu.edu.cn
Qingyang Zhao, School of Pharmaceutical Sciences (Shenzhen), Shenzhen Campus of Sun Yat-sen University, Shenzhen 518107, Guangdong, China. E-mail: zhaoqy26@mail.sysu.edu.cn
Chiral Chem. 2026;2:202511. 10.70401/cc.2025.0008
Received: November 08, 2025Accepted: December 17, 2025Published: December 26, 2025
Abstract
O-Silylated hemiaminals are utilized as elegant imine precursors in the formal asymmetric [4+2] annulation of indoles for the first time, wherein chiral phosphoric acid (CPA) acts (1) as a Brønsted acid catalyst to facilitate methanimine formation under mild conditions and then (2) as an anion-binding catalyst for dearomative annulation. This methodology exhibits a broad substrate scope with remarkable functional group tolerance and enantioselectivity (up to 97% yield and 99% ee), providing straightforward access to the challenging indoline-fused tetrahydroquinolines bearing multiple stereogenic centers. Mechanistic studies reveal the critical role of PhNHCO- groups in enhancing both reactivity and enantioselectivity, probably due to non-covalent interactions with CPA. The kinetic isotope effects experiment and negative linear Hammett correlation suggest a concerted process.
Graphical Abstract
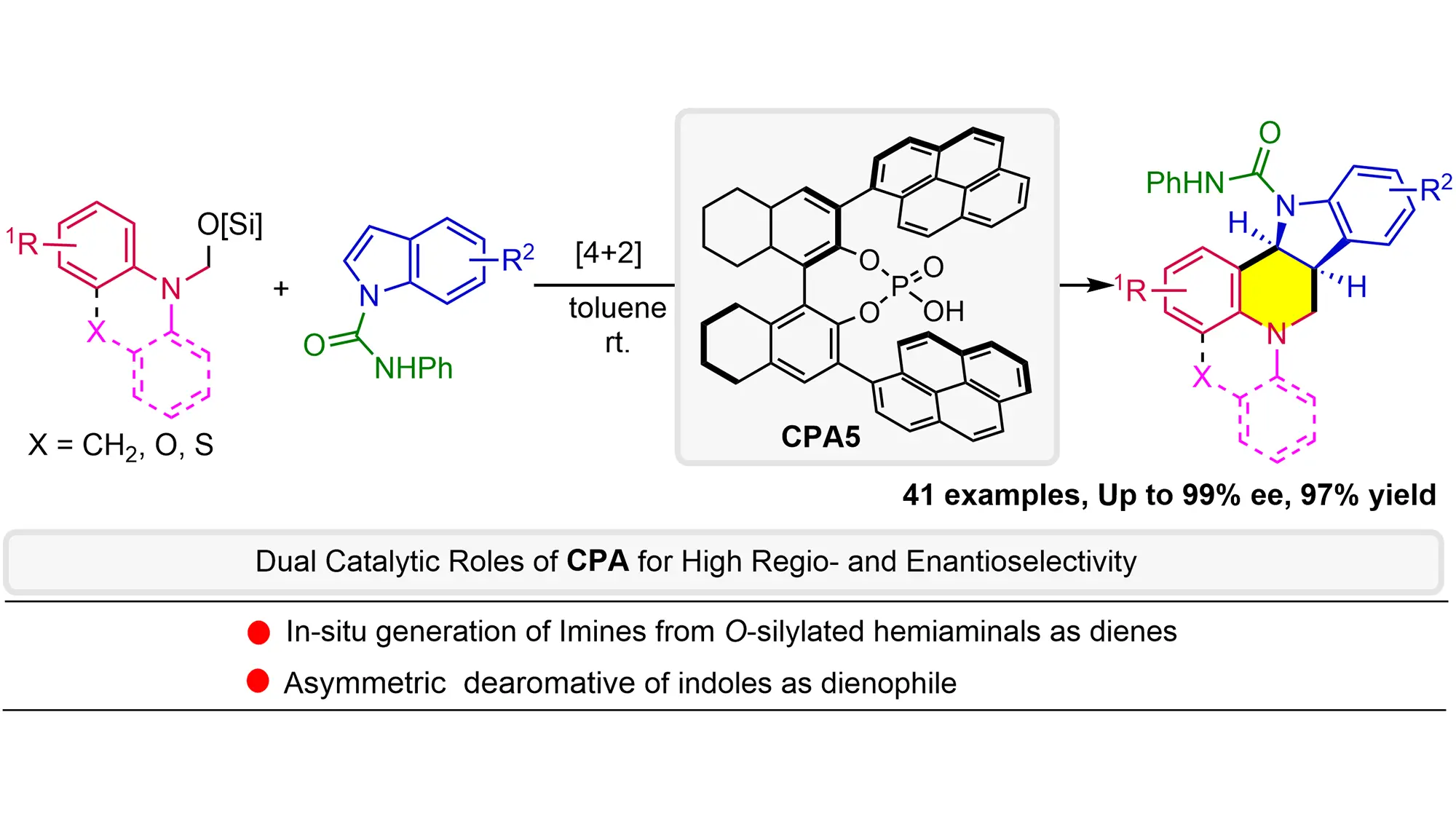
Keywords
Dearomatization, chiral phosphoric acid, hemiaminal, indole, tetrahydroquinoline
1. Introduction
Imines are important synthons in the asymmetric synthesis of chiral nitrogen heterocycles, which constitute a privileged structural motif prevalent in biologically active compounds. The rising percentage of nitrogen heterocycles in approved pharmaceuticals[1] underscores the urgent need to enhance structural diversity in these molecules and advance imine-based synthetic methodologies. In recent years, O-silylated hemiaminals have emerged as highly reactive imine precursors, which can in situ generate various methanimines and thus circumvent several issues inherent to conventional preformed imines (Figure 1A)[2-22]. However, to the best of our knowledge, the chemical space of these imine precursors remains constrained in asymmetric synthesis[23-28], particularly for constructing chiral nitrogen heterocycles[29-31].
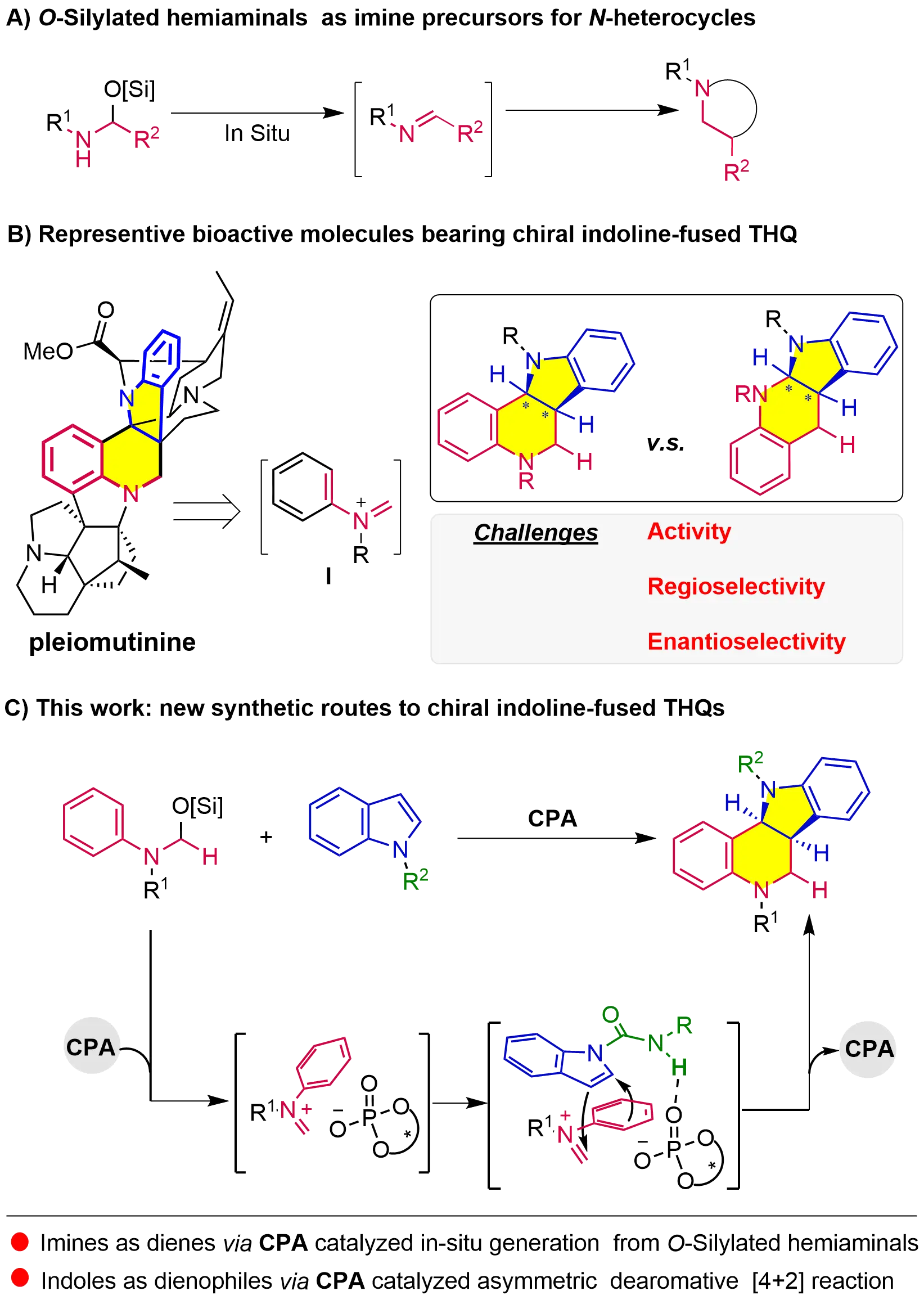
Figure 1. Asymmetric dearomative of indole with methanimines for THQs. THQ: Tetrahydroquinoline; CPA: chiral phosphoric acid.
The [4+2] cycloaddition reaction of N-aryl methanimines with dienophiles has proven effective for assembling the bioactive chiral tetrahydroquinolines (THQs) skeleton[32-43]. Nevertheless, indoles remain notably underexplored as dienophiles, whether in asymmetric versions or not, although this dearomative strategy[44-46] provides direct access to indoline-fused THQs[47-50]. Moreover, despite their unique potential for enhancing the molecular diversity of tetrahydroquinoline scaffolds, methaniminium cations have never been reported as diene partners for this asymmetric transformation, whether derived from O-silylated hemiaminals[51] or alternative precursors (Figure 1B)[52-55]. The primary challenges arise from the limited steric differentiation of such methanimines and the inherent complexity of the dearomative transformation, which hinders precise control over regio- and enantioselectivity and limits the improvement of catalytic efficiency.
Considering the excellent catalytic activity and selectivity of chiral phosphoric acids (CPAs) in asymmetric transformations involving imines and indoles[56-60], together with their significance in activating O-silylated hemiaminals, we reasoned that CPAs could serve as a dual-function catalyst to facilitate the dearomative [4+2] reaction of indoles with O-silylated hemiaminals (Figure 1C): (1) enabling the in-situ formation of the methaniminium cation from O-Silylated hemiaminals as a Brønsted acid; (2) inducing the stereochemical control of the upcoming dearomative cycloaddition reaction as a chiral anion through non-covalent interactions with indoles and the methaniminium cation. Herein, we describe the first example of CPA catalyzed asymmetric dearomative [4+2] reaction of indoles with O-silylated hemiaminals for the target-oriented synthesis of 2-unsubstituted indoline-fused THQs in excellent regio- and enantioselectivities.
2. Methods
2.1 Materials
Chemicals were commercially purchased from Adamas-beta, Tansoole, Bide Pharmatech Ltd., Aladdin, and Daicel Chiral Technologies (China) Co., and were directly used without further purification unless otherwise stated. Reactions were carried out under an atmosphere of nitrogen using a glovebox unless otherwise noted. Toluene and CH2Cl2 were freshly distilled from CaH2 and distilled three times prior to use.
2.2 Typical procedure for asymmetric formal [4+2] cycloaddition reaction of O-silylated hemiaminals and indoles
To a 10 mL tube which was charged with indole (0.1 mmol, 1.0 equiv.), CPA5 (5 mol%) and toluene (1.0 mL), was added the solution of O-Silylated hemiaminals in toluene (0.12M, 1.0 mL), and then the reaction mixture was stirred for 12 h at room temperature. After completion (detected by TLC), the reaction mixture was used directly for column chromatography separation (CH2Cl2/ethyl acetate = 20:1 to 8:1 as the eluent) to afford the corresponding pure products.
The corresponding racemic products were synthesized according to the above procedure by replacing the CPA5 with Copper(II) trifluoromethanesulphonate (3.3 mg, 10 mol%) as the catalyst.
2.3 Removal of the directing or protecting groups
2.3.1 Removal of the directing groups for 3f
To a 25 mL flask, 3f (35.5 mg, 0.1 mmol), KOH (aq.) (1 M, 1.0 mL), and ethanol (2 mL) were refluxed in a 100 oC oil bath for 12 h. After completion (detected by TLC), the reaction was cooled down to room temperature, and the resulting solution was neutralized with aqueous HCI and extracted with ethyl acetate (3 × 10 mL). The organic layers were dried over anhydrous Na2SO4 and evaporated in vacuo. The desired product 8 was obtained after purification by flash chromatography on silica gel (petroleum ether/ethyl acetate = 8:1) as a yellow oil (19.6 mg, 83% yield). The ee of compound 8 was determined by HPLC using an ICDaicel column (n-hexane/i-PrOH = 80/20, flow rate = 1.0 mL/min, λ = 254 nm, tminor = 4.9 min, tmajor = 5.5 min).
2.3.2 Removal of the protecting groups for 7a
To a 10 mL flask were charged 7a (43.1mg, 0.1 mmol), Pd/C (10 mg, 10 mol%, 10 wt%), and MeOH (2.0 mL). The flask was evacuated and backfilled with hydrogen (3 times) and then stirred under 1 atm (balloon pressure) of hydrogen at room temperature for 24 h. When the reaction was completed, the Pd/C catalyst was filtered before evaporation of solvent. The solid residue was purified by column chromatography to afford the product 9 as a white solid (31.4 mg, 92% yield) (eluent: petroleum ether/ethyl acetate = 3:1). The ee of compound 9 was determined by HPLC using an IADaicel column (n-hexane/i-PrOH = 70/30, flow rate = 1.0 mL/min, λ = 254 nm, tminor = 11.3 min, tmajor = 15.1 min).
To a 10 mL flask charged with the crude product 9, KOH (1 M, 1.0 mL) and ethanol (2 mL) were refluxed in a 100 oC oil bath for 12 h. After completion (detected by TLC), the reaction mixture was cooled down to room temperature, and the resulting solution was neutralized with aqueous HCI and extracted with ethyl acetate (3 x 10 mL). The organic layers were dried over anhydrous Na2SO4 and evaporated in vacuo. The solid residue was purified by column chromatography to afford the product 10 as a white solid (13.9 mg, 63% yield for two steps) (eluent: petroleum ether/ethyl acetate = 3:1). The ee of compound 10 was determined by HPLC using an IADaicel column (n-hexane/i-PrOH = 70/30, flow rate = 1.0 mL/min, λ = 254 nm, tminor = 10.1 min, tmajor = 16.4 min).
3. Results and Discussion
3.1 Optimize the reaction conditions
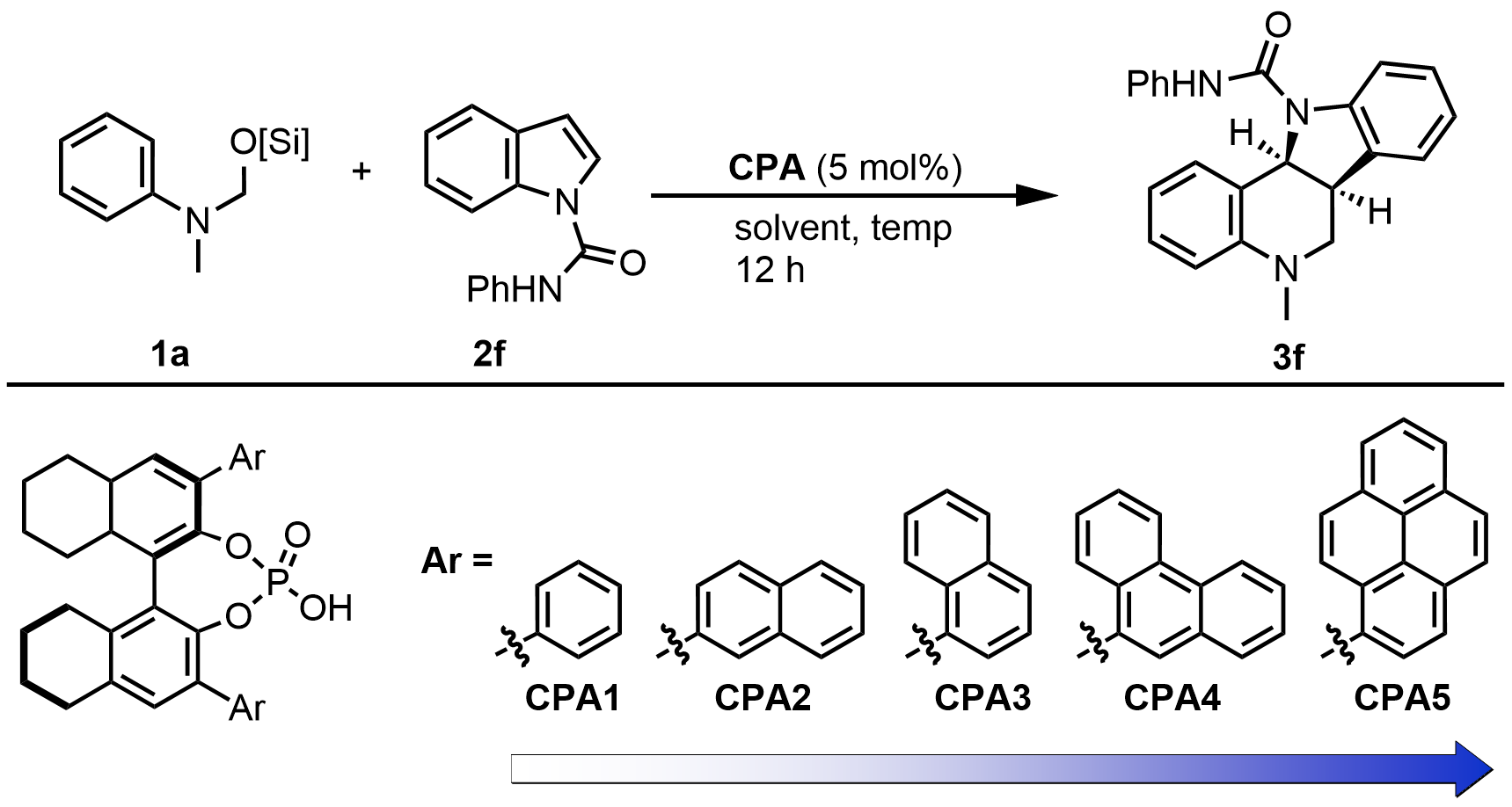
At the first step, we investigated the feasibility of utilizing hemiaminal silyl ether as a methanimine precursor with N-substituted indole in the presence of CPA. As we reported, the CPA1 catalyzed cycloaddition reaction of hemiaminal silyl ether 1a and simple indole 2a was not observed (Figure 2). The same results were obtained when common indole derivatives, including N-methyl indole 2b, N-acetyl indole 2c, N-Boc indole 2d, and N-Ts indole 2e, were employed in the reaction. Considering that an H atom on the N-atom of the indole moiety may provide a secondary binding site to interact with the Lewis basic site (P=O) on the phosphate, a series of indole-1-carboxamides were prepared and screened in the reaction[61]. It was delightful to observe that indole-1-carboxamides 2f-2i proceeded smoothly to produce the corresponding indoline-fused THQs 3f-3i in excellent yields (92-96% yield), albeit with low to moderate enantiomeric excess (ee) (33-49% ee). Consistent with our hypothesis, the H atom on the indole-1-carboxamide plays a vital role in the reaction. No reaction was observed when N-methyl-N-phenyl-1H-indole-1-carboxamide 2j was employed as the substrate under the same conditions.

Figure 2. Optimization of the N-substituted indoles. 0.12 mmol of 1a was added to the solution of 2 (0.1 mmol), CPA1 (5 mol%) in toluene (1.0 mL). Isolated yields. ee was determined by chiral HPLC. CPA: chiral phosphoric acid; HPLC: high-performance liquid chromatography.
Encouraged by the above initial results, the conditions were further optimized using methanimine precursor O-silylated hemiaminal 1a and N-phenyl-indole-1-carboxamide 2f. As depicted in Table 1, toluene was a better solvent than DCM, DCE, THF, and 1,4-dioxane (entries 1-5). Subsequently, various H8-BINOL-derived CPA catalysts with different steric properties were screened in the reaction (entries 6-9). It was found that the enantioselectivity was strongly responsive to the extent of the substitutions on the 3,3’-positions of the H8-BINOL-framework, with the 1-pyrenyl catalyst CPA5 being optimal (97% yield, 97% ee). The steric demand of the catalyst, together with the potential aryl–aryl interactions with the substrate, was presumably thought to match the less sterically demanding N-phenylmethaniminium substrate and create a tailored microenvironment for asymmetric annulation. In addition, lowering the temperature to 0 °C did not improve the reaction (entry 10).
Table 1. Optimization of the reactiona.
| entry | catalyst | solvent | T (oC) | yield(%) | ee (%) |
| 1 | CPA1 | toluene | rt | 95 | 49 |
| 2 | CPA1 | CH2Cl2 | rt | 89 | 32 |
| 3 | CPA1 | DCE | rt | 87 | 29 |
| 4 | CPA1 | THF | rt | 85 | 39 |
| 5 | CPA1 | dioxane | rt | 92 | 47 |
| 6 | CPA2 | toluene | rt | 94 | 60 |
| 7 | CPA3 | toluene | rt | 96 | 86 |
| 8 | CPA4 | toluene | rt | 95 | 75 |
| 9 | CPA5 | toluene | rt | 97 | 97 |
| 10 | CPA5 | toluene | 0 | 95 | 96 |
a: 0.12 mmol 1a was added to the solution of 2f (0.1 mmol), CPA (5 mol%) in toluene (1.0 mL). Isolated yields. ee was determined by chiral HPLC; CPA: chiral phosphoric acid; HPLC: high-performance liquid chromatography.
3.2 Expand the reaction substrates
With the optimal conditions established, we focused on the investigation of indole ring variants (Figure 3). Notably, the reaction demonstrated excellent tolerance toward various functional groups in the indole moiety, irrespective of their electronic properties. A wide range of groups, including methyl, methoxy, fluoro, chloro, and bromo, were well compatible with this asymmetric dearomative cycloaddition reaction, regardless of their position at the 4-, 5-, or 6-position of the indole. As a result, the corresponding indoline-fused tetrahydroquinoline products 4a-4p were obtained in high to excellent yields and enantioselectivities (82 to 97% yield, 88 to 99% ee). It is noted that the product 4q, bearing an all-carbon quaternary stereocenter, was also prepared from 3-methyl indole, although both the yield and ee were reduced presumably due to the steric effects.
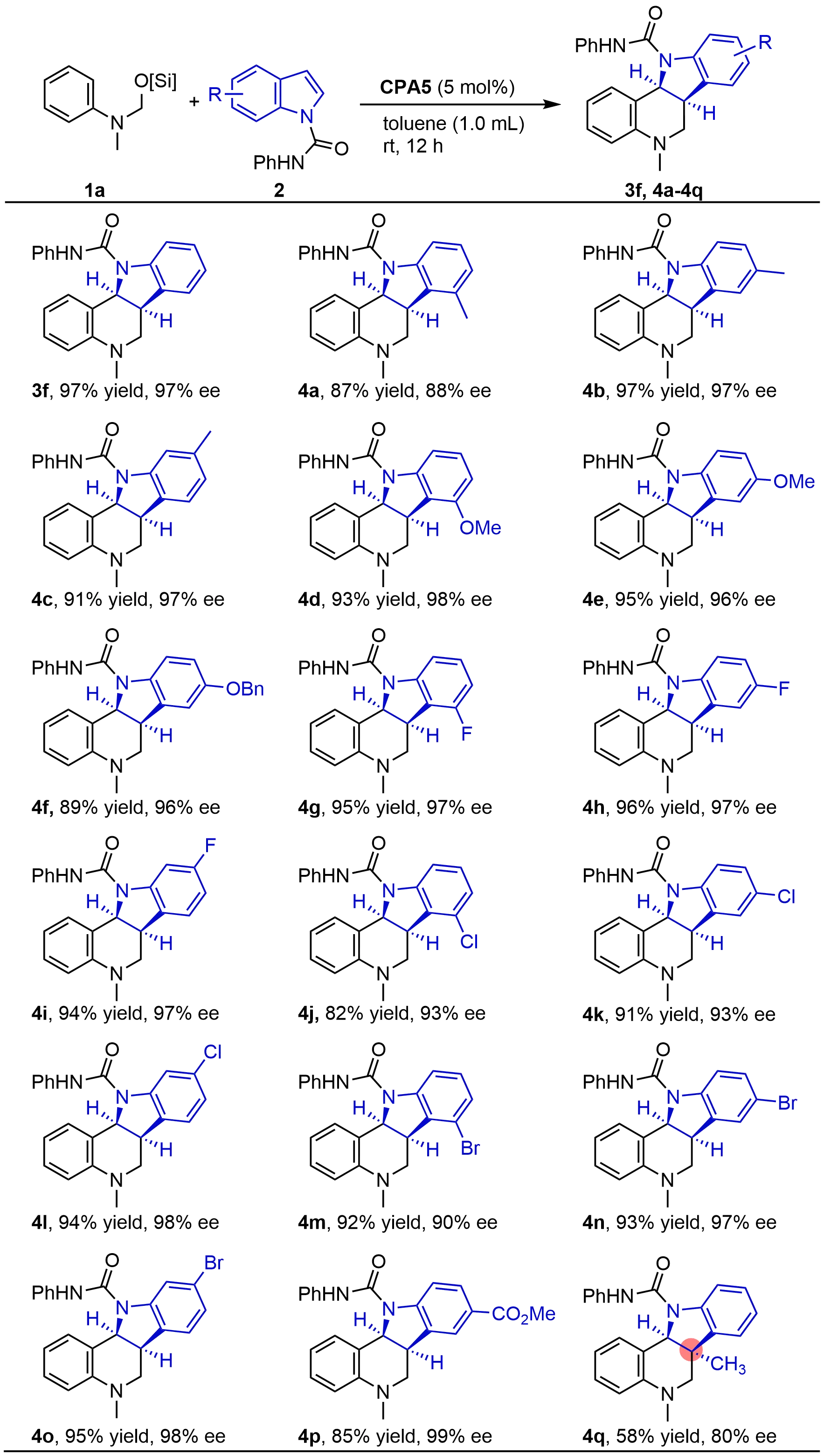
Figure 3. Asymmetric dearomative cycloaddition with various indoles. Isolated yields. ee was determined by chiral HPLC. CPA: chiral phosphoric acid; HPLC: high-performance liquid chromatography.
Subsequently, the scope of O-silylated hemiaminals was investigated (Figure 4). Hemiaminals with electron-donating substituents, such as methyl, iso-propyl, tert-butyl, methoxy and ethoxy, underwent the reaction smoothly to afford the indoline-fused THQs with excellent enantioselectivities in high yields (5a-5g, 95 to 98% ee, 88 to 97% yield). Halide substituents including F, Cl, Br and I were well compatible with the reaction to give the corresponding halide-substituted indoline-fused tetrahydroquinolines in high yields and enantioselectivities (5h-5k, 91 to 94% ee, 81 to 87% yield). As expected, 3,5-disubstituted or 3,4,5-trisubstituted N-methyl hemiaminals proceeded smoothly under the conditions to produce the desired indoline-fused tetrahydroquinolines 5l-5n with high enantioselectivities in high yields. Specifically, hemiaminals containing a meta-substituent (methyl and chloro) gave rise to single regioselective products 5o (97% ee, 82% yield) and 5p (96% ee, 93% yield), respectively. Moreover, a 2-naphthyl hemiaminal also produced exclusively tetrahydro-7H-benzo[f]indolo[3,2-c]quinoline 5q with 98% ee and 96% yield. The absolute configuration of 5i was determined by X-ray crystallographic analysis (CCDC 2189145), and other products were assigned by analogy.
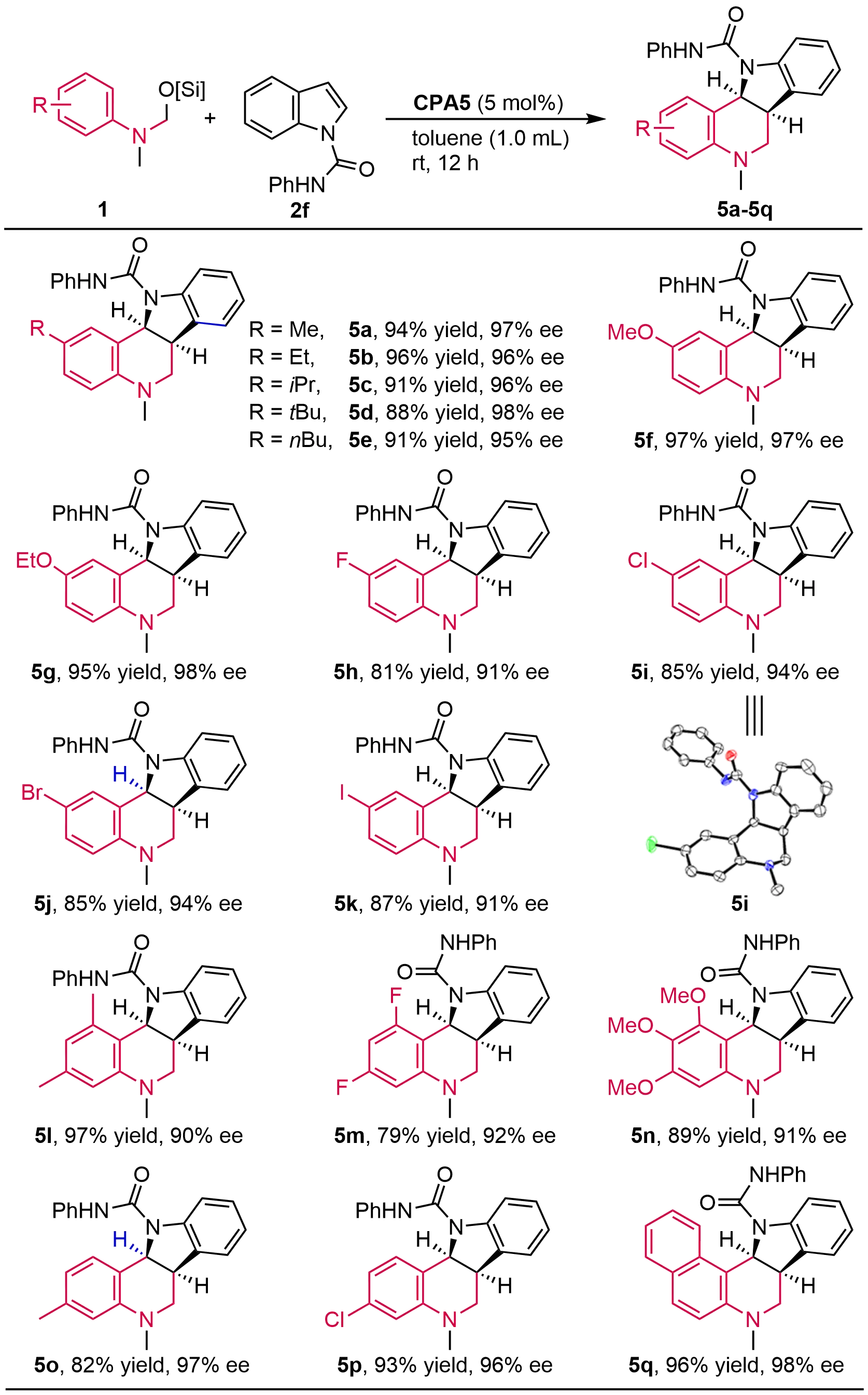
Figure 4. Asymmetric dearomative cycloaddition with various hemiaminals. Isolated yields. ee was determined by chiral HPLC. CPA: chiral phosphoric acid; HPLC: high-performance liquid chromatography.
Furthermore, substituent effects of O-silylated hemiaminals on the N atom were also investigated (Figure 5). Remarkably, 6a bearing the removable benzyl (Bn) group showed similar reactivity and enantioselectivity to 1a, giving 7a with 95% ee and 97% yield. Diphenyl substitution was compatible with the reaction to give the corresponding 7b with 92% ee and 85% yield. In addition, the cyclic O-silylated hemiaminals, including 6c bearing tetrahydroquinoline, 6d bearing benzomorpholine, 6e bearing phenoxazine and 6f bearing phenothiazine, were also examined in the reaction. It was found that these substrates were well-compatible with the reaction to afford the polycyclic products 7c-7f with 90-98% ee and 63-96% yields.

Figure 5. Asymmetric dearomative cycloaddition with different substituents on the nitrogen atom of O-silylated hemiaminals. Isolated yields. ee was determined by chiral HPLC. CPA: chiral phosphoric acid; HPLC: high-performance liquid chromatography.
3.3 Synthetic transformation
For further synthetic transformation, the directing group PhNHCO- at the indole moiety was conveniently removed in 83% yield and 97% ee (Figure 6). Notably, PhNHCO- at the indole moiety and Bn- at the tetrahydroquinoline moiety of the final products could be selectively removed to afford indoline-fused tetrahydroquinolines bearing free NH groups in high yields without loss of enantiomeric purity, which are more valuable from the perspective of molecular complexity in synthetic chemistry and drug discovery. This merit would also pave the way for late-stage derivatization in more complex polycyclic frameworks.
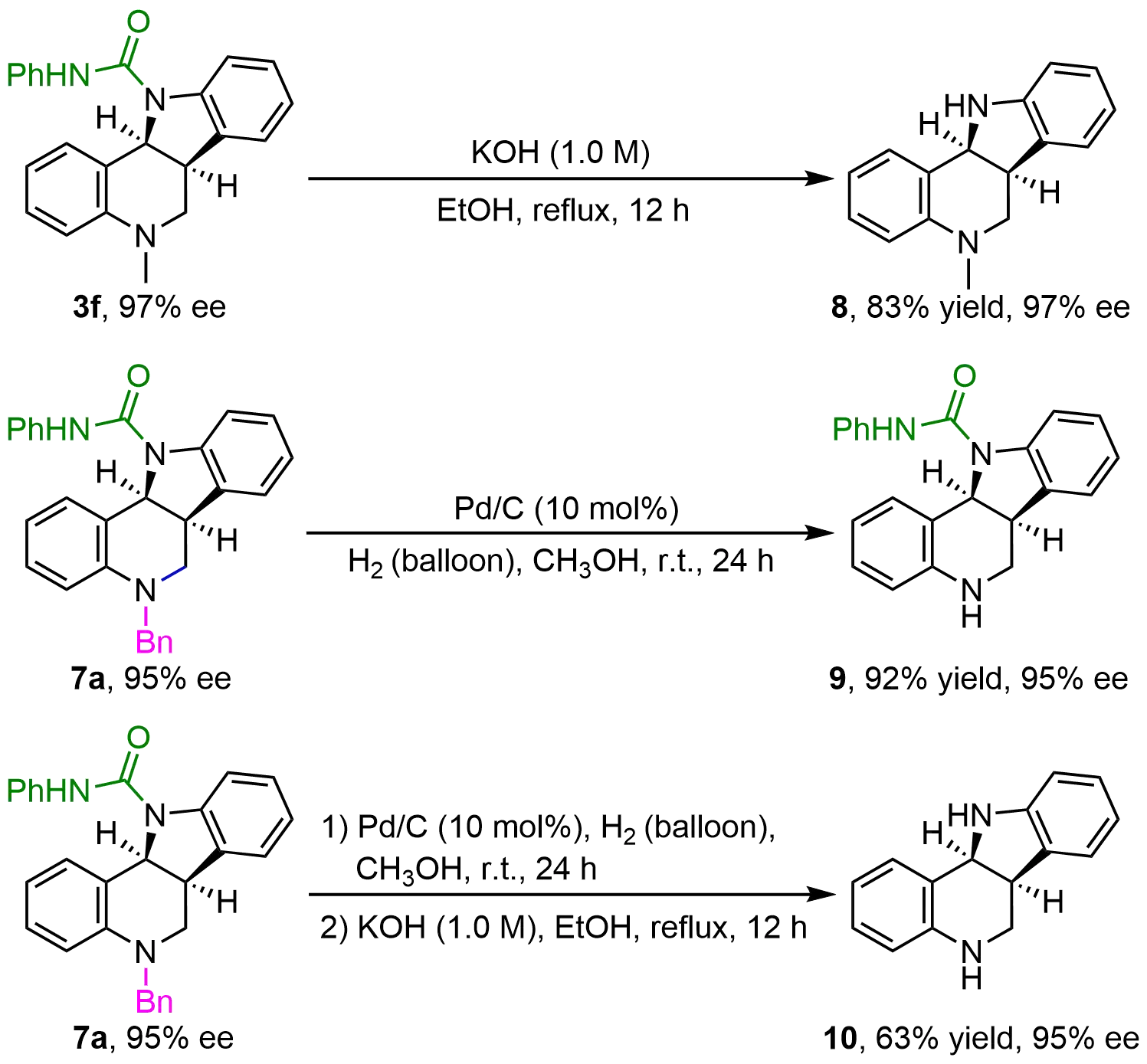
3.4 Mechanistic studies
To gain mechanistic insight into the catalytic cycle, we conducted a series of control experiments. The reaction of hemiaminal silyl ether 1a with 2f did not proceed in the absence of phosphoric acid, but the formal cycloaddition occurred in the presence of p-toluenesulfonic acid (Figure 7A). These experiments suggest that the CPA plays a vital role in the desilanolation of hemiaminal silyl ether 1a to generate the active iminium phosphate for the cycloaddition reaction. In principle, both the stepwise process (a, b and d) and the concerted pathway (c and d) are possible for the formal [4+2] cycloaddition of N-aryl imines and electron-rich olefins (Figure 7B). To gain further insight into the mechanism, kinetic studies were performed. Notably, primary kinetic isotope effect (KIE) was not observed for the hydrogens of the aniline group of D-1a (step d) (Figure 7C, Figure S1).

Besides, the analysis of the effect of para substituents on the aniline group on the reaction rate reveals a negative linear Hammett correlation (ρ = -2.4349 ± 0.2284, Figure S2). Similarly, the analysis of the effect of substituents on indoles on the reaction rate also shows a negative linear Hammett correlation (ρ = -1.9203 ± 0.2117, Figure S3). These results are consistent with a concerted process.
4. Conclusion
In summary, we have developed an efficient and modular asymmetric dearomative formal [4+2] reaction for synthesizing enantioenriched indoline-fused tetrahydroquinolines. This method employs O-silylated hemiaminals as precursors to in situ generate highly reactive methaniminium salts in the presence of CPA, which then catalyze the sequential dearomative cycloaddition of methaniminium salts and diverse substituted indoles under mild conditions. The reaction tolerates a wide range of functional groups with high efficiency (up to 97% yield and 99% ee). The PhNHCO- group at the indole moiety plays a crucial role in both reactivity and enantioselectivity, probably through non-covalent interactions with CPA. In addition, a concerted mechanism was implied by the analysis of KIE experiments and Hammett correlation.
Supplementary materials
The supplementary material for this article is available at: Supplementary materials.
Authors contribution
Guan Z: Conceptualization, supervision, formal analysis, writing-original draft.
Zhao Q, Ren Z: Formal analysis, writing-original draft, writing-review & editing.
Mo N, Zhang Y: Methodology, investigation.
Conflicts of interest
The authors declare no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
The data reported in this paper are available in the main text or Supplementary Information, including methods, NMR data, HRMS data, HPLC spectra Crystal data and NMR spectra. Crystallographic data for the structures reported in this article have been deposited at the Cambridge Crystallographic Data Centre, under deposition numbers CCDC 2189145 (5i).
Funding
This work was supported by the financial support from the National Natural Science Foundation of China (Grant Nos. NSFC 22171224, 21971204, 21602172), the Fund of Education Department of Shaanxi Provincial Government (Grant No. 24JP187), and the Medical Science and Technology Foundation of Guangdong Province (Grant No. A2025079).
Copyright
© The Author(s) 2025.
References
-
1. Marshall CM, Federice JG, Bell CN, Cox PB, Njardarson JT. An update on the nitrogen heterocycle compositions and properties of U.S. FDA-approved pharmaceuticals (2013-2023). J Med Chem. 2024;67(14):11622-11655.[DOI]
-
2. Matheau-Raven D, Gabriel P, Leitch JA, Almehmadi YA, Yamazaki K, Dixon DJ. Catalytic reductive functionalization of tertiary amides using vaska’s complex: Synthesis of complex tertiary amine building blocks and natural products. ACS Catal. 2020;10(15):8880-8897.[DOI]
-
3. Feng M, Zhang H, Maulide N. Challenges and breakthroughs in selective amide activation. Angew Chem Int Ed. 2022;61(49):e202212213.[DOI]
-
4. Iwasaki T, Nozaki K. Counterintuitive chemoselectivity in the reduction of carbonyl compounds. Nat Rev Chem. 2024;8(7):518-534.[DOI]
-
5. Huang PQ, Ou W, Han F. Chemoselective reductive alkynylation of tertiary amides by Ir and Cu(I) bis-metal sequential catalysis. Chem Commun. 2016;52(80):11967-11970.[DOI]
-
6. Fuentes de Arriba Á L, Lenci E, Sonawane M, Formery O, Dixon DJ. Iridium-catalyzed reductive strecker reaction for late-stage amide and lactam cyanation. Angew Chem Int Ed. 2017;56(13):3655-3659.[DOI]
-
7. Ou W, Han F, Hu XN, Chen H, Huang PQ. Iridium-catalyzed reductive alkylations of secondary amides. Angew Chem Int Ed. 2018;57(35):11354-11358.[DOI]
-
8. Gabriel P, Gregory AW, Dixon DJ. Iridium-catalyzed Aza-spirocyclization of indole-tethered amides: An interrupted pictet-spengler reaction. Org Lett. 2019;21(17):6658-6662.[DOI]
-
9. Biallas P, Yamazaki K, Dixon DJ. Difluoroalkylation of tertiary amides and lactams by an iridium-catalyzed reductive reformatsky reaction. Org Lett. 2022;24(10):2002-2007.[DOI]
-
10. Chen J, Lim JW, Ong DY, Chiba S. Iterative addition of carbon nucleophiles to N,N-dialkyl carboxamides for synthesis of α-tertiary amines. Chem Sci. 2021;13(1):99-104.[DOI]
-
11. Wang XG, Ou W, Liu MH, Liu ZJ, Huang PQ. Tandem-catalysis-enabled highly chemoselective deoxygenative alkynylation and alkylation of tertiary amides: A versatile entry to functionalized α-substituted amines. Org Chem Front. 2022;9(12):3237-3246.[DOI]
-
12. Shennan BDA, Sánchez-Alonso S, Rossini G, Dixon DJ. 1,2-redox transpositions of tertiary amides. J Am Chem Soc. 2023;145(40):21745-21751.[DOI]
-
13. Shi Q, Liu WH. Reactivity umpolung of tertiary amide enabled by catalytic reductive stannylation. Angew Chem Int Ed. 2023;62(37):e202309567.[DOI]
-
14. Wu X, Wang Y, Yang L, Xie X, Zhang Z. Transition-metal-free chemoselective catalytic hydrosilylation of tertiary amides to hemiaminals by Me2SiH2 generated from controllable disproportionation of 1,1,3,3-tetramethyldisiloxane. Org Chem Front. 2023;10(5):1198-1205.[DOI]
-
15. Venditto NJ, Boerth JA. Deoxy-arylation of amides via a tandem hydrosilylation/radical–radical coupling sequence. Org Lett. 2024;26(17):3617-3621.[DOI]
-
16. Pijper B, Martín R, Huertas-Alonso AJ, Linares ML, López E, Llaveria J, et al. Fully automated flow protocol for C(sp3)–C(sp3) bond formation from tertiary amides and alkyl halides. Org Lett. 2024;26(14):2724-2728.[DOI]
-
17. Almehmadi YA, McGeehan J, Guzman NJ, Christensen KE, Yamazaki K, Dixon DJ. Iridium-catalysed synthesis of C,N,N-cyclic azomethine imines enables entry to unexplored nitrogen-rich 3D chemical space. Nat Synth. 2024;3(9):1168-1175.[DOI]
-
18. Lu GS, Ruan ZL, Wang Y, Lü JF, Ye JL, Huang PQ. Catalytic reductive amination and tandem amination–alkylation of esters enabled by a cationic iridium complex. Angew Chem Int Ed. 2025;64(12):e202422742.[DOI]
-
19. Tian H, Jiang F, Wang X. Merging SOMO activation with transition metal catalysis: Deoxygenative functionalization of amides to β-aryl amines. Sci Adv. 2025;11(3):eadt4187.[DOI]
-
20. Shi Q, Yang R, Liu WH. Catalytic reductive aza-boron-wittig reaction. ACS Catal. 2025;15(5):3741-3750.[DOI]
-
21. Almehmadi YA, Passmore AJ, Gabriel P, Dixon DJ. Iridium-catalyzed reductive deoxygenation of esters for the synthesis of sterically hindered ethers. Angew Chem Int Ed. 2025;64(40):e202508301.[DOI]
-
22. Xu Q, Ou W, Wang Q, Tao Y, Chen T, Liao Z, et al. Photocatalyzed reductive amination for multiple deuteration of amines. ACS Catal. 2025;15(23):20065-20074.[DOI]
-
23. Chen DH, Sun WT, Zhu CJ, Lu GS, Wu DP, Wang AE, et al. Enantioselective reductive cyanation and phosphonylation of secondary amides by iridium and chiral thiourea sequential catalysis. Angew Chem Int Ed. 2021;60(16):8827-8831.[DOI]
-
24. Li Z, Zhao F, Ou W, Huang PQ, Wang X. Asymmetric deoxygenative alkynylation of tertiary amides enabled by iridium/copper bimetallic relay catalysis. Angew Chem Int Ed. 2021;60(51):26604-26609.[DOI]
-
25. Chen H, Wu ZZ, Shao DY, Huang PQ. Multicatalysis protocol enables direct and versatile enantioselective reductive transformations of secondary amides. Sci Adv. 2022;8(47):eade3431.[DOI]
-
26. Agrawal T, Perez-Morales KD, Cort JA, Sieber JD. Asymmetric synthesis of propargylic α-stereogenic tertiary amines by reductive alkynylation of tertiary amides using Ir/Cu tandem catalysis. J Org Chem. 2022;87(9):6387-6392.[DOI]
-
27. Xu FF, Chen JQ, Shao DY, Huang PQ. Catalytic enantioselective reductive alkynylation of amides enables one-pot syntheses of pyrrolidine, piperidine and indolizidine alkaloids. Nat Commun. 2023;14(1):6251.[DOI]
-
28. Deng X, Jiang F, Wang X. Asymmetric deoxygenative functionalization of secondary amides with vinylpyridines enabled by a triple iridium-photoredox-chiral phosphoric acid system. Org Lett. 2024;26(12):2483-2488.[DOI]
-
29. Li J, Gu Z, Zhao X, Qiao B, Jiang Z. Asymmetric aerobic decarboxylative Povarov reactions of N-aryl α-amino acids with methylenephthalimidines via cooperative photoredox and chiral brønsted acid catalysis. Chem Commun. 2019;55(86):12916-12919.[DOI]
-
30. Mo NF, Yu L, Zhang Y, Yao YH, Kou X, Ren ZH, et al. Asymmetric spirocyclization enabled by iridium and brønsted acid-catalyzed formal reductive cycloaddition. CCS Chem. 2021;3(7):1775-1786.[DOI]
-
31. Ji KL, He SF, Xu DD, He WX, Zheng JF, Huang PQ. Concise total synthesis of (–)-quinocarcin enabled by catalytic enantioselective reductive 1,3-dipolar cycloaddition of secondary amides. Angew Chem Int Ed. 2023;62(25):e202302832.[DOI]
-
32. Kouznetsov VV. Recent synthetic developments in a powerful imino Diels–Alder reaction (Povarov reaction): Application to the synthesis of N-polyheterocycles and related alkaloids. Tetrahedron. 2009;65(14):2721-2750.[DOI]
-
33. Sridharan V, Suryavanshi PA, Menéndez JC. Advances in the chemistry of tetrahydroquinolines. Chem Rev. 2011;111(11):7157-7259.[DOI]
-
34. Jiang X, Wang R. Recent developments in catalytic asymmetric inverse-electron-demand Diels–Alder reaction. Chem Rev. 2013;113(7):5515-5546.[DOI]
-
35. Masson G, Lalli C, Benohoud M, Dagousset G. Catalytic enantioselective [4+2]-cycloaddition: A strategy to access aza-hexacycles. Chem Soc Rev. 2013;42(3):902-923.[DOI]
-
36. Ghashghaei O, Masdeu C, Alonso C, Palacios F, Lavilla R. Recent advances of the Povarov reaction in medicinal chemistry. Drug Discov Today Technol. 2018;29:71-79.[DOI]
-
37. Muthukrishnan I, Sridharan V, Menéndez JC. Progress in the chemistry of tetrahydroquinolines. Chem Rev. 2019;119(8):5057-5191.[DOI]
-
38. Vinogradov MG, Turova OV, Zlotin SG. Catalytic asymmetric aza-Diels–Alder reaction: Pivotal milestones and recent applications to synthesis of nitrogen-containing heterocycles. Adv Synth Catal. 2021;363(6):1466-1526.[DOI]
-
39. Clerigué J, Ramos MT, Menéndez JC. Enantioselective catalytic Povarov reactions. Org Biomol Chem. 2022;20(8):1550-1581.[DOI]
-
40. Masdeu C, de Los Santos JM, Palacios F, Alonso C. The intramolecular Povarov tool in the construction of fused nitrogen-containing heterocycles. Top Curr Chem. 2023;381(4):20.[DOI]
-
41. Kant K, Naik P, Patel CK, Some S, Banerjee S, Aljaar N, et al. Organocatalytic approaches towards the synthesis of asymmetric Tetrahydroquinoline (THQ) derivatives. Synthesis. 2025;57(2):296.[DOI]
-
42. Brion A, Martini V, Lombard M, Retailleau P, Della Ca N, Neuville L, et al. Phenolic dienes as highly selective dienophiles in the asymmetric organocatalyzed three-component vinylogous Povarov reaction. J Org Chem. 2024;89(17):12298-12306.[DOI]
-
43. Jeong HM, Jung HS, Kim DG, Kim JY, Ryu DH. Enantio- and diastereoselective tandem giese addition/homolytic aromatic substitution reaction via visible-light photoredox catalysis. ACS Catal. 2025;15(6):4579-4585.[DOI]
-
44. Sheng FT, Wang JY, Tan W, Zhang YC, Shi F. Progresses in organocatalytic asymmetric dearomatization reactions of indole derivatives. Org Chem Front. 2020;7(23):3967-3998.[DOI]
-
45. Liu YZ, Song H, Zheng C, You SL. Cascade asymmetric dearomative cyclization reactions via transition-metal-catalysis. Nat Synth. 2022;1(3):203.[DOI]
-
46. Pandit S, Majumdar N. Catalytic intermolecular [4+2]-cycloaddition toward the stereoselective C2–C3 annulation of indoles. Synthesis. 2024;57(2):362-379.[DOI]
-
47. Desimoni G, Faita G, Mella M, Toscanini M, Boiocchi M. Multicomponent reactions of indole, ethyl glyoxylate and anilines: From Friedel–Crafts to aza-Diels–Alder reactions catalysed by scandium triflate. Eur J Org Chem. 2009;2009(16):2627-2634.[DOI]
-
48. Wang XS, Yin MY, Wang W, Tu SJ. A stereoselective Povarov reaction leading to exo-tetrahydroindolo[3,2-c]quinoline derivatives catalyzed by iodine. Eur J Org Chem. 2012;2012(25):4811-4818.[DOI]
-
49. Chen Z, Wang B, Wang Z, Zhu G, Sun J. Complex bioactive alkaloid-type polycycles through efficient catalytic asymmetric multicomponent aza-Diels–Alder reaction of indoles with oxetane as directing group. Angew Chem Int Ed. 2013;52(7):2027-2031.[DOI]
-
50. Gao HJ, Miao YH, Sun WN, Zhao R, Xiao X, Hua YZ, et al. Diversity-oriented catalytic asymmetric dearomatization of indoles with o-quinone diimides. Adv Sci. 2023;10(35):e2305101.[DOI]
-
51. Huang YH, Wang SR, Wu DP, Huang PQ. Intermolecular dehydrative [4+2] aza-annulation of N-arylamides with alkenes: A direct and divergent entrance to aza-heterocycles. Org Lett. 2019;21(6):1681-1685.[DOI]
-
52. Song Z, Zhao YM, Zhai H. One-step construction of tetrahydro-5H-indolo[3,2-c]quinolines from benzyl azides and indoles via a cascade reaction sequence. Org Lett. 2011;13(24):6331-6333.[DOI]
-
53. Nishino M, Hirano K, Satoh T, Miura M. Copper-catalyzed oxidative direct cyclization of N-methylanilines with electron-deficient alkenes using molecular oxygen. J Org Chem. 2011;76(15):6447-6451.[DOI]
-
54. Zhao MN, Yu L, Hui RR, Ren ZH, Wang YY, Guan ZH. Iron-catalyzed dehydrogenative [4+2] cycloaddition of tertiary anilines and enamides for the synthesis of tetrahydroquinolines with amido-substituted quaternary carbon centers. ACS Catal. 2016;6(6):3473-3477.[DOI]
-
55. Bush TS, Yap GPA, Chain WJ. Transformation of N,N-dimethylaniline N-oxides into diverse tetrahydroquinoline scaffolds via formal Povarov reactions. Org Lett. 2018;20(17):5406-5409.[DOI]
-
56. Liu H, Dagousset G, Masson G, Retailleau P, Zhu J. Chiral brønsted acid-catalyzed enantioselective three-component Povarov reaction. J Am Chem Soc. 2009;131(13):4598-4599.[DOI]
-
57. Parmar D, Sugiono E, Raja S, Rueping M. Complete field guide to asymmetric BINOL-phosphate derived Brønsted acid and metal catalysis: History and classification by mode of activation; Brønsted acidity, hydrogen bonding, ion pairing, and metal phosphates. Chem Rev. 2014;114(18):9047-9153.[DOI]
-
58. Xia ZL, Xu-Xu QF, Zheng C, You SL. Chiral phosphoric acid-catalyzed asymmetric dearomatization reactions. Chem Soc Rev. 2020;49(1):286-300.[DOI]
-
59. Woldegiorgis AG, Han Z, Lin X. Recent advances in chiral phosphoric acid catalyzed asymmetric organic reactions: An overview. J Mol Struct. 2024;1297:136919.[DOI]
-
60. Adams HK, Kadarauch M, Hodson NJ, Lit AR, Phipps RJ. Design approaches that utilize ionic interactions to control selectivity in transition metal catalysis. Chem Rev. 2025;125(5):2846-2907.[DOI]
-
61. Dagousset G, Retailleau P, Masson G, Zhu J. Chiral phosphoric acid-catalyzed enantioselective three-component povarov reaction using cyclic enethioureas as dienophiles: Stereocontrolled access to enantioenriched hexahydropyrroloquinolines. Chemistry. 2012;18(19):5869-5873.[DOI]
Copyright
© The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Publisher’s Note
Science Exploration remains a neutral stance on jurisdictional claims in published
maps
and institutional affiliations. The views expressed in this article are solely those
of
the author(s) and do not reflect the opinions of the Editors or the publisher.
Share And Cite



Science Exploration Style
Mo NF, Zhang Y, Ren ZH, Zhao Q, Guan ZH. Catalytic asymmetric dearomative formal [4+2] annulation of indoles with O-Silylated hemiaminals as dienes: The dual role of chiral phosphoric acid. Chiral Chem. 2026;2:202511. https://doi.org/10.70401/cc.2025.0008
Tips
Copy completed.
Submit a Manuscript
Author Instructions
Cite this Article
Article Metrics
0
View
0
Download
Cited
Article Updates

Science Exploration Style
Mo NF, Zhang Y, Ren ZH, Zhao Q, Guan ZH. Catalytic asymmetric dearomative formal [4+2] annulation of indoles with O-Silylated hemiaminals as dienes: The dual role of chiral phosphoric acid. Chiral Chem. 2026;2:202511. https://doi.org/10.70401/cc.2025.0008
copy
Share Link
copy