Catalytic construction of P-stereogenic centers through asymmetric hydrophosphination of unsaturated C–C bonds
*Correspondence to:
Yu Lu, School of Multidisciplinary Science, Key Laboratory of Medical Molecule Science and Pharmaceutical Engineering, Ministry of Industry and Information Technology, School of Chemistry and Chemical Engineering, Beijing Institute of Technology, Beijing 100081, China.
E-mail: 6120220270@bit.edu.cn
Xiao-Hui Yang, School of Multidisciplinary Science, Key Laboratory of Medical Molecule Science and Pharmaceutical Engineering, Ministry of Industry and Information Technology, School of Chemistry and Chemical Engineering, Beijing Institute of Technology, Beijing 100081, China. E-mail: xhyang@bit.edu.cn
Xiao-Hui Yang, School of Multidisciplinary Science, Key Laboratory of Medical Molecule Science and Pharmaceutical Engineering, Ministry of Industry and Information Technology, School of Chemistry and Chemical Engineering, Beijing Institute of Technology, Beijing 100081, China. E-mail: xhyang@bit.edu.cn
Chiral Chem. 2026;2:202603. 10.70401/cc.2026.0014
Received: January 28, 2026Accepted: March 04, 2026Published: March 10, 2026
Abstract
P-Stereogenic centers are important structural motifs prevalent in natural products, bioactive molecules, and high-performance ligands. Their presence confers significant value across medicinal chemistry, materials science, and asymmetric catalysis. In recent years, catalytic asymmetric hydrophosphination has emerged as a powerful and efficient strategy for constructing such P-stereogenic compounds. Distinguished by high atom economy, broad substrate compatibility, and excellent stereocontrol under mild conditions, these transformations align closely with the principles of sustainable and green synthesis. This review summarizes recent advances in the catalytic asymmetric hydrophosphination of unsaturated C–C bonds for the synthesis of P-stereogenic centers. It covers substrates including alkenes bearing electron-withdrawing groups, alkynes, as well as specialized systems such as enynes, allenes, and conjugated dienes. Emphasis is placed on the design of catalytic systems, encompassing transition-metal catalysts (e.g., Pd, Ni, Cu, Co, Mn) and organocatalysts, along with their mechanisms. Current challenges, such as the low reactivity of unactivated or sterically hindered substrates and difficulties in achieving stereochemical differentiation between phosphorus substituents, are also discussed. This review aims to provide a reference for further innovation and methodological development in the synthesis of P-stereogenic molecules.
Graphical Abstract
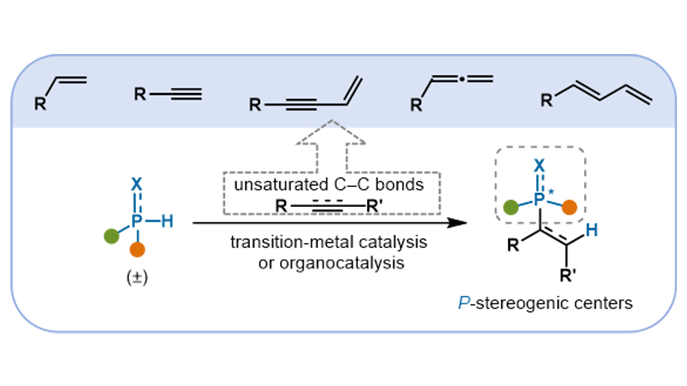
Keywords
P-stereogenic centers, asymmetric hydrophosphination, unsaturated C–C bond, transition-metal catalysis, enantioselective synthesis
1. Introduction
P-Stereogenic centers are common in natural products, bioactive molecules, and high-performance ligands, which give them significant value for applications in medicinal chemistry, materials science, and asymmetric catalysis (Scheme 1a)[1-3]. Consequently, the efficient and highly enantioselective construction of such compounds remains a focal point in synthetic chemistry[4]. In recent years, catalytic asymmetric hydrophosphination reactions have emerged as a powerful strategy for synthesizing P-stereogenic molecules[5-7]. Compared to traditional methods such as chiral resolution and chiral auxiliary-induced strategies, these catalytic transformations not only provide high atom economy, broad substrate scope, and streamlined synthetic routes, but also enable excellent enantiocontrol under mild conditions[8-12].
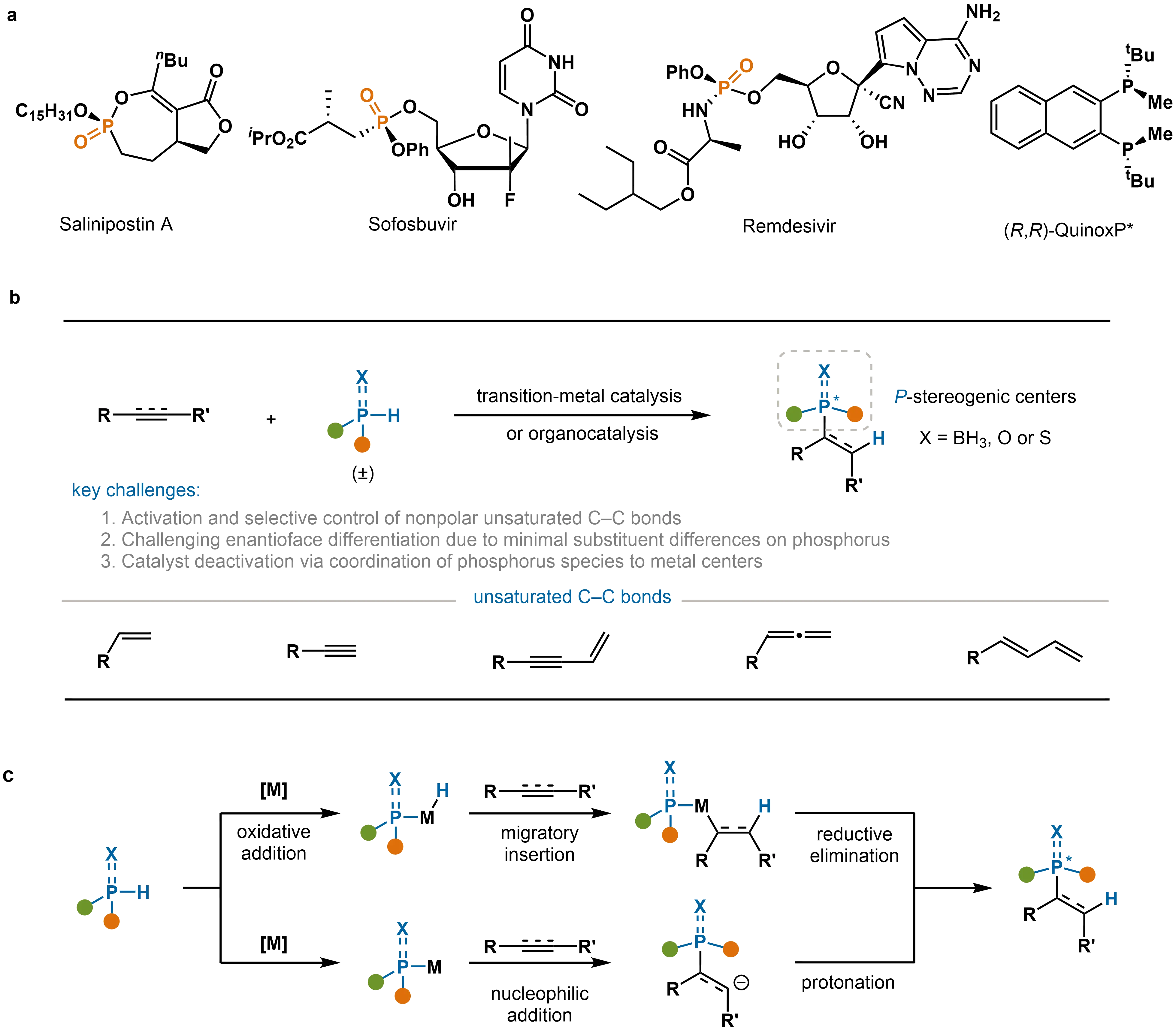
Scheme 1. (a) Representative P-stereogenic functional molecules; (b) Catalytic asymmetric hydrophosphination of unsaturated C–C bonds for P-stereogenic center construction; (c) Two general mechanistic manifolds.
Significant advances have been achieved in the asymmetric hydrophosphination of polar π-bonds (e.g., aldehydes, ketones, and imines)[13]. In contrast, analogous reactions employing nonpolar unsaturated C–C bonds (such as alkenes, alkynes, and conjugated enynes) have been challenging due to issues such as low reactivity and poor control over regioselectivity and stereoselectivity (Scheme 1b). The construction of P-stereogenic centers is particularly challenging in these systems because the stereoelectronic differences between substituents on phosphorus (e.g., diaryl, alkyl-aryl) are often subtle. This makes it difficult for catalytic systems to effectively differentiate between the enantiotopic faces of the phosphorus atom, leading to low enantioselectivity. Recently, advances in transition-metal catalysis (e.g., Pd, Cu, Ni, Co, Mn)[14,15] and the development of strategies such as cooperative catalysis and organocatalysis[16] have enabled a series of efficient and highly selective hydrophosphination reactions. These methods now allow for the precise synthesis and stereocontrol of P-chiral centers, ranging from simple alkene and alkyne substrates to more complex conjugated systems (Scheme 1b). Currently, several mechanistic pathways have been established for this transformation. Two of the most common involve (Scheme 1c): (1) activation of the P–H bond via oxidative addition, followed by migratory insertion and reductive elimination to afford the desired product; and (2) nucleophilic attack of the phosphorus lone pair on an activated alkene, with subsequent protonation yielding the product.
Although several reviews have documented the construction of P-stereogenic centers via strategies such as desymmetrization, kinetic resolution, and chiral auxiliary induction[4,10,11,16,17], a dedicated summary of the construction of these compounds via the hydrophosphination of unsaturated C–C bonds is lacking. To fill this gap, this review summarizes recent advances in the catalytic asymmetric synthesis of P-stereogenic compounds through the hydrophosphination of unsaturated C–C bonds. It highlights the design concepts, catalyst structures, key intermediates, and mechanisms. Furthermore, current challenges and prospective research directions in this field are discussed, with the goal of providing a useful reference for further methodological innovation and expansion in this rapidly evolving area.
For clarity and consistency in this discussion, we adopt the umbrella term “hydrophosphination” to refer to the addition reactions of both trivalent (P(III)) and pentavalent phosphorus (P(V)) reagents across the C–C π bonds of alkenes or alkynes. Strictly speaking, this terminology encompasses both hydrophosphination (with P(III) nucleophiles) and hydrophosphorylation (with P(V) nucleophiles). Its broad application here, however, facilitates a unified and comparative analysis of methodologies that construct C–P bonds through alkene and alkyne functionalization. This inclusive definition enables a systematic examination of the catalytic strategies, stereocontrol mechanisms, and substrate scopes relevant to these valuable transformations.
2. Asymmetric Hydrophosphination of Alkenes Bearing Electron-Withdrawing Groups
Activated alkenes, bearing electron-withdrawing groups directly attached to their unsaturated C–C bonds, have emerged as the predominant substrates for constructing P-stereogenic centers through asymmetric hydrophosphination. Representative examples include α,β-unsaturated carbonyl compounds (such as ketones, amides, and esters), vinyl sulfones, and phosphine sulfides. The electron-withdrawing groups in these substrates can effectively lower the lowest unoccupied molecular orbital (LUMO) energy level of the unsaturated C–C bond[18,19], which enhances its electrophilicity and facilitates nucleophilic attack. Additionally, the defined spatial and electronic environment provided by these groups offers favorable opportunities for stereoselectivity control. To date, various catalytic systems have been developed to accommodate these activated alkenes, including transition-metal catalysts based on copper, palladium, and manganese, as well as organocatalytic approaches. By employing distinct activation modes and stereoregulation mechanisms, these systems enable the efficient and highly enantioselective construction of P-stereogenic centers across a range of structurally diverse activated alkene substrates.
2.1 Ni- or Pd-catalyzed hydrophosphination of alkenes
In 2014, Duan and coworkers developed an NCN-type bisimidazoline Pd-catalyst (Catalyst 1) and a PCP-type Pd-catalyst (Catalyst 2). These catalysts successfully enabled the highly enantioselective 1,4-addition of racemic alkylphenylphosphines to α,β-unsaturated ketones, providing access to chiral phosphorus compounds featuring both P-stereogenic and C-stereogenic centers (Scheme 2a,b)[20]. Subsequently, Duan, Wang and co-workers also designed a novel chiral bisphosphine (PCP′) pincer nickel complex (Catalyst 3) in 2021 (Scheme 2c)[21]. This catalyst enabled the construction of P-stereogenic centers through the asymmetric Michael addition of primary phosphines to α,β-unsaturated ketones. Notably, this approach circumvented the challenge that chiral secondary phosphines are hard to be synthesized due to their low inversion barrier of phosphorus atoms and easy racemization. These systems demonstrated broad substrate scope, including a range of α,β-unsaturated ketones with β-aryl, β-heteroaryl, and β-alkyl substituents, as well as α,β-unsaturated N-acylpyrroles, all of which reacted smoothly under the developed conditions.
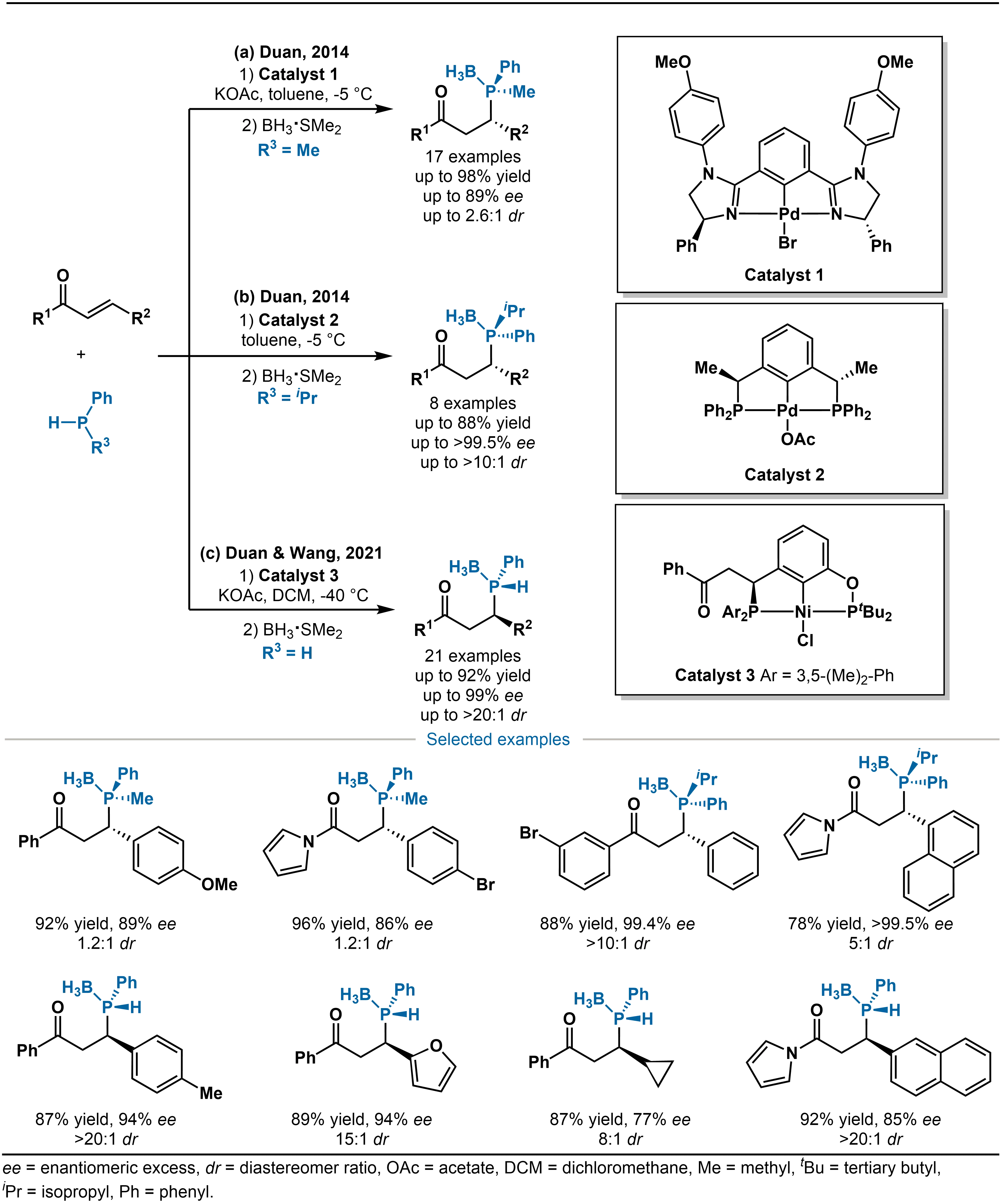
Scheme 2. (a-b) Pd-catalyzed asymmetric hydrophosphination of alkenes; (c) Ni-catalyzed asymmetric hydrophosphination of alkenes.
The proposed catalytic cycle is summarized in Scheme 3. The precatalyst 1 undergoes anion exchange with potassium acetate (KOAc) to generate the active nickel species 2. Subsequently, phenylphosphine participates in a transphosphination process with intermediate 2, during which the acetate ion acts as a base to deprotonate the phosphine and form the key nickel phosphido intermediate 3. The subsequent step is the enantioselectivity-determining step: the nucleophilic addition of nickel phosphido intermediate 3 to the β-carbon of the enone (4) takes place under the stereodirecting influence of the chiral PCP′ ligand. The bulky substituents (such as tert-butyl) on the ligand shield one prochiral face of the enone, thereby directing the phosphido attack predominantly to the less hindered opposite face. This process simultaneously establishes a C-stereogenic center and fixes the configuration of the resulting P-stereogenic center. Following the nucleophilic addition, the resulting alkoxide-nickel species 5 is protonated by acetic acid present in the reaction medium. This releases the secondary phosphine product 6 and regenerates the active nickel-acetate catalyst. It is noteworthy that the free secondary phosphine product must be immediately protected with borane after completion of the reaction. Borane coordination effectively stabilizes the P-stereogenic center, preventing its racemization upon warming and facilitating isolation as well as downstream transformations.
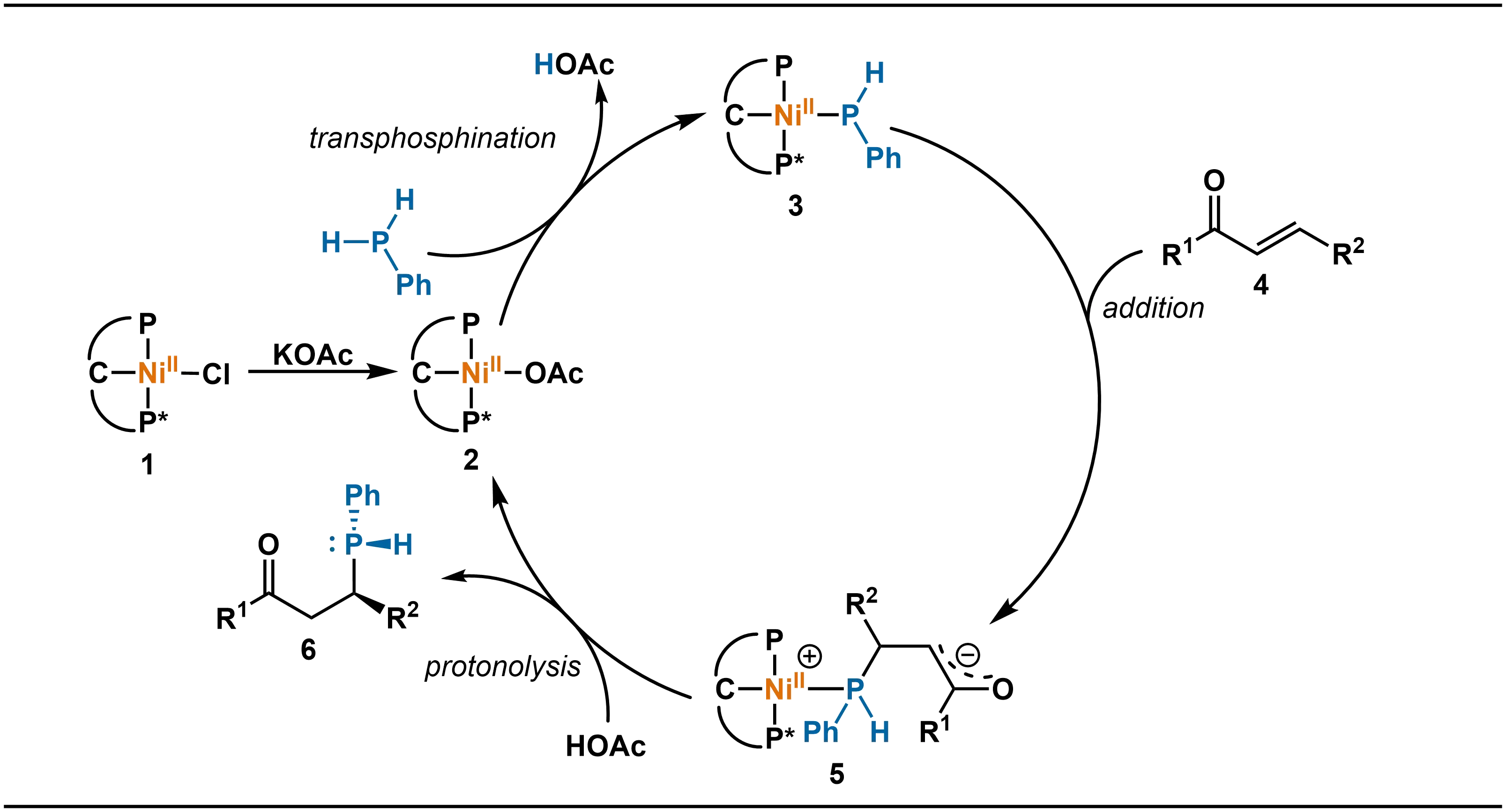
2.2 Co-catalyzed hydrophosphination of alkenes
In 2021, Duan and coworkers reported a cobalt-catalyzed (Catalyst 4) and pyridinyl-directed approach for the enantioselective hydrophosphination of acrylate derivatives with secondary phosphine oxides (SPOs) (Scheme 4)[22]. This cobalt catalytic system is not only applicable to acrylates but also exhibits good universality for alkenes containing cyano or amide groups, as well as more electron-deficient substrates such as sulfonates and aryl sulfoxides. In all cases, the corresponding P-stereogenic tertiary phosphine oxides were obtained in excellent yields (up to 99%) and enantioselectivities (up to 99.5% ee). It should be noted that the pyridinyl directing group is essential for the transformation, as no reaction was observed in its absence.
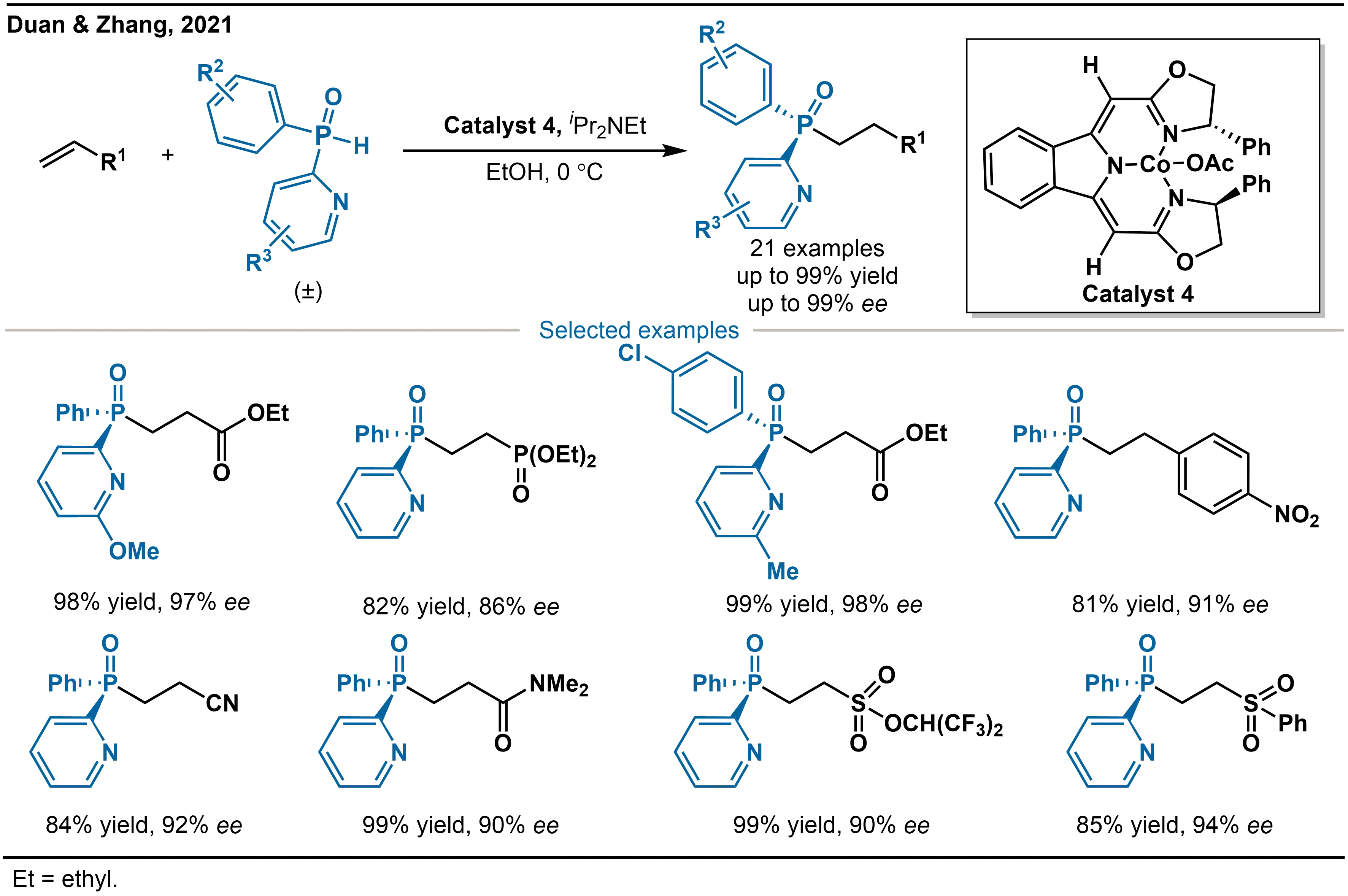
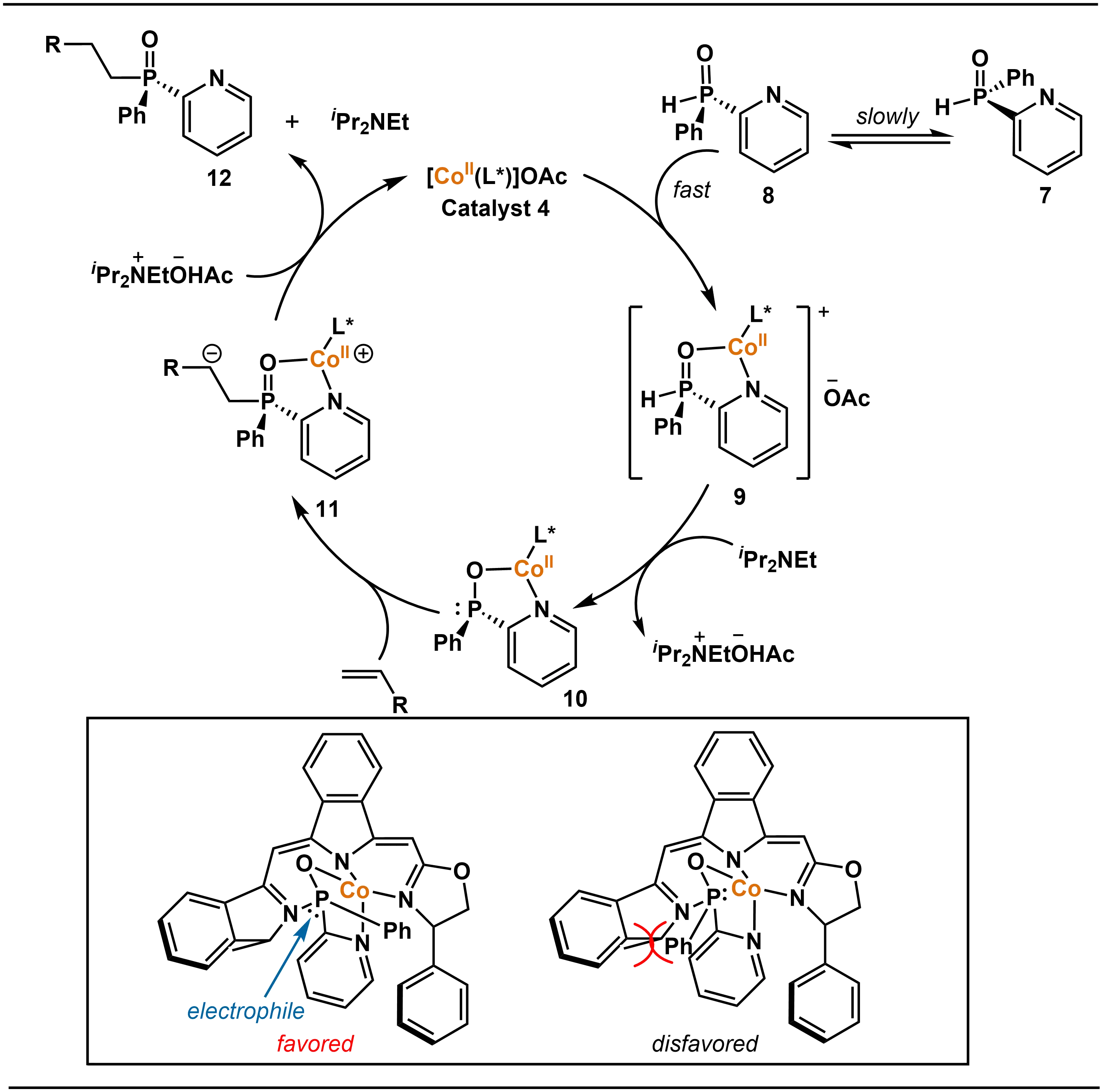
Mechanistic investigations revealed that the pyridinyl group functions as a directing moiety in this transformation. By engaging in bidentate coordination with the cobalt catalyst, it effectively differentiates the two sterically similar aryl substituents on the phosphorus atom of the secondary phosphine oxide, thereby enabling high enantiocontrol. As illustrated in Scheme 5, the proposed catalytic cycle involves the following steps: First, the chiral cobalt Catalyst 4 selectively coordinates with the (R)-configured secondary phosphine oxide 8 to form a stable pentavalent phosphorus intermediate 9, a step that already displays significant enantioselectivity. Subsequently, in the presence of base, intermediate 9 is converted into the trivalent phosphorus nucleophilic species 10. Due to the steric constraints from the ligand structure, nucleophilic attack occurs preferentially from the less hindered face, which affords intermediate 11. Finally, protonation of 11 releases the (R)-tertiary phosphine oxide product 12 and regenerates the catalyst.
2.3 Mn-catalyzed hydrophosphination of alkenes
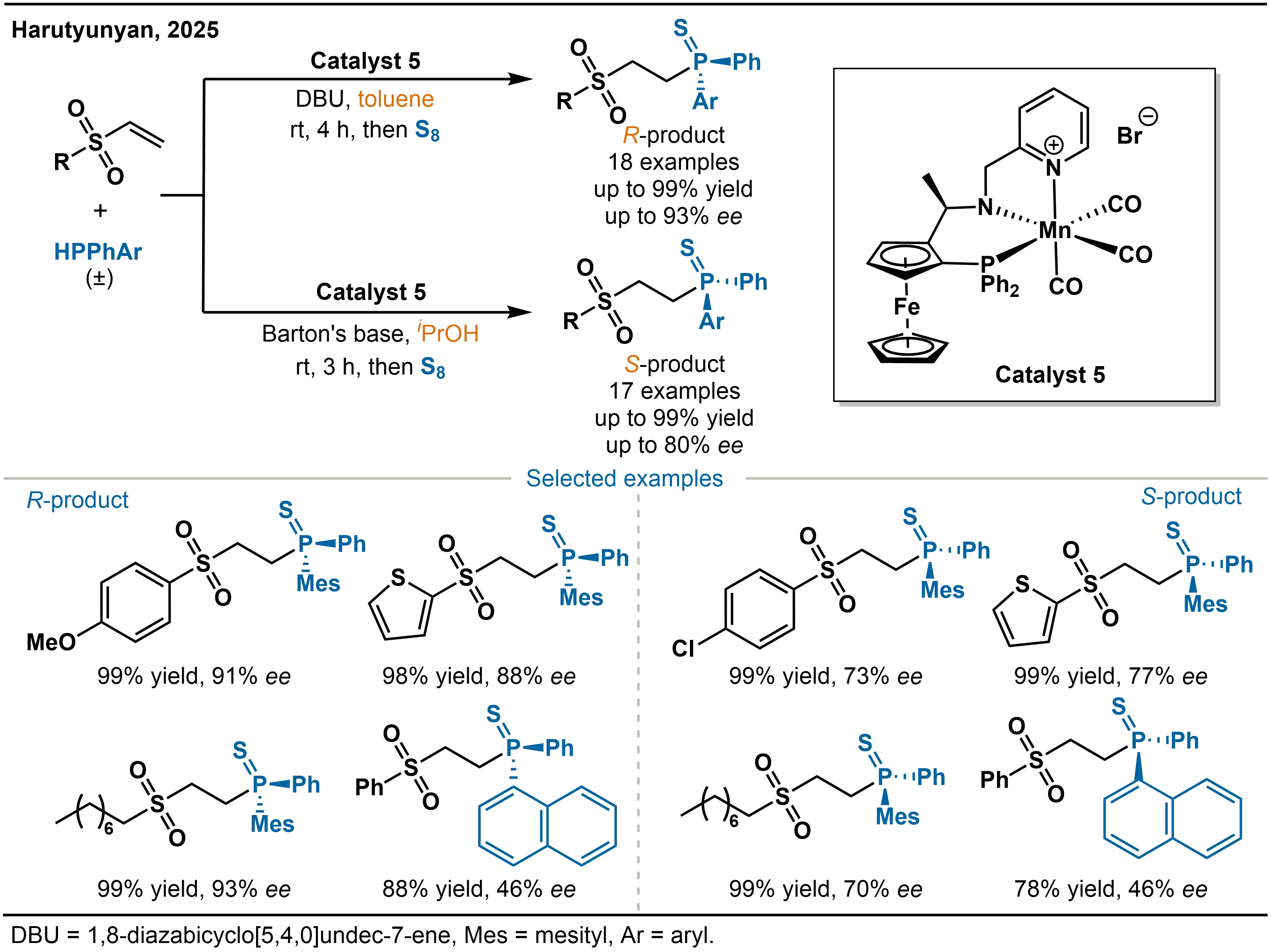
α,β-Unsaturated sulfones, such as vinyl sulfones, possess strong electron-withdrawing properties that significantly enhance the reactivity of carbon-carbon double bonds. Harutyunyan and co-workers exploited this structural feature to develop an efficient catalytic system based on a manganese(I) complex (Catalyst 5) with 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) or 2-tert-butyl-1,1,3,3-tetramethylguanidine (Barton's base) as additives (Scheme 6)[23]. By adjusting the reaction solvent (toluene or protic alcohols), this system achieves highly enantioselective hydrophosphination of vinyl sulfones with racemic diarylphosphines, thereby providing an efficient route to P-stereogenic phosphine derivatives. A notable feature of this Mn-catalyzed asymmetric hydrophosphination is the solvent-dependent stereochemical outcome. In the aprotic solvent toluene, the reaction predominantly affords (R)-configured P-stereogenic phosphines with ee values up to 93% under optimized conditions. Switching the solvent to protic solvents such as iPrOH and tBuOH results in complete inversion of product configuration, leading mainly to the (S)-configured products. Further addition of Barton’s base and optimization of catalyst loading allow the ee values of the inverted configuration to be enhanced to a maximum of 80%, which highlights the pivotal role of solvent regulation in governing stereoselectivity.
2.4 Cu-catalyzed hydrophosphination of alkenes
α,β-Unsaturated amides exhibit notably higher LUMO energy levels compared to the corresponding aldehydes, esters, and ketones due to conjugation between the nitrogen lone pair and the carbonyl group. This electronic feature leads to reduced electrophilicity and poses a significant challenge for conventional phosphorus nucleophiles to undergo direct addition.
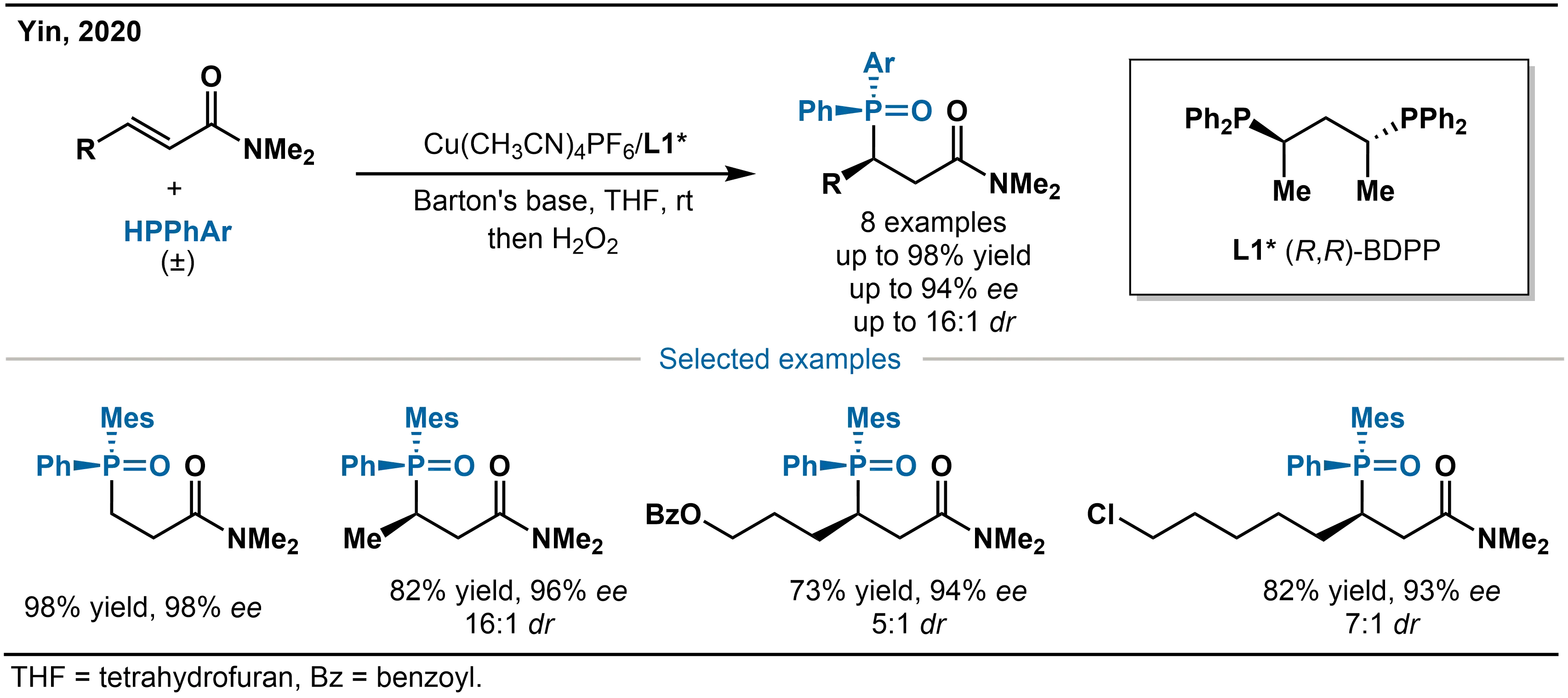
To address this challenge, Yin and co-workers developed in 2020 a Cu(I)-catalyzed asymmetric hydrophosphination of α,β-unsaturated amides (Scheme 7)[24]. By using the high nucleophilicity of in situ generated Cu(I)-PPh2 species, this system effectively activates α,β-unsaturated amides and enables their conversion into chiral phosphine derivatives with high enantioselectivity. Moreover, by employing unsymmetric diarylphosphines as nucleophiles in combination with the (R,R)-BDPP chiral ligand ((R,R)-bis(diphenylphosphino)pentane, L1*), they achieved the synthesis of the corresponding P-stereogenic phosphine compounds with excellent enantioselectivity (up to 98% ee) and good diastereoselectivity (up to 16:1 dr).
Chiral phosphine ligands are key components in asymmetric catalysis. Among them, 1,2-bisphosphines exhibit outstanding performance in numerous reactions owing to their rigid chelating scaffolds. To enable efficient synthesis of such ligands, Yin and co-workers further developed a Cu-catalyzed conjugate addition of diphenylphosphine to α,β-unsaturated phosphine sulfides (Scheme 8)[25]. The study was subsequently extended to employ racemic diarylphosphine, investigating its catalytic asymmetric addition to α,β-unsaturated phosphine sulfides. By employing chiral ligands such as (R,R)-Ph-BPE (1,2-bis(2,5-diphenylphospholano)ethane, L2*) or (R,R)-BDPP (L1*), dynamic kinetic resolution of racemic diarylphosphines was successfully achieved, enabling the efficient generation of P-stereogenic phosphines. This strategy overcame several intrinsic challenges: (1) the inherently low reactivity of α,β-unsaturated phosphine sulfides; (2) potential catalyst deactivation resulting from product coordination as a bidentate ligand; (3) erosion of enantioselectivity due to ligand exchange during the reaction; and (4) the subtle steric differentiation between the two aryl groups in diarylphosphines, which complicates efficient dynamic kinetic resolution.
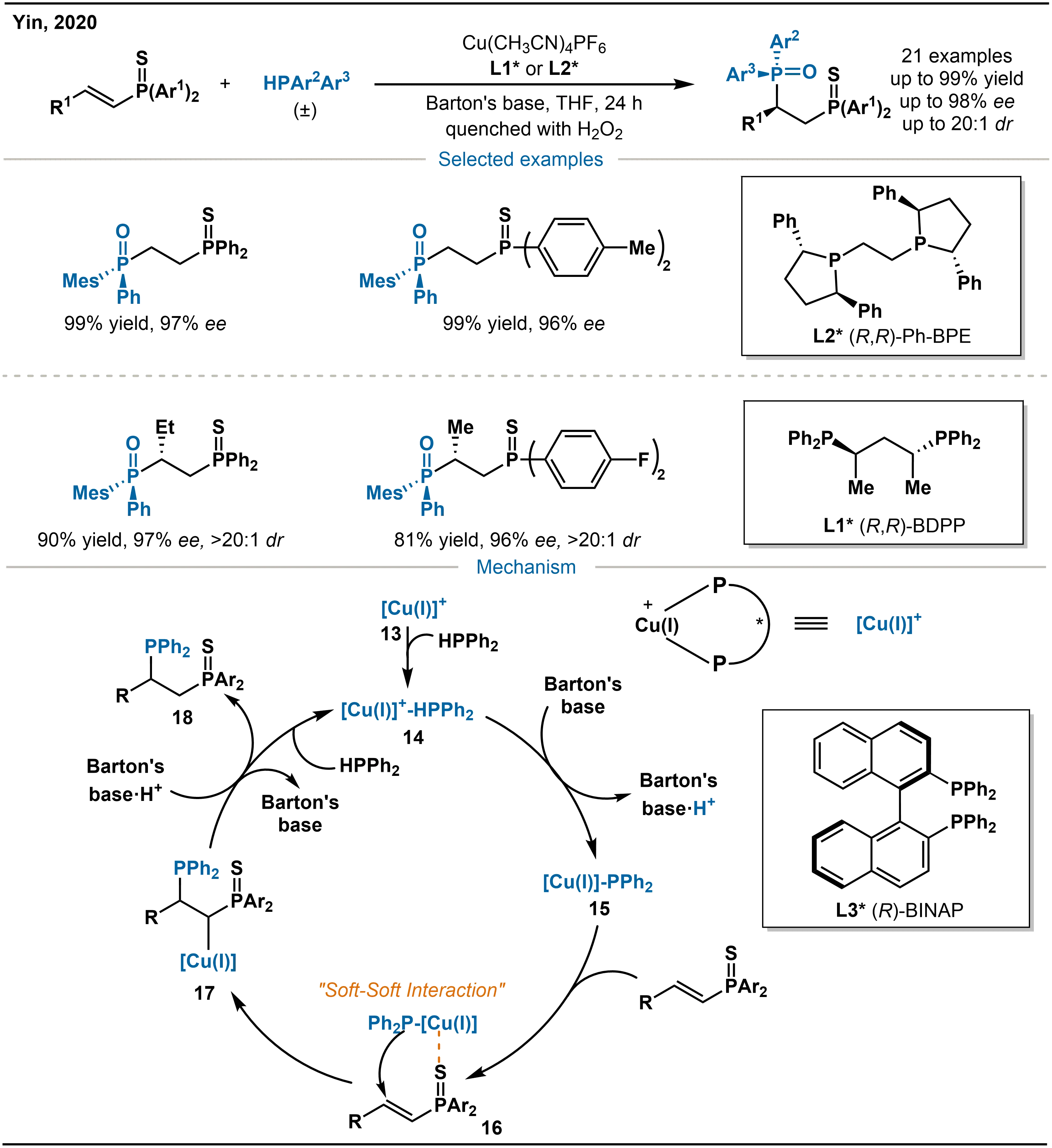
Scheme 8. Mechanism of Cu-catalyzed asymmetric hydrophosphination of α,β-unsaturated phosphine sulfides.
With diphenylphosphine as the phosphorus source, the proposed catalytic cycle is as follows: HPPh2 coordinates with Cu(I)-(R)-BINAP (2,2'-bis(diphenylphosphino)-1,1'-binaphthyl) to form complex 14, which promotes the deprotonation of HPPh2 to give the nucleophilic Cu(I)-diphenylphosphido species 15. This intermediate 15 then coordinates with α,β-unsaturated phosphine sulfides through the “soft-soft interaction” between Cu(I) and sulfur atoms, this interaction originates from the inherent affinity of the soft Lewis acidic Cu(I) center for the soft Lewis basic sulfur atoms, thereby promoting the subsequent nucleophilic addition to afford intermediate 17. Protonation of 17 releases the product and regenerates the copper catalyst, which re-associates with HPPh₂ to close the catalytic cycle through the re-formation of 14. It is worth noting that the deprotonation of HPPh2 by Barton’s base does not proceed efficiently in the absence of the copper complex, underscoring the essential role of metal coordination in activating the phosphorus nucleophile.
Building on the aforementioned findings, in 2024, Yin and co-workers reported a Cu(I)-catalyzed diastereodivergent addition of phosphinothioates to α,β-unsaturated thioamides (Scheme 9)[26]. This transformation, facilitated by the “soft-soft” interaction between the copper center and sulfur atoms, efficiently constructs phosphinate derivatives bearing vicinal P- and C-stereogenic centers with high diastereoselectivity and enantioselectivity. Investigations revealed that the ligand structure critically controls the diastereomeric outcome of the reaction. The use of (R,R)-Ph-BPE ligand (L2*) exclusively affords one diastereomer, whereas switching to (R,Rp)-TANIAPHOS ligand (L4*) leads selectively to the other configuration, demonstrating complete stereodivergence driven by ligand choice. The reaction exhibits broad substrate generality. A wide range of α,β-unsaturated thioamides and phosphinothioates, bearing aryl, heteroaryl, or alkyl substituents with diverse substitution patterns, are well tolerated, consistently delivering the corresponding products in good to excellent yields and with high enantioselectivity. The observed diastereodivergence originates from the distinct chiral environments created by the two ligands, which precisely steer the reaction along different stereochemical trajectories through tailored “soft-soft” coordination, ultimately yielding diastereomers with defined and complementary configurations.
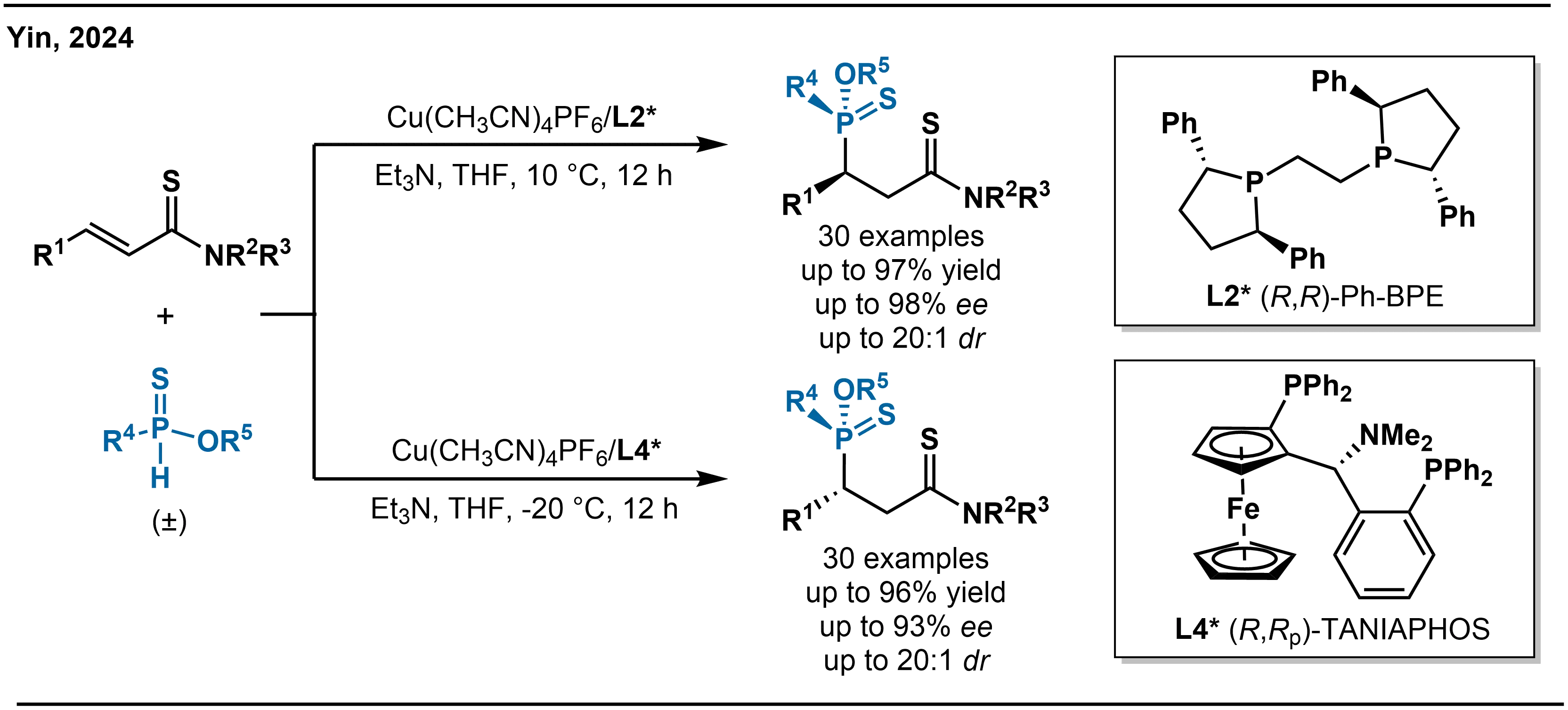
Scheme 9. Cu-catalyzed asymmetric diastereodivergent hydrophosphination of α,β-unsaturated thioamides.
Yin and co-workers have achieved the efficient construction of P-stereogenic phosphine compounds via a copper-catalyzed nucleophilic addition pathway. In contrast, Gong and co-workers confronted head-on the core challenge that phosphorus-centered radicals have long been inaccessible for asymmetric catalysis due to their inherent configurational lability, low inversion barrier, and distinctive reactivity profiles, and thus developed a novel copper-photoredox synergistic catalytic system. They accomplished for the first time the visible light-driven kinetic resolution of racemic H-phosphinates with α-trifluoromethyl styrenes, efficiently affording a diverse array of fluorine-containing P-chiral phosphinates with significant pharmaceutical potential (Scheme 10)[27]. This method successfully overcomes the long-standing issue of the facile racemization of phosphorus-centered radicals and broadens the application prospects of metallaphotoredox catalysis in heteroatom-centered stereocontrol.
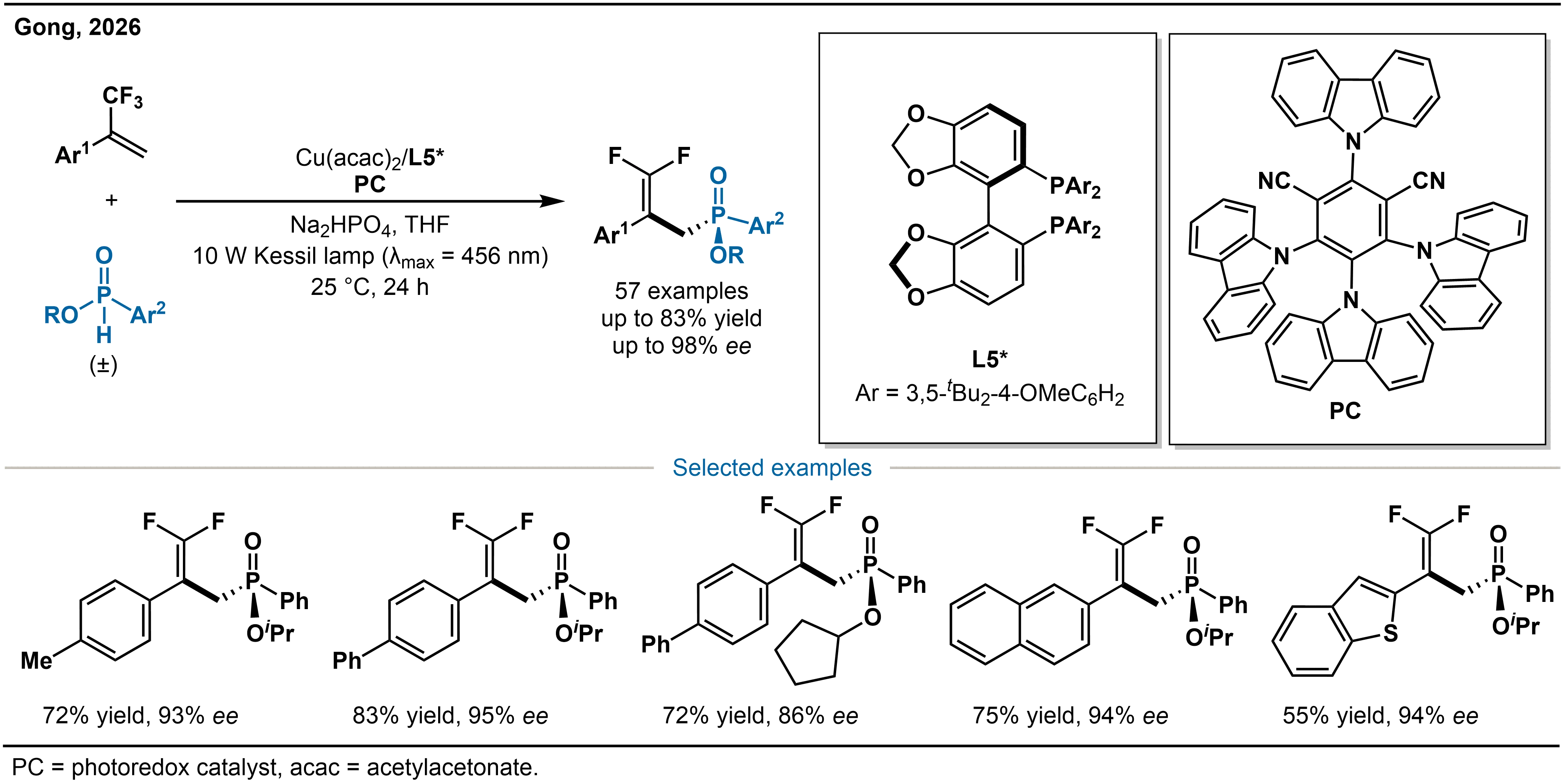
Scheme 10. Cu-catalyzed phosphorus radical transformations for the assembly of P-stereogenic architectures.
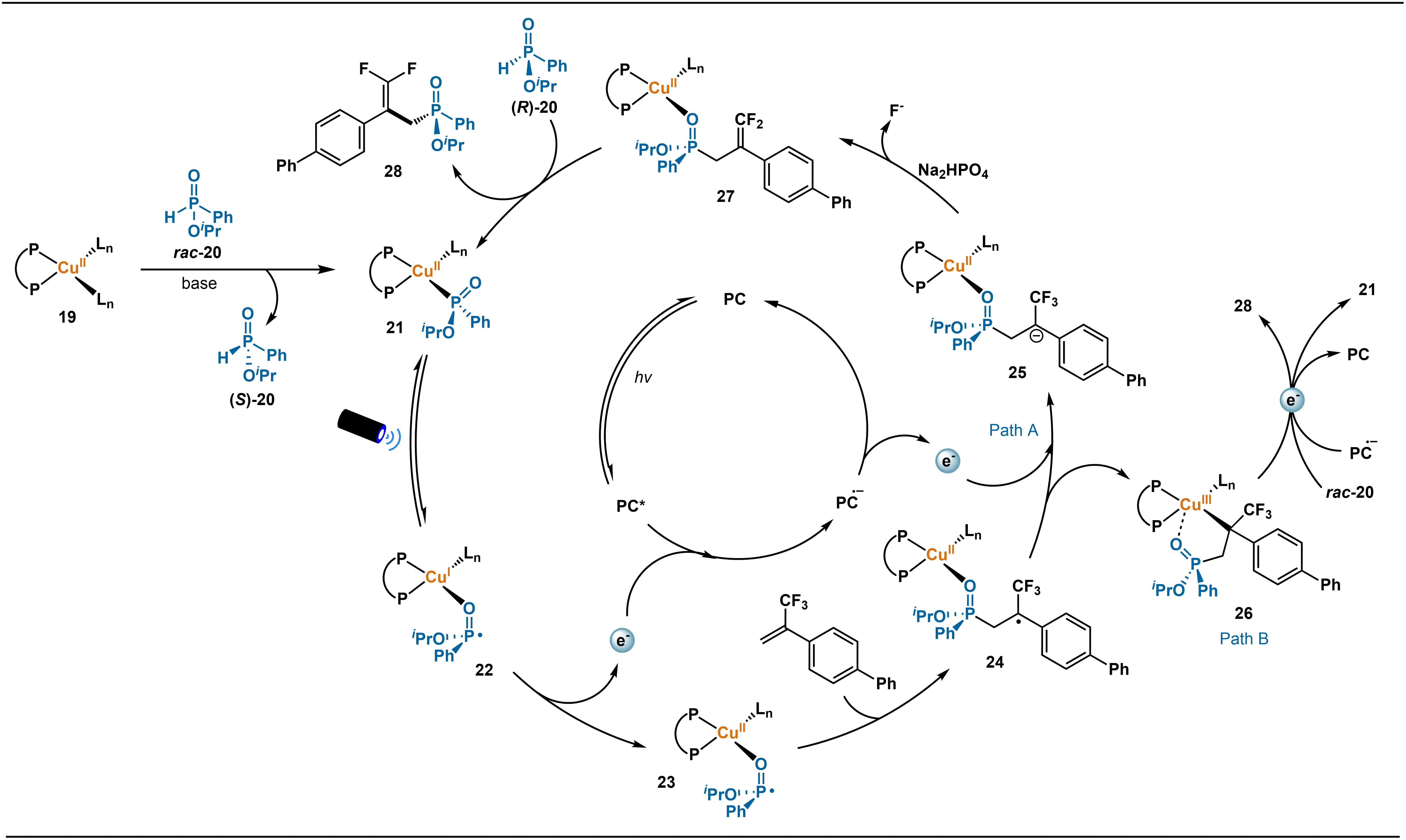
The authors propose a Cu-photoredox synergistic catalytic strategy for stereocontrol, the core of which lies in a sequential “stereochemical relay” process (Scheme 11). The catalytic cycle begins with base-assisted ligand exchange between the Cu catalyst 19 and the H-phosphinate substrate (rac-20), affording the chiral Cu(II) intermediate 21. Due to superior steric matching, the (R)-configured substrate reacts more rapidly, enabling efficient kinetic resolution. Subsequently, under visible-light irradiation, intermediate 21 undergoes a ligand-to-metal charge transfer (LMCT) process that triggers homolytic cleavage of the P–O bond, generating a key, configurationally stable Cu(I)-phosphorus radical intermediate 22. This metal-bound P-centered radical retains its configuration under mild photochemical conditions and does not readily racemize. Concurrently, the photocatalyst PC absorbs a photon to reach its excited state PC*. The metal-bound phosphorus radical is then oxidized by the excited photocatalyst and adds stereospecifically to the α-trifluoromethylstyrene. The resulting alkyl radical intermediate 24 undergoes single-electron reduction, followed by β-fluorine elimination and ligand exchange, to deliver the enantiomerically enriched P-chiral phosphinate product 28 and regenerate the catalyst (PathA). An alternative pathway (PathB) may also operate, in which 24 is intercepted intermolecularly to form an alkyl-Cu(III) intermediate 26. Subsequent β-fluorine elimination, single-electron transfer from PC•–, and ligand exchange with rac-20 under basic conditions likewise afford product 28 and regenerate the catalyst. Both pathways are consistent with experimental observations and together provide a comprehensive mechanistic framework for the transformation.
2.5 Organocatalytic asymmetric hydrophosphination of alkenes
In addition to the aforementioned metal catalytic systems, Jiang, Yin, Ban and co-workers developed an organocatalytic approach using chiral Brønsted acid catalysis (Scheme 12)[28]. This method enables the highly enantioselective hydrophosphinylation of 2-vinylazaarenes with SPOs, providing efficient access to P-stereogenic 2-azaarylethylphosphine oxides, valuable scaffolds in asymmetric metal catalysis. The reaction employs the chiral spirocyclic phosphoric acid (Catalyst 6) and is performed in a mixed solvent of toluene and cyclohexane. The catalytic system exhibits broad substrate tolerance, accommodating various alkyl-aryl-substituted SPOs as well as a wide range of 2-vinylazaarenes, including pyridines, isoquinolines, and benzothiazoles. High enantioselectivity is consistently maintained regardless of whether electron-donating or electron-withdrawing substituents are present on the azaarene aromatic rings. Notably, the authors further expanded the substrate scope to diaryl-substituted SPOs, achieving the first highly enantioselective transformation of such derivatives, thereby enhancing the synthetic utility of the methodology.
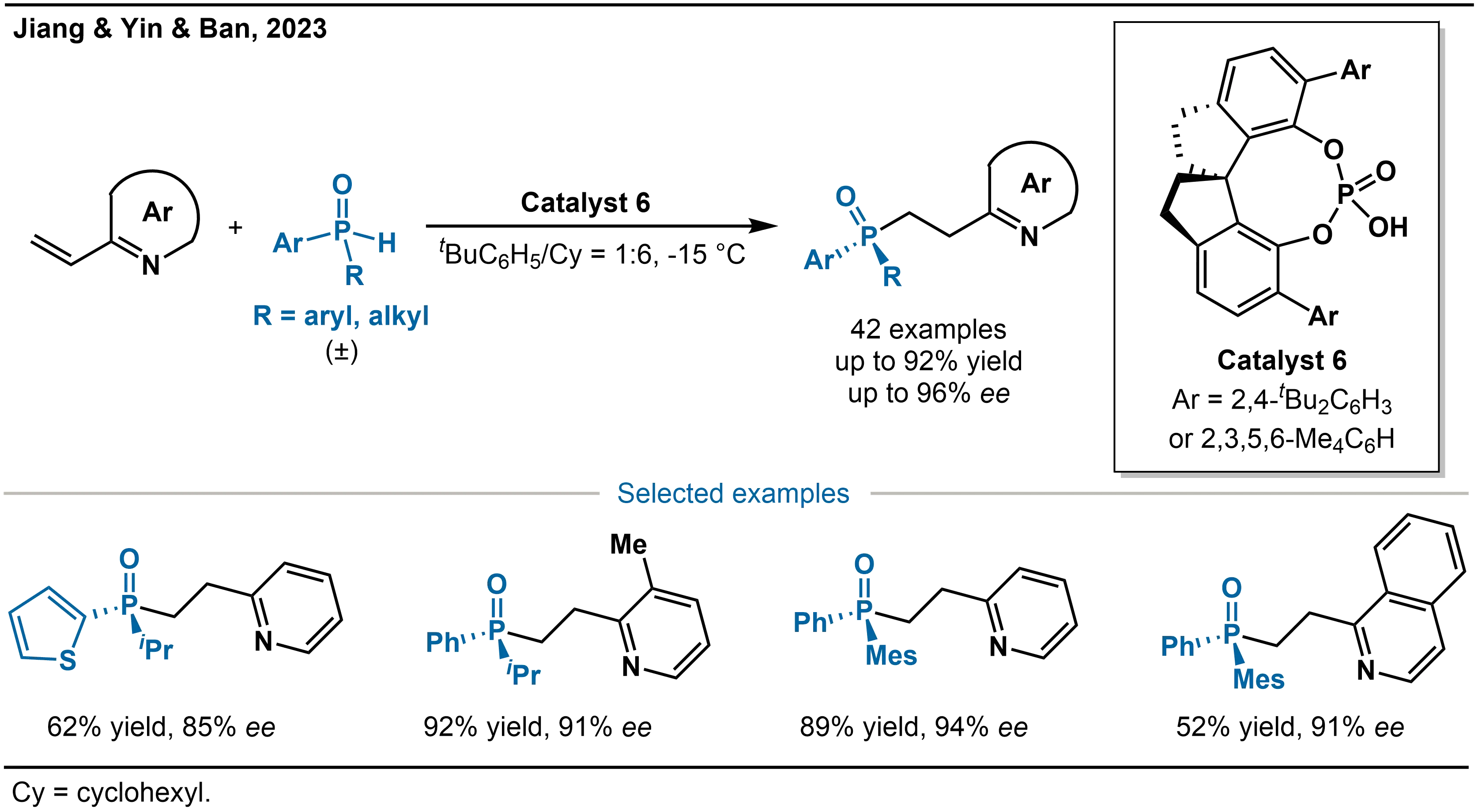
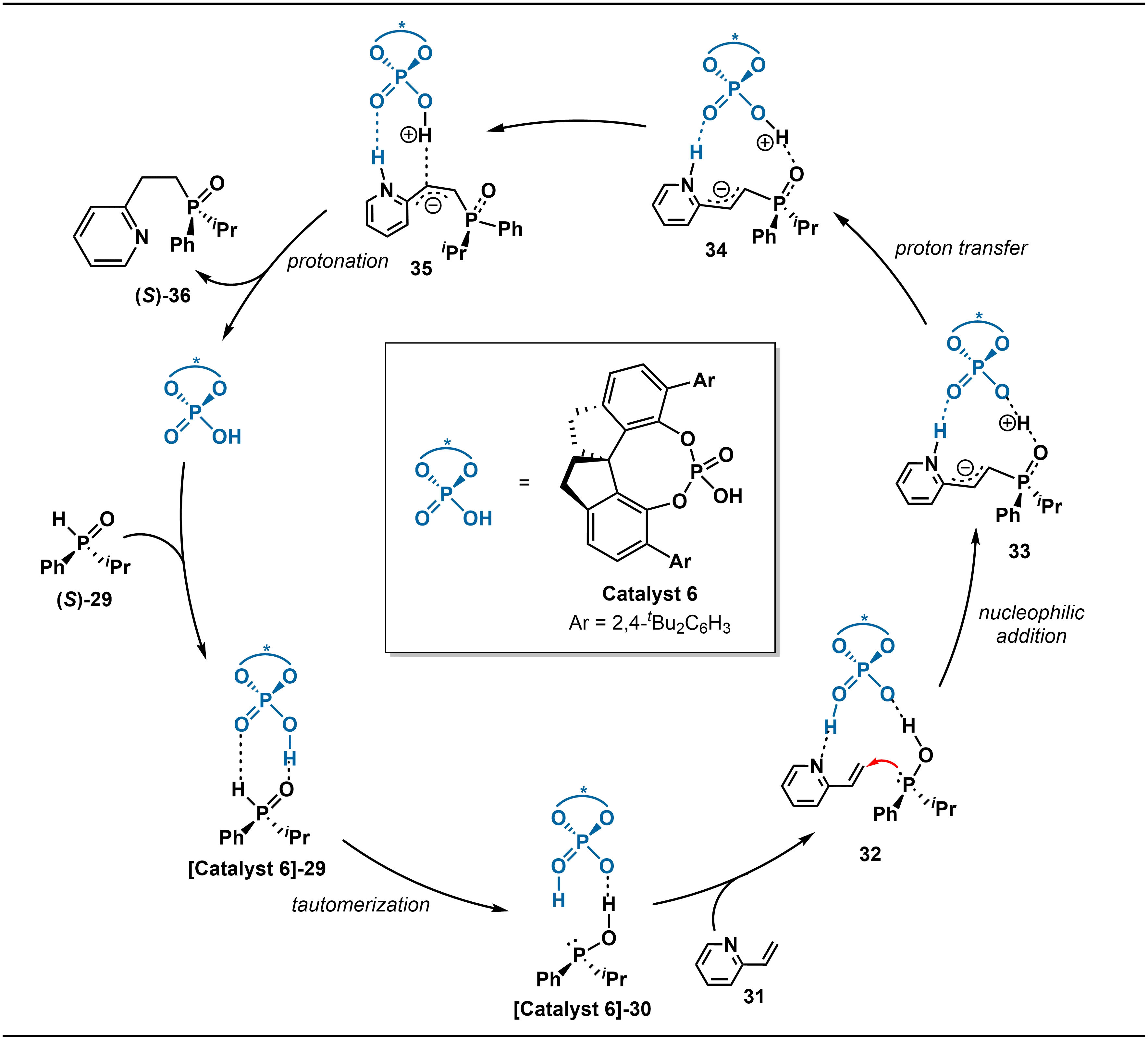
The catalytic cycle of the chiral Brønsted acid Catalyst 6 for this asymmetric hydrophosphination begins with the activation of the secondary phosphine oxide 29 (Scheme 13). The catalyst significantly lowers the energy barrier for the tautomerization of 29 to the highly reactive trivalent phosphinous intermediate 30 through hydrogen-bonding interactions. Subsequently, Catalyst 6 simultaneously binds both 30 and the 2-vinylazaarene 31 to form a pre-reactive chiral complex 32 stabilized by dual hydrogen bonds. Within this complex, the trivalent phosphinous intermediate 30 undergoes nucleophilic attack on the carbon-carbon double bond of 31, which is the enantioselectivity-determining step. After the addition, the hydroxyl proton of 30 is transferred back to the catalyst to yield intermediate 34, which then undergoes conformational adjustment through rotation of the C–C and P–C bonds to form intermediate 35. Finally, the α-carbanion in 35 is protonated to afford the final product, the P-chiral tertiary phosphine oxide 36, while the catalyst is regenerated, thus closing the catalytic cycle.
3. Asymmetric Hydrophosphination of Alkynes
Alkynes, as readily available and abundant unsaturated building blocks, offer a versatile platform for catalytic asymmetric hydrophosphination. This reaction has emerged as an efficient route to alkenylphosphine derivatives containing P-stereogenic centers. Such products integrate the synthetic versatility of the alkenyl moiety with the stereochemical directing capability of the phosphorus chiral center, rendering them valuable synthetic intermediates. These compounds can be further elaborated into functional skeletons, including bidentate phosphine ligands and precursors to bioactive molecules, thus bearing significant potential in asymmetric catalysis, medicinal chemistry, and polymer synthesis.
The efficient implementation of this transformation faces several intertwined challenges: precise control over regioselectivity (Markovnikov vs. anti-Markovnikov addition) is required; enantioselectivity must be induced through tailored steric and electronic matching between the chiral catalyst and the substrate; the structural diversity and reaction compatibility of phosphorus reagents, including secondary phosphines, phosphine oxides, and phosphinates, remain to be expanded; and the activation of steric hindrance or aliphatic alkynes continues to pose significant difficulties. In recent years, advances in transition-metal catalysis (e.g., Pd, Ni, Cu) combined with rational chiral ligand design have provided key strategies to address these challenges. Through careful modulation of the electronic properties of the metal center and the steric structure of the ligand, highly selective hydrophosphination of diverse alkynes has been realized.
3.1 Pd-catalyzed asymmetric hydrophosphination of alkynes
Palladium catalysis represents a prominent approach for constructing P-stereogenic centers via asymmetric hydrophosphination of alkynes. Leveraging the distinct coordination and insertion properties of palladium, combined with the steric and electronic modulation offered by chiral bidentate phosphine ligands, Pd-catalysis enables precise control over both regioselectivity and enantioselectivity while maintaining broad compatibility with diverse phosphorus reagents and alkyne substrates. As a result, palladium catalysis has become one of the core catalytic platforms in this field.
The difficulty in achieving dynamic kinetic resolution with phosphinate substrates, due to their high configurational stability, has hampered efficient access to multifunctional P-stereogenic phosphinates. To overcome this limitation, in 2020, Wang and co-workers reported a catalytic system consisting of palladium and the chiral ligand L6*, which effectively addresses both reactivity and enantiocontrol challenges in the hydrophosphination of alkynes with racemic phosphinates (Scheme 14)[29]. This system accommodates a wide range of alkyne substrates, including aryl, heteroaryl, aliphatic, and internal alkynes, as well as phosphinates bearing various ester substituents, affording the corresponding P-stereogenic alkenylphosphinates in good yields (up to 85%) and high enantioselectivities (up to 86% ee). The method also permits gram-scale synthesis and subsequent diversification of the products.
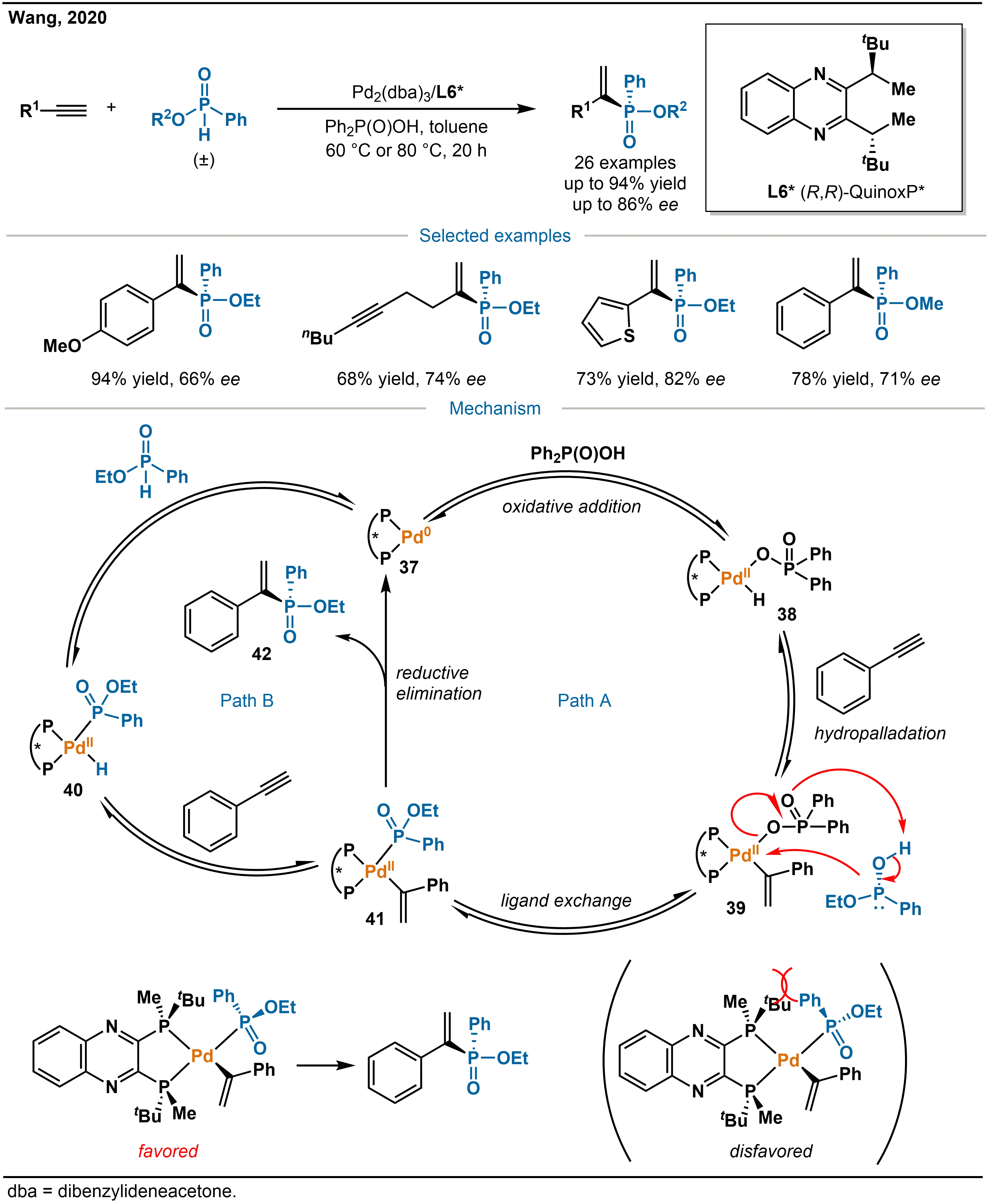
Scheme 14. Pd-catalyzed asymmetric hydrophosphination of alkynes and the mechanism.
Regarding the reaction mechanism, the authors propose two competing or parallel pathways: Path A and Path B. In Path A, the chiral Pd catalyst37 undergoes oxidative addition of the O–H bond of the additive Ph2P(O)OH to generate the intermediate 38. Subsequent Markovnikov-type hydropalladation of the alkyne yields alkenylpalladium species 39, which then undergoes ligand exchange with the phosphinate substrate to give the key phosphinylpalladium intermediate 41. Reductive elimination from 41 releases the alkenylphosphinate product and regenerates the catalyst. The observation that the reaction efficiency decreases in the absence of Ph2P(O)OH supports the involvement of this additive in the catalytic cycle via PathA. Alternatively, Path B involves direct oxidative addition of the P–H bond of the phosphinate substrate to Pd catalyst 37, forming the phosphinyl hydridopalladium intermediate 40. Subsequent hydropalladation of the alkyne leads to the same intermediate 41 as in Path A, followed by an identical reductive elimination step. Deuterium-labeling experiments indicate that the P–H bond insertion is reversible, providing evidence consistent with the feasibility of Path B.
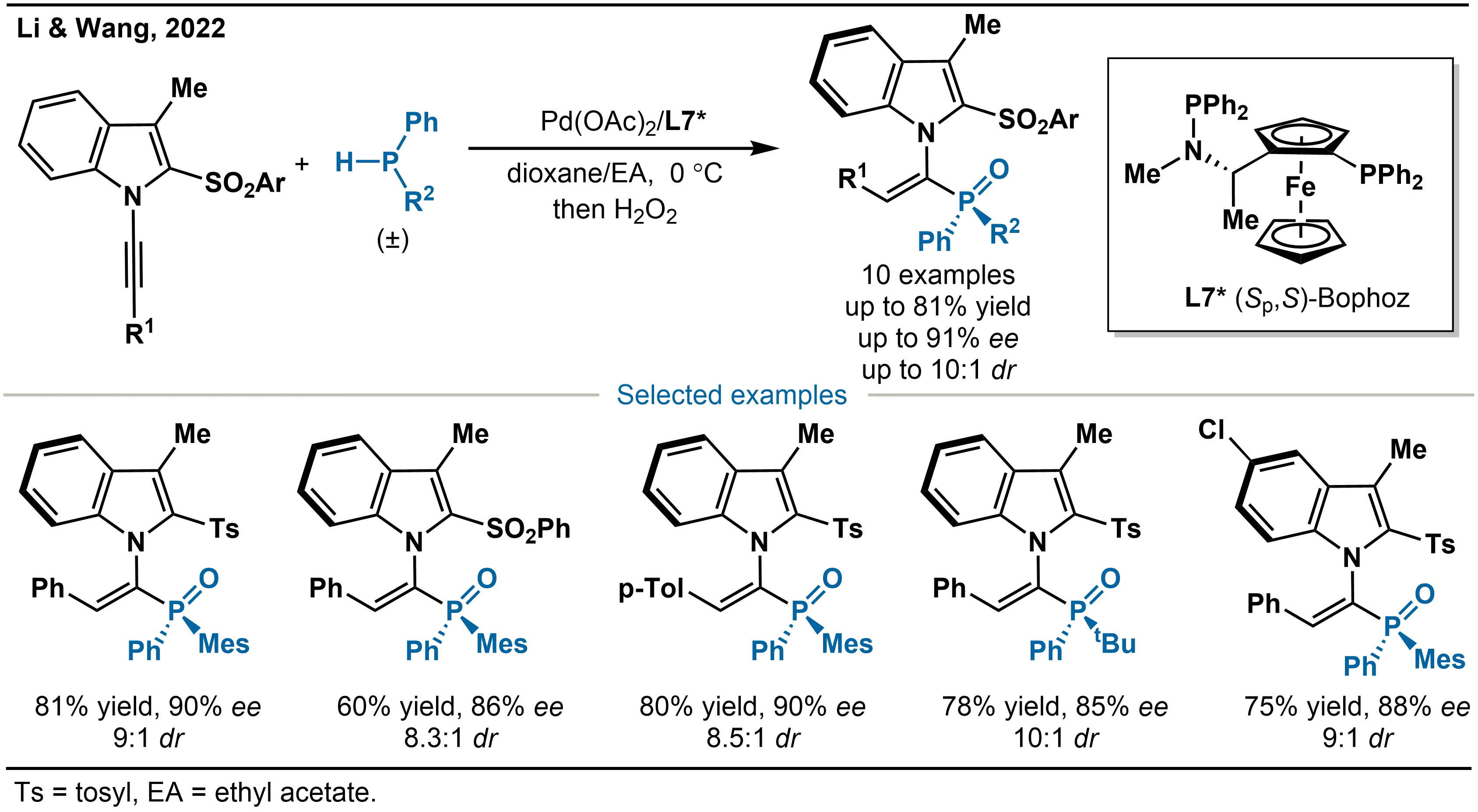
Aiming to address the challenge of stereoselective hydrophosphination of internal alkynes, Li and co-workers employed 1-alkynylindoles as electronically activated yet sterically hindered internal alkyne substrates (Scheme 15)[30]. Using unsymmetric secondary phosphines such as phenylmesitylphosphine or phenyl-tert-butylphosphine as phosphorus sources, together with a catalytic system based on Pd(OAc)2 and the chiral (Sp,S)-Bophoz ligand (L7*), they achieved the catalytic asymmetric hydrophosphination of internal alkynes. This innovative strategy enables the simultaneous construction of axial chirality and P-stereogenicity, delivering vinylphosphine oxide products with high diastereo- and enantio-selectivity. A central difficulty in this transformation is overcoming the substrate inhibition. By employing an electron-rich bidentate phosphine ligand, the team successfully observed both high reactivity and enantiocontrol. The method thus offers an efficient and novel route for constructing a library of multifunctional chiral phosphine ligands that incorporate both axial and P-centered chirality.
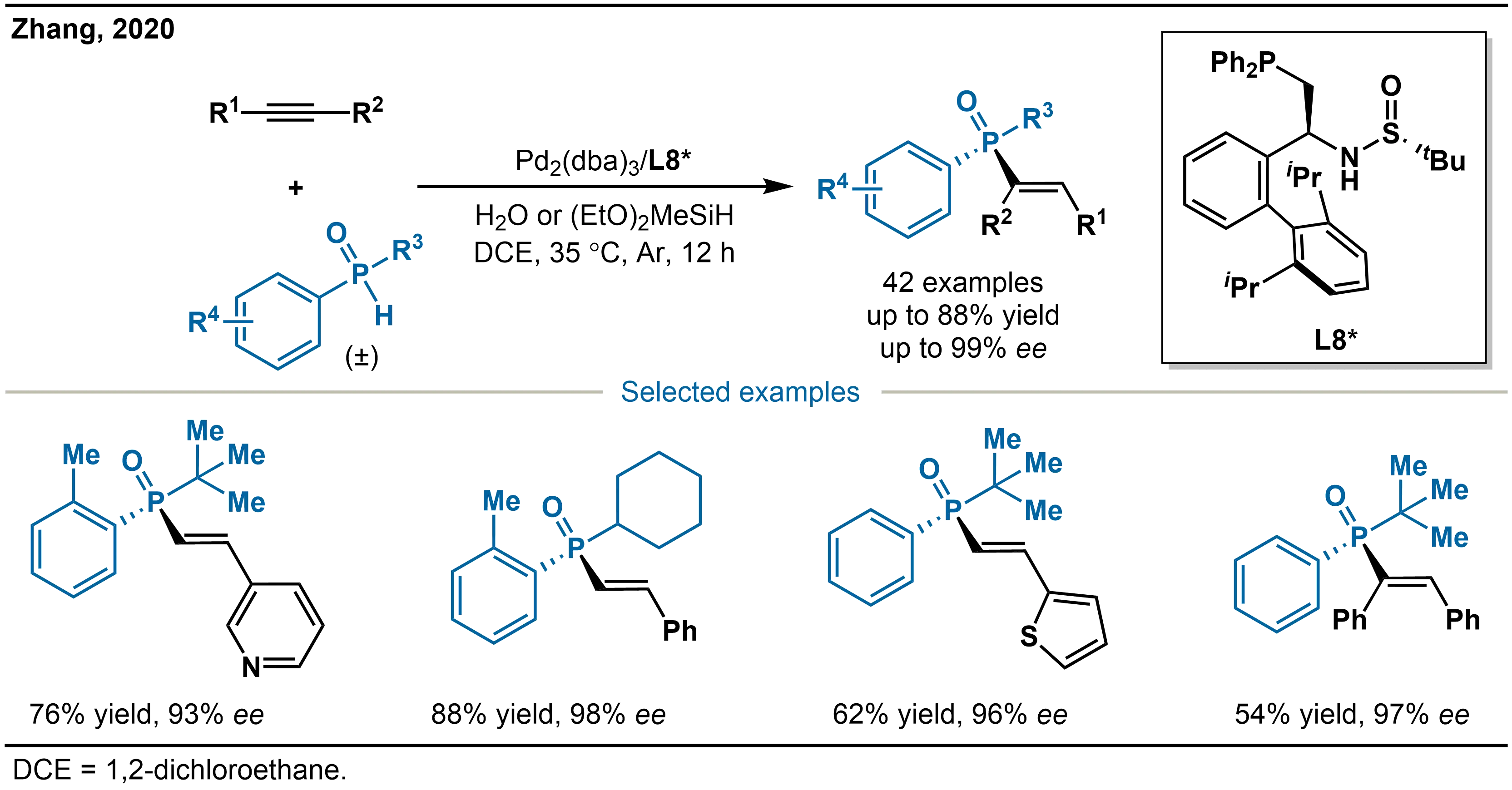
Beyond strategies that integrate multiple chiral elements, universal catalytic systems targeting both terminal and internal alkynes have also attracted considerable attention. Zhang and co-workers reported a Pd/L8* catalytic system, which enables the highly selective asymmetric anti-Markovnikov hydrophosphinylation of alkynes (Scheme 16)[31]. The method displays broad substrate scope, effectively accommodating a variety of (hetero)aryl- and alkyl-substituted alkynes, including both terminal and internal alkynes, as well as diversely substituted SPOs, with good tolerance toward sterically demanding groups. Functionalities such as amines, halides, esters, heteroaryl units, and ferrocenyl groups are all well tolerated. In the case of unsymmetrical alkynes, the reaction proceeds with high regioselectivity, while alkyl terminal alkynes can undergo double addition to afford bis-phosphinylated products. The use of additives further enhances both reaction efficiency and selectivity.
3.2 Ni-catalyzed asymmetric hydrophosphination of alkynes
Nickel, as an earth-abundant metal, offers catalytic systems that combine low cost with high activity. The accessible Ni0/NiII redox couple provides unique opportunities for modulating reaction pathways. Through synergistic interplay between the nickel center and chiral ligands, precise control over regioselectivity and enantioselectivity in alkyne hydrophosphination has been achieved. Notably, nickel catalysis has shown excellent performance with challenging substrates such as aliphatic and internal alkynes.
In 2021, to circumvent the instability associated with air-sensitive free secondary phosphines, Zhang and co-workers developed a catalytic system comprising [Ni(COD)2] and the chiral bisphosphine ligand (S,S)-BDPP (L9*), with (PhO)2PO2H as an additive (Scheme 17)[32]. At -30 °C, stable secondary phosphine oxides were reduced in situ by PhSiH3 to generate free secondary phosphines, which then participated in the highly enantioselective hydrophosphination of unactivated alkynes. This protocol efficiently delivers P-stereogenic tertiary phosphines without requiring direct handling of air-sensitive and toxic secondary phosphines. The products are obtained with excellent regioselectivity (up to > 20:1 rr (regioselectivity ratio)) and enantioselectivity (up to 99% ee).
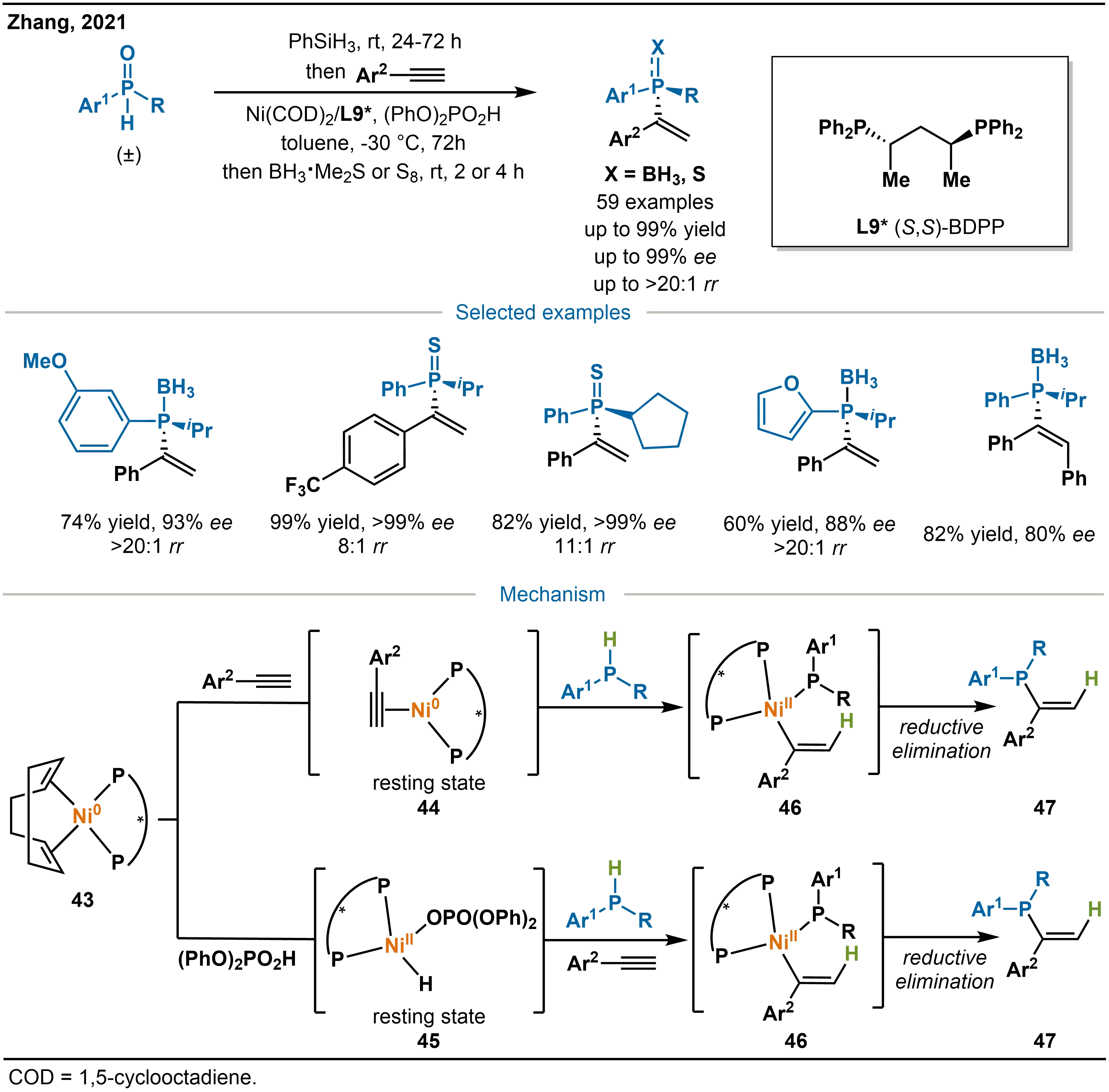
Scheme 17. Ni-catalyzed asymmetric hydrophosphination of alkynes with secondary phosphine oxides and the mechanism.
The proposed mechanism involves a chiral nickel catalyst formed in situ from Ni(COD)2 and (S,S)-BDPP, with the additive (PhO)2PO2H altering the resting state of the catalyst. Initially, SPOs are reduced by PhSiH3 to the corresponding secondary phosphines under mild conditions. In the absence of (PhO)2PO2H, the Ni-BDPP-COD complex 43 undergoes ligand exchange with the alkyne to form a nickel-alkyne adduct 44, followed by hydronickelation with the secondary phosphine to yield a vinyl-nickel intermediate 46. When (PhO)2PO2H is present, oxidative addition of the additive to the Ni(0) species 43 occurs first, generating a Ni(II) intermediate 45; subsequent ligand exchange introduces the secondary phosphine and alkyne, and hydronickelation then affords the same vinyl-nickel intermediate 46. The hydronickelation follows Markovnikov selectivity, with nickel adding to the less hindered alkyne carbon. Finally, reductive elimination releases the P-stereogenic tertiary phosphine and regenerates the Ni(0) catalyst.
The method described above bypasses the direct handling of secondary phosphines by employing in situ reduction of SPOs. This strategy successfully enables the highly selective synthesis of structurally diverse tertiary phosphines and offers a practical and innovative approach to constructing P-stereogenic centers. Based on this pioneering work, Zhang and co-workers further advanced the control of selectivity in nickel-catalyzed alkyne hydrophosphination in 2023 (Scheme 18)[33]. To address the challenge that regioselectivity in conventional systems often depends on ligands, metals, or additives and remains difficult to predict, they introduced a novel strategy based on tuning the oxidation state of the nickel catalyst. Specifically, they developed a Ni(II)-catalyzed asymmetric anti-Markovnikov hydrophosphination of unactivated alkynes. At 0 °C, the reaction delivers alkenylphosphines with high anti-Markovnikov regioselectivity (> 20:1 rr) and enantioselectivity (up to 91% ee). Notably, the catalyst loading can be reduced to 2.5 mol%, and the reaction is amenable to gram-scale synthesis.
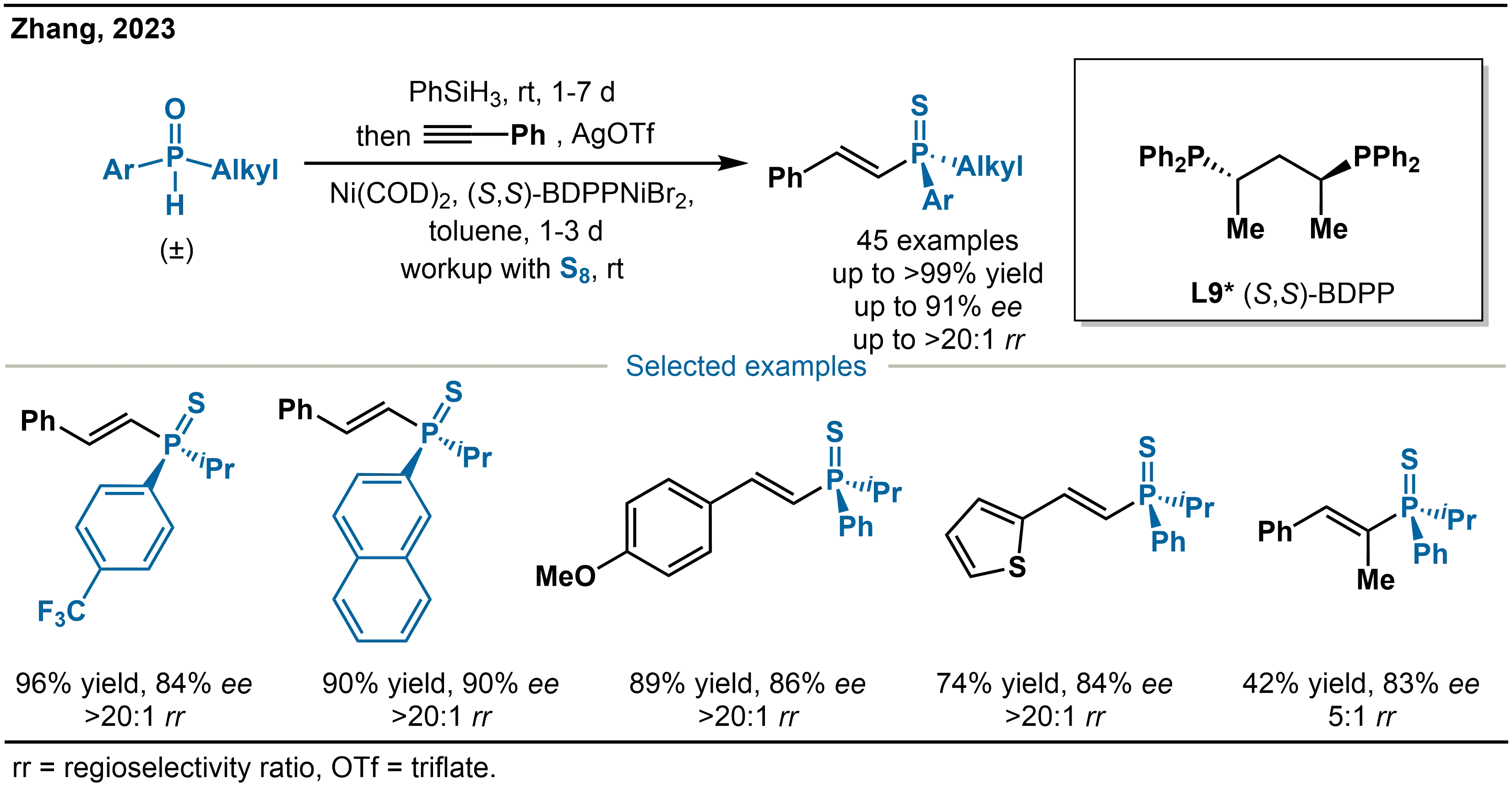
Mechanistic studies indicate that cationic Ni-phosphido complexes are the key active catalytic species in the NiII catalytic system (Scheme 19). The reaction proceeds through phosphido migratory insertion into the alkyne, followed by protonation to achieve anti-Markovnikov addition with high regioselectivity and enantioselectivity. In contrast, in the Ni0 catalytic system, promoted by acid additives, the reaction follows a pathway involving alkyne coordination, protonation, ligand exchange, and reductive elimination, which exclusively yields Markovnikov products. By switching between Ni0 and NiII valence states, these two distinct pathways can be precisely controlled, enabling the systematic and selective synthesis of both Markovnikov and anti-Markovnikov products.
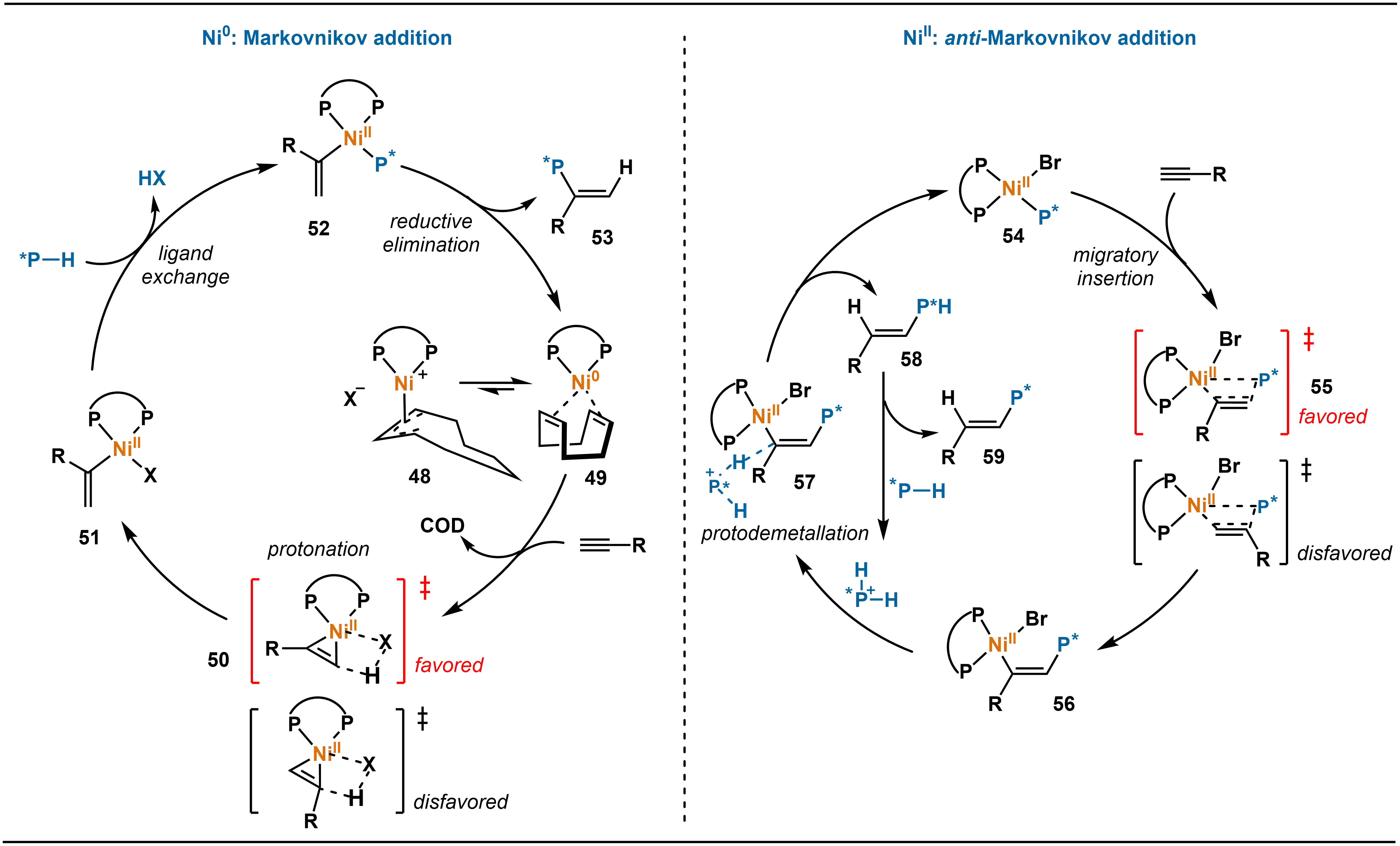
Scheme 19. Mechanism of Ni-catalyzed asymmetric anti-Markovnikov hydrophosphination of alkynes.
In 2025, Zhang and co-workers further expanded the scope of Ni-catalyzed hydrophosphination by addressing the intrinsic instability of primary phosphine oxides (PPOs), which tend to undergo disproportionation. They designed sterically hindered PPOs as stable substrates, enabling the asymmetric synthesis of ambiphilic secondary alkenyl phosphine oxides (Scheme 20)[34]. The catalytic system employs Ni(COD)2 as the pre-catalyst, combined with suitable chiral ligands: (R,R)-Ph-BPE (L2*) for terminal alkynes and (S,Sp)-iPr-Fox (L10*) for internal alkyne. The reaction is performed in mesitylene with phenylphosphinic acid as an additive under a nitrogen atmosphere at room temperature, using secondary phosphines generated in situ from secondary phosphine oxides as the phosphorus source. This strategy allows regioselective functionalization of a broad range of alkynes, further enriching the synthetic pathways and application of P-stereogenic phosphine compounds.
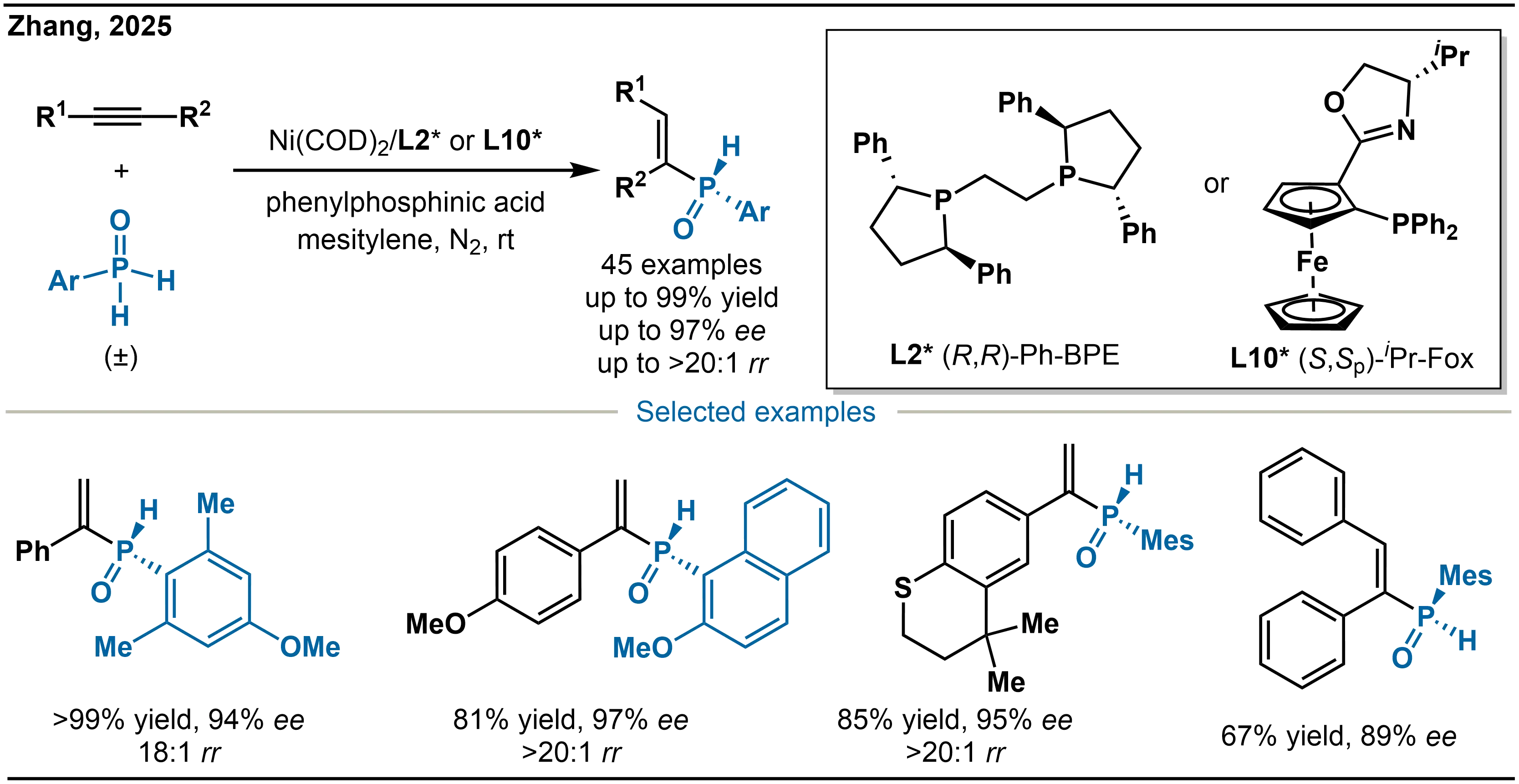
Scheme 20. Ni-catalyzed asymmetric hydrophosphination of alkynes with primary phosphine oxides.
Building on the advances in nickel-catalyzed hydrophosphination of alkynes with secondary phosphines and phosphine oxides, Hu and co-workers turned their attention to the challenging compatibility of H-phosphinate phosphorus sources (Scheme 21)[35]. They developed a catalytic system consisting of Ni(COD)2 and chiral ligand L11*, enabling highly regioselective and enantioselective hydrophosphination of unactivated alkynes with H-phosphinates. This work further broadens the scope of phosphorus reagents.
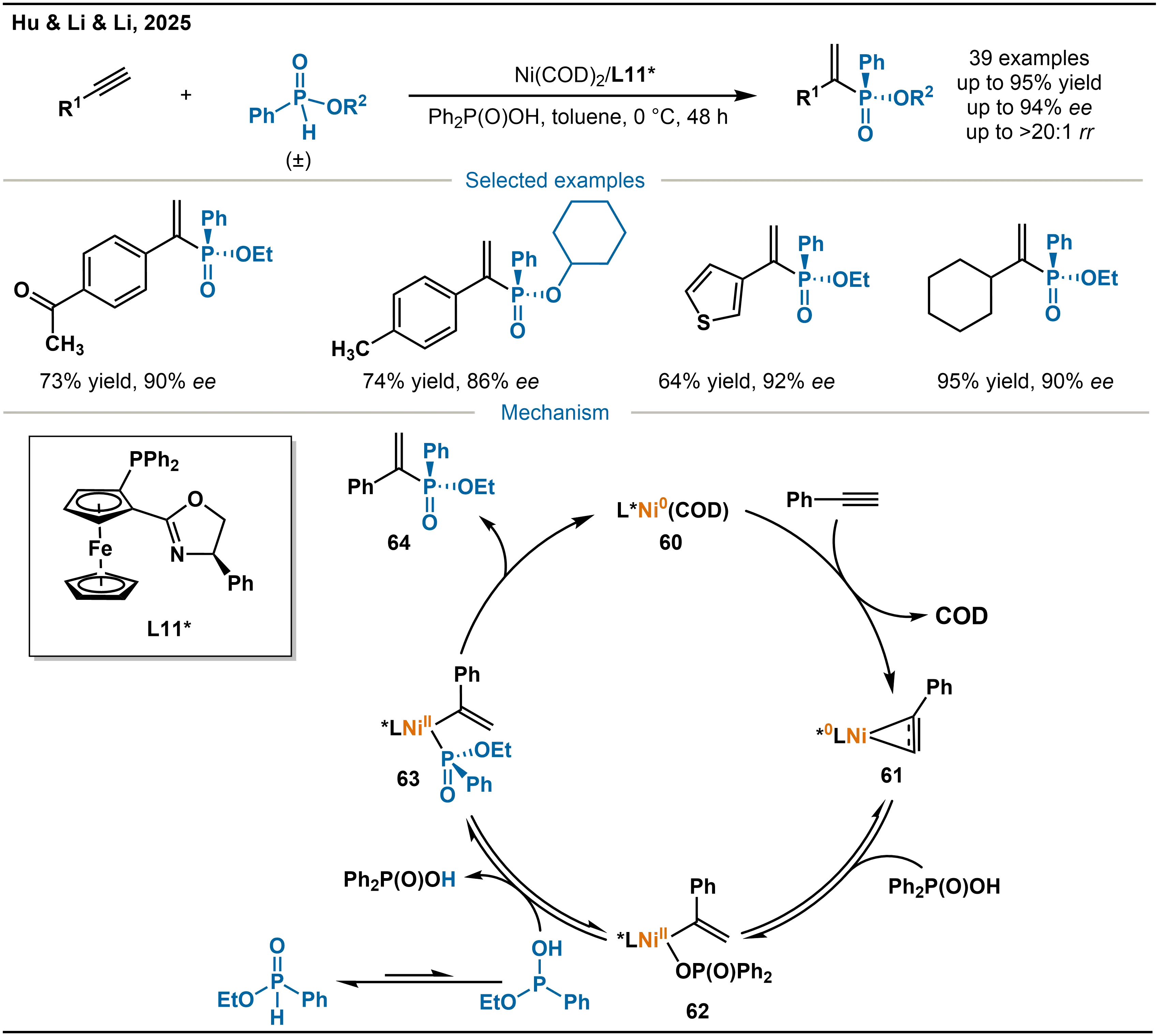
Scheme 21. Ni-catalyzed asymmetric hydrophosphination of alkynes with phosphinate and the mechanism.
The proposed reaction mechanism illustrated as (Scheme 21). Initially, complex 60 undergoes ligand exchange between the alkyne and the COD ligand, forming the more stable Ni-alkyne intermediate 61. Intermediate 61 then reacts with Ph3P(O)OH, and subsequent protonation yields the vinyl-nickel resting-state species 62. The isomerized phosphinate undergoes ligand exchange, releasing Ph3P(O)OH and generating intermediate 63. Finally, reductive elimination from 63 forms the C–P bond, delivering the product and regenerating complex 60 to complete the catalytic cycle.
3.3 Cu-catalyzed asymmetric hydrophosphination of alkynes
Compared to Pd- and Ni-catalysts, copper catalytic systems offer distinctive advantages in dynamic kinetic resolution by enabling efficient conversion of racemic phosphorus reagents. In 2025, Su and co-workers reported a CuI/L12* catalyzed highly enantioselective hydrophosphination of unactivated terminal alkynes with racemic pentavalent hydrophosphoryl compounds (Scheme 22)[36]. To the best of our knowledge, this transformation constitutes the first DYKAT (dynamic kinetic asymmetric transformation)-type hydrophosphination of unsaturated C–C bonds.
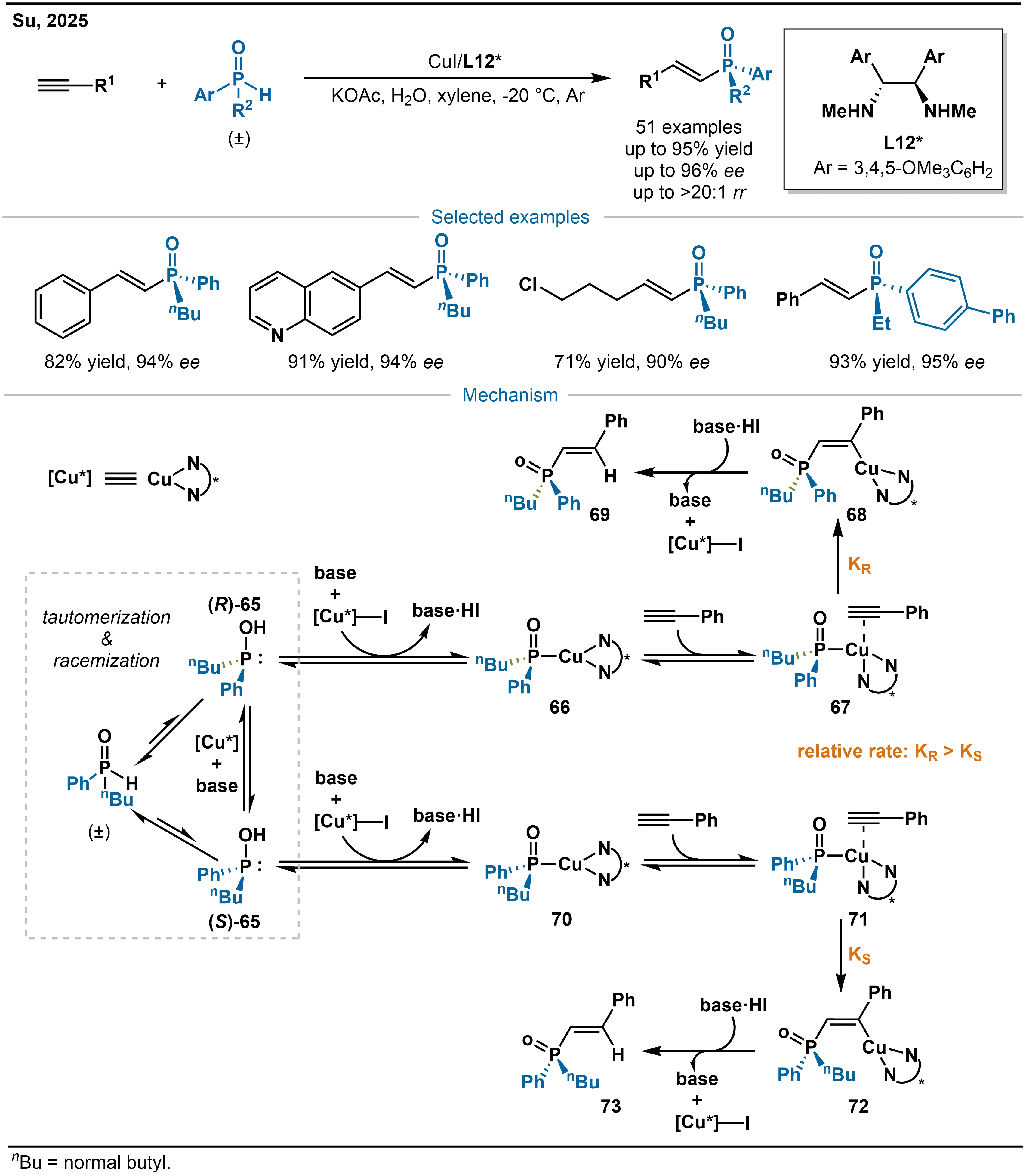
Scheme 22. Cu-catalyzed asymmetric hydrophosphination of alkynes and the proposed mechanism.
The proposed mechanism involves a chiral Cu-catalyst formed in situ from CuI and the chiral 1,2-diamine ligand L12*. The racemic pentavalent hydrophosphoryl substrate undergoes tautomerization to give trivalent phosphinous acid enantiomers (S)-65 and (R)-65. A key feature is the copper- and KOAc-promoted racemization of these phosphinous acids, both components are indispensable, while water enhances the solubility of KOAc and accelerates the process. (R)-65 reacts with the chiral copper species [Cu]-I in the presence of base to form the phosphoryl-copper intermediate (S)-P(O)-[Cu] (66). The alkyne then coordinates to this intermediate 66 and undergoes exclusive cis-addition in an anti-Markovnikov fashion to produce an alkenyl-[Cu] species 68; this step is the enantiodetermining step. Protonation of the alkenyl-[Cu] intermediate 68 releases the P-stereogenic alkenylphosphine oxide product 69 and regenerates the active [Cu*]-I species. Notably, (R)-65 is the preferred enantiomeric substrate, reacting significantly faster than (S)-65.
4. Asymmetric Hydrophosphination of Special Substrates
Beyond simple alkenes and alkynes, special substrates containing unsaturated carbon-carbon bonds, such as conjugated enynes, allenes, and conjugated dienes, have demonstrated unique value in the research on constructing P-stereogenic centers by means of catalytic asymmetric hydrophosphination due to their structural diversity and controllable reaction sites. By precisely regulating regioselectivity and enantioselectivity, these substrates can efficiently synthesize structurally novel P-stereogenic organophosphorus compounds, providing diversified pathways for the preparation of chiral ligands, catalysts, and drug intermediates, and thus have become a research hotspot in this area.
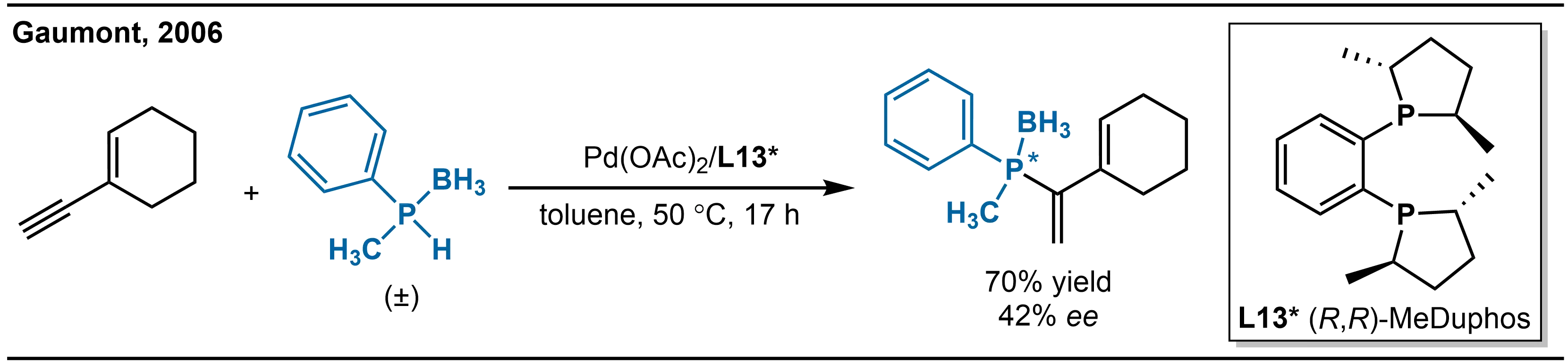
Conjugated enynes, which contain both alkene and alkyne functionalities, have drawn significant attention as unique substrates for asymmetric hydrophosphination. The development of catalytic systems for such transformations and elucidation of their mechanisms represent important research directions. In 2006, Gaumont and co-workers reported the highly selective asymmetric hydrophosphination of conjugated enynes with secondary phosphine-boranes, marking the first example of Pd-catalyzed asymmetric hydrophosphination of conjugated enynes (Scheme 23)[37].
In 2022, Zhang and co-workers reported Ni(COD)2/(S,S)-BDPP (L9*)-catalyzed highly regio- and enantioselective asymmetric hydrophosphination of conjugated enynes with SPOs (Scheme 24)[38]. This method efficiently delivers P-stereogenic alkenylphosphine oxides and provides an important benchmark for the catalytic functionalization of conjugated enyne substrates.
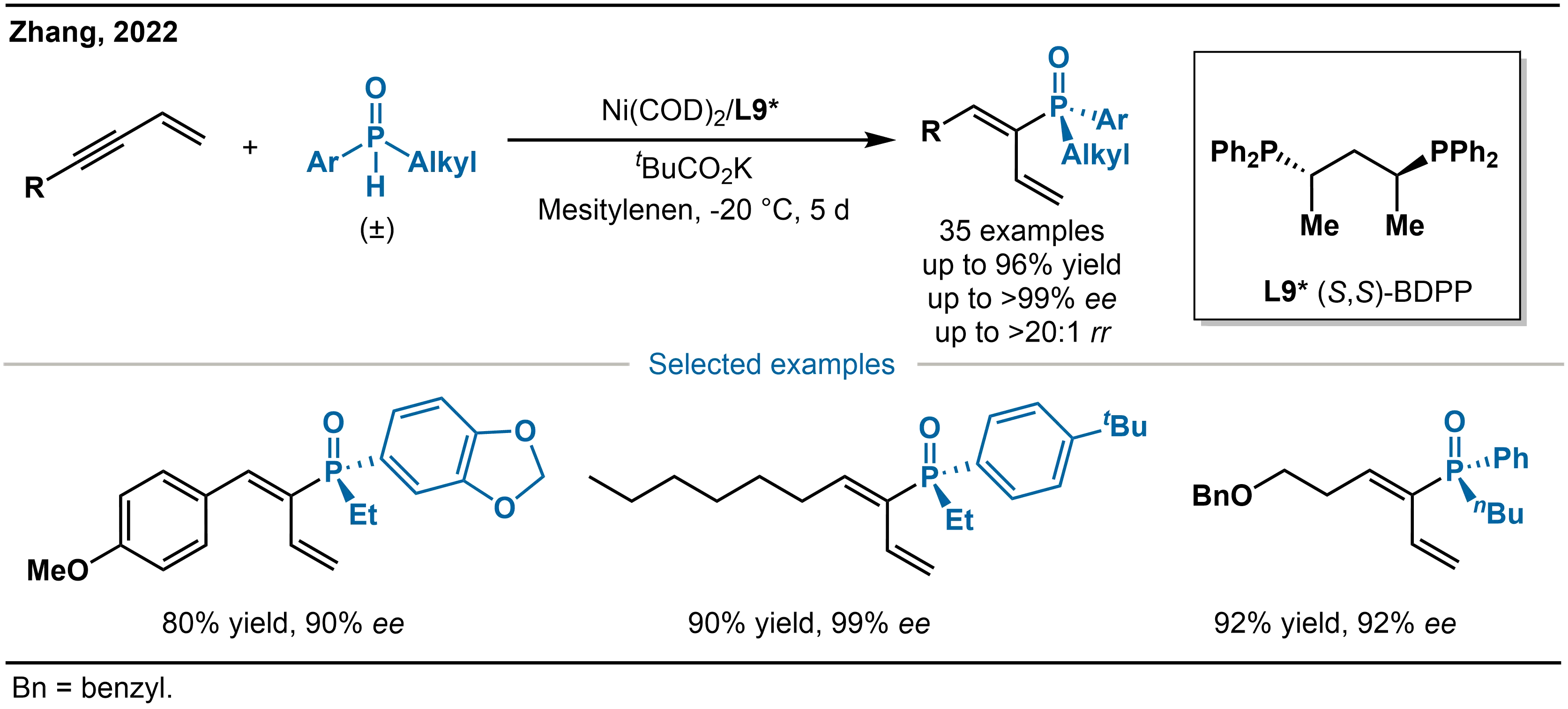
On the basis of this finding, Wang, Yang, and co-workers further developed a Pd(OAc)2/L5*-catalyzed system for the highly regio- and enantio-selective hydrophosphination of conjugated enynes with phosphinates or SPOs (Scheme 25)[39]. Mechanistically, the chiral palladium complex 74, formed in situ from Pd(OAc)2 and L5*, initiates the reaction via two parallel pathways. In Path A, complex 74 undergoes oxidative addition of the O–H bond of Ph2P(O)OH to generate the hydropalladation species 75. Species 75 then adds across the conjugated enyne to form the η1-butadienylpalladium intermediate 76. Subsequent ligand exchange with the phosphorus nucleophile (phosphinate or SPO) affords the phosphorylpalladium intermediate 78, which undergoes reductive elimination to release the product and regenerate catalyst 74. In Path B, the P–H bond of the phosphorus nucleophile directly undergoes oxidative addition to complex 74, yielding an alternative hydropalladation intermediate 77. This species then follows a sequence analogous to that in Path A, insertion into the conjugated enyne, ligand exchange, and reductive elimination, to complete the catalytic cycle.
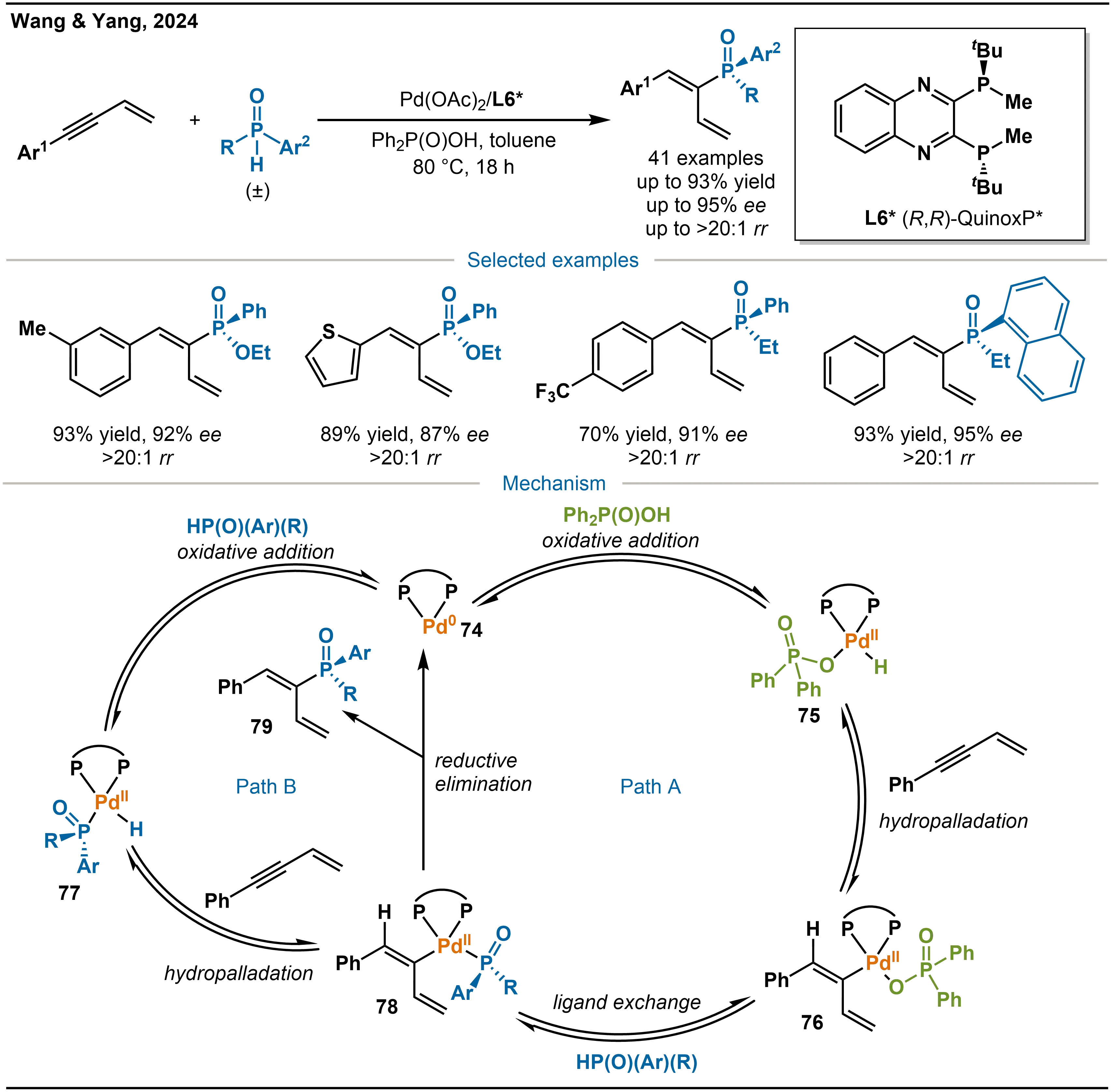
Scheme 25. Pd-catalyzed asymmetric hydrophosphination of conjugated enynes and the mechanism.
Although single-metal catalysis can efficiently transform specific substrates, synergistic control over structurally diverse unsaturated C–C bond systems, such as conjugated enynes, allenes, and conjugated dienes, remains challenging, often compromising either generality or selectivity. To overcome this limitation, He and co-workers innovatively developed an asymmetric formal hydrophosphination system based on Pd/Co synergistic catalysis (Scheme 26)[40]. The system employs [Pd(allyl)Cl]2/L14* and Co(OAc)2/L15* as the catalysts and has been successfully applied to the efficient functionalization of allenes. By simply switching the ligands, the same catalytic platform can be adapted to conjugated enynes and conjugated dienes with high efficiency. Notably, by employing different ligand combinations, the stereodivergent hydrophosphination of conjugated enynes was achieved for the first time, which enabled the precise synthesis of all four phosphorus-containing stereoisomers.
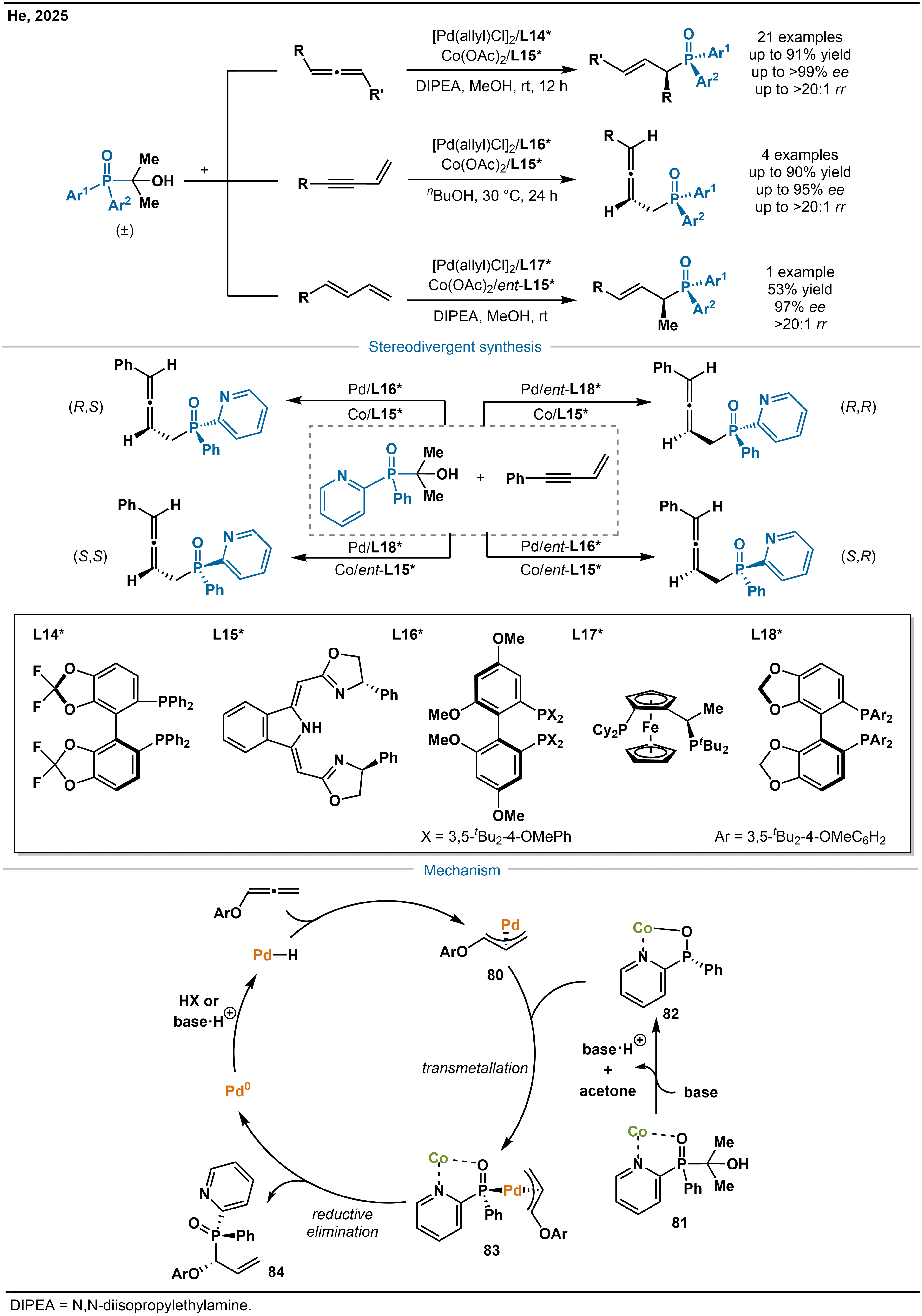
Scheme 26. Pd/Co-cooperative catalyzed asymmetric formal hydrophosphination and the mechanism.
Using allenes as an illustrative example, they proposed the mechanism. After generation of a Pd–H species, hydropalladation of the allene occurs to afford an η3-allylpalladium intermediate 80. Concurrently, the Co unit coordinates with a masked P(V) reagent (an α-hydroxyalkylphosphonate derivative, 81) and, upon deprotonation by a base (N,N-Diisopropylethylamine, DIPEA), forms the nucleophilic Co–phosphorus species 82. These two intermediates then undergo transmetallation to give intermediate 83, a step that is strictly controlled by the stereochemical match between the two chiral ligands; high selectivity is achieved only when L14* and L15* are configurationally matched. Finally, reductive elimination releases the product and regenerates both catalytic species.
Subsequently, building on previous research, He and co-workers further focused on the formal hydrophosphinylation of conjugated enynes, with a specific focus on constructing fully C(sp2)-substituted P(V) stereogenic centers. Such structures are synthetically challenging owing to the subtle steric and electronic differences among their substituents. To address this, they developed a highly efficient catalytic system composed of [Pd(allyl)Cl]2/L19* and Co(OAc)2/L20* (Scheme 27)[41]. This system achieves exclusive 1,2-asymmetric hydrophosphination of conjugated enynes, effectively suppressing the 1,4-addition products often formed in conventional catalytic systems and thereby significantly enhancing regioselectivity. Mechanistic studies suggest that a Pd–H species selectively undergoes hydropalladation with the alkyne unit of the conjugated enyne. The ensuing outer-sphere allylic substitution step is rate-determining. The newly developed Boxmi ligand, with a sterically blocked left side and an open right side, precisely directs the trajectory of nucleophilic attack, while the Co catalyst governs the configuration of the resulting P-stereogenic center. Through this synergistic interplay, exclusive 1,2-addition is achieved.
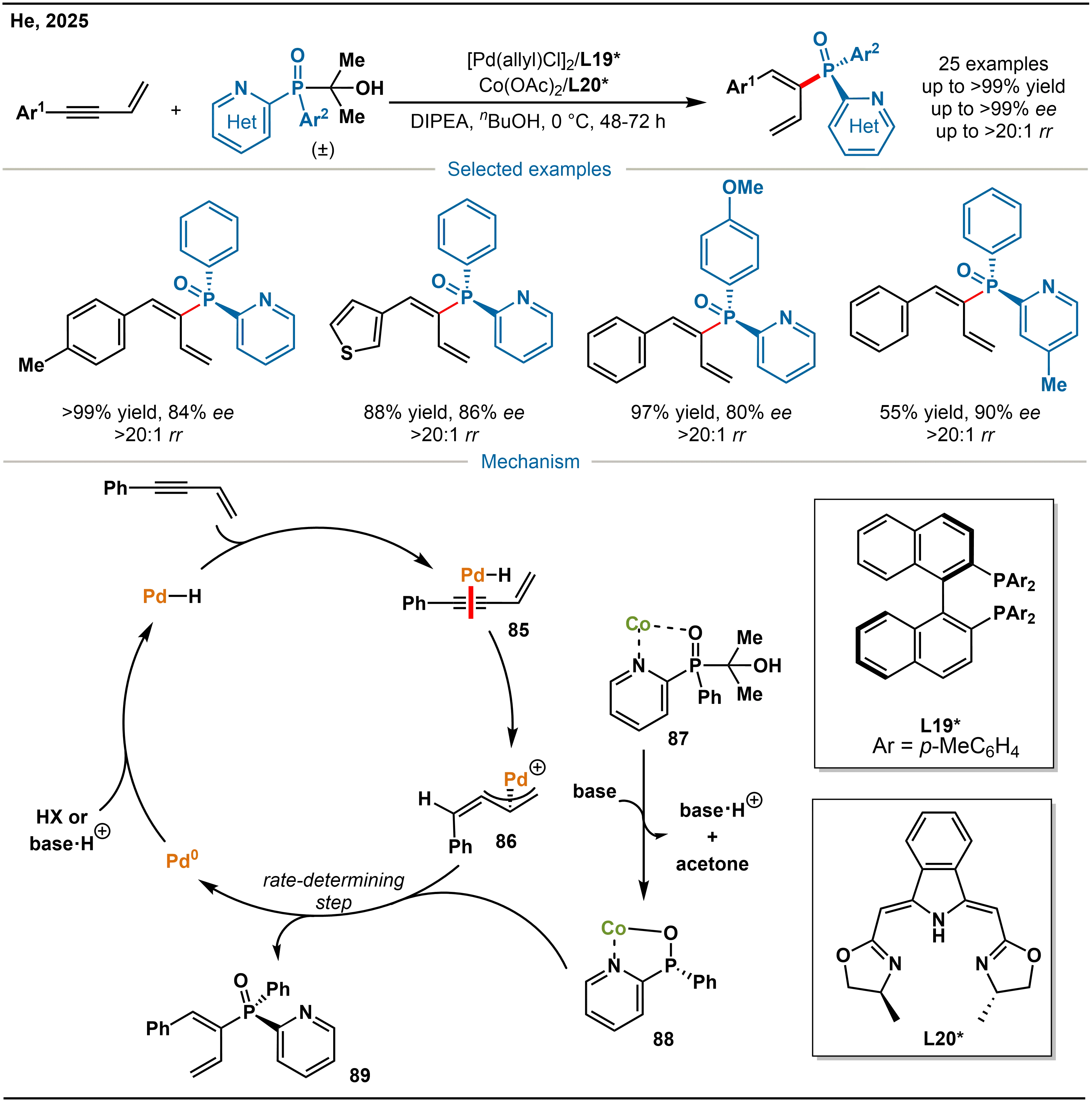
Scheme 27. Pd/Co cooperative catalyzed asymmetric formal hydrophosphination of conjugated enynes and the mechanism.
5. Conclusion and Outlook
In summary, significant advances have been made in the catalytic asymmetric hydrophosphination of unsaturated C–C bonds for the construction of P-stereogenic centers. A broad array of catalytic systems, including transition-metal catalysts such as Pd, Ni, Cu, Co, and Mn, as well as organocatalytic approaches, have been developed to achieve high levels of enantioselectivity and regioselectivity across diverse substrates, including alkenes, alkynes, and other unsaturated C–C bond motifs. These methods are notable for excellent atom economy, functional group compatibility, and efficient access to structurally varied P-stereogenic phosphines, phosphine oxides, and phosphinates. The strategic implementation of chiral ligands, dynamic kinetic resolution, and synergistic catalytic platforms has further refined stereocontrol and expanded the substrate scope. Together, these developments firmly establish asymmetric hydrophosphination as a versatile and powerful methodology in contemporary synthetic chemistry.
Despite these impressive achievements, several challenges and opportunities persist. A key area for future innovation is the activation of unactivated and sterically encumbered substrates, such as aliphatic alkenes and internal alkynes, which remain challenging for existing catalytic systems. Furthermore, achieving high enantioselectivity and regioselectivity with challenging phosphorus nucleophiles, particularly those with minimal steric or electronic differentiation, is another important goal. Notably, while the catalytic synthesis of C-stereogenic phosphonates via asymmetric hydrophosphonylation of phosphodiesters [e.g., HP(O)(OR)2] is well documented, the analogous transformation to afford P-stereogenic centers remains an unmet target. Future research should also focus on mechanistic elucidation through integrated experimental and computational studies, to develop more efficient and sustainable catalytic systems, and explore novel applications for P-stereogenic compounds in asymmetric catalysis and medicinal chemistry. Incorporating emerging activation paradigms, such as photoredox, electrocatalytic, or biocatalytic strategies, could unlock innovative pathways for the stereoselective synthesis of P-stereogenic molecules, thereby enriching the toolbox for asymmetric synthesis.
Authors contribution
Wang BL, Xing LK, Wang YX: Investigation, writing-original draft, writing-review & editing.
Lu Y, Yang XH: Conceptualization, writing-review & editing.
Conflicts of interest
The authors declare no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
The authors are thankful for financial support from the National Natural Science Foundation of China (Nos. 22371016 & 22201018), the Beijing Natural Science Foundation (No. 2262029), and the National Key R&D Program of China (No. 2021YFA1401200).
Copyright
© The Author(s) 2026.
References
-
1. Dutartre M, Bayardon J, Jugé S. Applications and stereoselective syntheses of P-chirogenic phosphorus compounds. Chem Soc Rev. 2016;45(20):5771-5794.
-
2. Xu G, Senanayake CH, Tang W. P-chiral phosphorus ligands based on a 2, 3-dihydrobenzo [d][1, 3] oxaphosphole motif for asymmetric catalysis. Acc Chem Res. 2019;52(4):1101-1112.[DOI]
-
4. Ye X, Peng L, Bao X, Tan CH, Wang H. Recent developments in highly efficient construction of P-stereogenic centers. Green Synth Catal. 2021;2(1):6-18.[DOI]
-
5. Pullarkat S. Recent progress in palladium-catalyzed asymmetric hydrophosphination. Synthesis. 2016;48(4):493-503.[DOI]
-
6. Yuan Y, Darcel C. Earth abundant transition metal catalysts: New and efficient tools for hydrophosphination and oxyphosphination of alkenes and alkynes. ChemCatChem. 2024;16(18):e202400703.[DOI]
-
7. Sun T, Sun G, Sun W, Peng X, Liao J, You Y, et al. Advances in catalytic asymmetric hydrogen-phosphine/phosphorus functionalization of unsaturated carbon-carbon bonds. Chin J Org Chem. 2024;44(12):3647.[DOI]
-
9. Liu BX, Feng F, Long J, Gu SX. Recent advances in transition metal-catalyzed asymmetric synthesis of P-chiral organophosphorus compounds (2020–present). Chin J Chem. 2025;43(17):2228-2244.[DOI]
-
10. Lemouzy S, Giordano L, Hérault D, Buono G. Introducing chirality at phosphorus atoms: An update on the recent synthetic strategies for the preparation of optically pure P-stereogenic molecules. Eur J Org Chem. 2020;2020(23):3351-3366.[DOI]
-
11. Dong B, Zhang J, Ye XY, Huang X, Chi YR. Catalytic construction of P-stereogenic center via phosphorus-centered nucleophilic substitution. Chin Chem Lett. 2025;36(9):111052.[DOI]
-
12. Li H, Yin L. Research progress on copper-catalyzed asymmetric synthesis of chiral organophosphorus compounds. Chin J Org Chem. 2024;44(12):3575.[DOI]
-
13. Ding K, Zou JX, Su B. Metal-catalyzed enantioselective hydrophosphorylation reactions. Asian J Org Chem. 2025;14(11):e00597.[DOI]
-
14. Ding K, Su B. Synthesis of P-stereogenic compounds by transition metal-catalyzed asymmetric transformation of H–P(O) compounds: Progress, challenges, and prospects. Eur J Org Chem. 2024;27(4):e202301160.[DOI]
-
15. Luan C, Yang CJ, Liu L, Gu QS, Liu XY. Transition metal-catalyzed enantioselective C–P coupling reactions for the construction of P-stereogenic centers. Chem Catal. 2022;2(11):2876-2888.[DOI]
-
16. Liu J, Chen H, Wang M, He W, Yan JL. Organocatalytic asymmetric synthesis of P-stereogenic molecules. Front Chem. 2023;11:1132025.[DOI]
-
17. Zhang JM, Ma YM, He ZT. Asymmetric synthesis of P(V) skeleton via catalytic desymmetric substitution. Angew Chem Int Ed. 2026;65(2):e21470.[DOI]
-
19. Li Z, Duan W. Recent advances in the asymmetric conjugate addition reactions of phosphorus nucleophiles to electron-deficient alkenes. Chin J Org Chem. 2016;36(8):1805.[DOI]
-
20. Li C, Bian QL, Xu S, Duan WL. Palladium-catalyzed 1, 4-addition of secondary alkylphenylphosphines to α, β-unsaturated carbonyl compounds for the synthesis of phosphorus- and carbon-stereogenic compounds. Org Chem Front. 2014;1(5):541-545.
-
23. Wan B, Harutyunyan SR. P-stereogenic phosphines via Mn(I)-catalyzed asymmetric hydrophosphination of vinyl sulfones: A case of solvent-induced stereoinversion. Adv Synth Catal. 2025;367(24):e70198.[DOI]
-
26. Li YB, Li Y, Yin L. Copper(I)-catalyzed diastereodivergent construction of vicinal P-chiral and C-chiral centers facilitated by dual “soft-soft” interaction. Chin Chem Lett. 2024;35(7):109294.[DOI]
-
30. Ji D, Jing J, Wang Y, Qi Z, Wang F, Zhang X, et al. Palladium-catalyzed asymmetric hydrophosphination of internal alkynes: Atroposelective access to phosphine-functionalized olefins. Chem. 2022;8(12):3346-3362.[DOI]
-
33. Wang WH, Wu Y, Qi PJ, Zhang QW. NiII-catalyzed enantioselective Anti-Markovnikov hydrophosphination of unactivated alkynes. ACS Catal. 2023;13(10):6994-7001.[DOI]
-
35. Xu B, Gao Z, Liu C, Zhang J, Li J, Li X, et al. Asymmetric access to phosphinates via nickel-catalyzed hydrophosphorylation of alkynes. ACS Catal. 2025;15(21):18623-18630.[DOI]
-
38. Zhang YQ, Han XY, Wu Y, Qi PJ, Zhang Q, Zhang QW. Ni-catalyzed asymmetric hydrophosphinylation of conjugated enynes and mechanistic studies. Chem Sci. 2022;13(14):4095-4102.
-
39. Jiang Y, Cheng KW, Yang Z, Wang JJ. Pd-catalyzed enantioselective and regioselective asymmetric hydrophosphorylation and hydrophosphinylation of enynes. Chin Chem Lett. 2025;36(5):110231.[DOI]
-
41. Ren ZY, Tang MQ, Lin GQ, He ZT. Pd/co-catalyzed asymmetric hydrophosphinylation of conjugated enynes to access fully C(sp2)-substituted P(V) center. Chin J Chem. 2025;43(24):3469-3475.[DOI]
Copyright
© The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Publisher’s Note
Science Exploration remains a neutral stance on jurisdictional claims in published
maps
and institutional affiliations. The views expressed in this article are solely those
of
the author(s) and do not reflect the opinions of the Editors or the publisher.
Share And Cite



Science Exploration Style
Wang BL, Xing LK, Wang YX, Lu Y, Yang XH. Catalytic construction of P-stereogenic centers through asymmetric hydrophosphination of unsaturated C–C bonds. Chiral Chem. 2026;2:202603. https://doi.org/10.70401/cc.2026.0014
Tips
Copy completed.
Submit a Manuscript
Author Instructions
Cite this Article
Article Metrics
0
View
0
Download
Cited
Article Updates

- Abstract
- Keywords
- 1. Introduction
- 2. Asymmetric Hydrophosphination of Alkenes Bearing Electron-Withdrawing Groups
- 3. Asymmetric Hydrophosphination of Alkynes
- 4. Asymmetric Hydrophosphination of Special Substrates
- 5. Conclusion and Outlook
- Authors contribution
- Conflicts of interest
- Ethical approval
- Consent to participate
- Consent for publication
- Availability of data and materials
- Funding
- Copyright
Science Exploration Style
Wang BL, Xing LK, Wang YX, Lu Y, Yang XH. Catalytic construction of P-stereogenic centers through asymmetric hydrophosphination of unsaturated C–C bonds. Chiral Chem. 2026;2:202603. https://doi.org/10.70401/cc.2026.0014
copy
Share Link
copy