Pd(II)-catalyzed atroposelective and enantioselective C−H olefination of [n]metacyclophanes
Xin-En Yan
Jia Li
Ziyang Dong
Hongxuan Zhai
Quan Tang
*
,
Changgui Zhao
*

*Correspondence to:
Quan Tang, College of Chemistry, Beijing Normal University, Beijing 100875, China.
E-mail: tangquan@bnu.edu.cn
Changgui Zhao, College of Chemistry, Beijing Normal University, Beijing 100875, China. E-mail: cgzhao@bnu.edu.cn
Changgui Zhao, College of Chemistry, Beijing Normal University, Beijing 100875, China. E-mail: cgzhao@bnu.edu.cn
Chiral Chem. 2026;2:202606. 10.70401/cc.2026.0016
Received: February 05, 2026Accepted: March 20, 2026Published: March 24, 2026
Abstract
A Pd(II)-catalyzed C–H olefination strategy has been established for the enantioselective synthesis of [n]metacyclophanes. The method demonstrates excellent functional group tolerance across a series of olefins and metacyclophane precursors with varying ansa-chain lengths, achieving high stereocontrol through kinetic or dynamic kinetic resolution. Racemization experiments were conducted to investigate the relationship between molecular structure and conformational stability. Furthermore, the synthetic utility of this approach was demonstrated through gram-scale synthesis and the application of a resulting bifunctional thiourea catalyst in asymmetric Michael additions, highlighting its potential in asymmetric synthesis.
Graphical Abstract
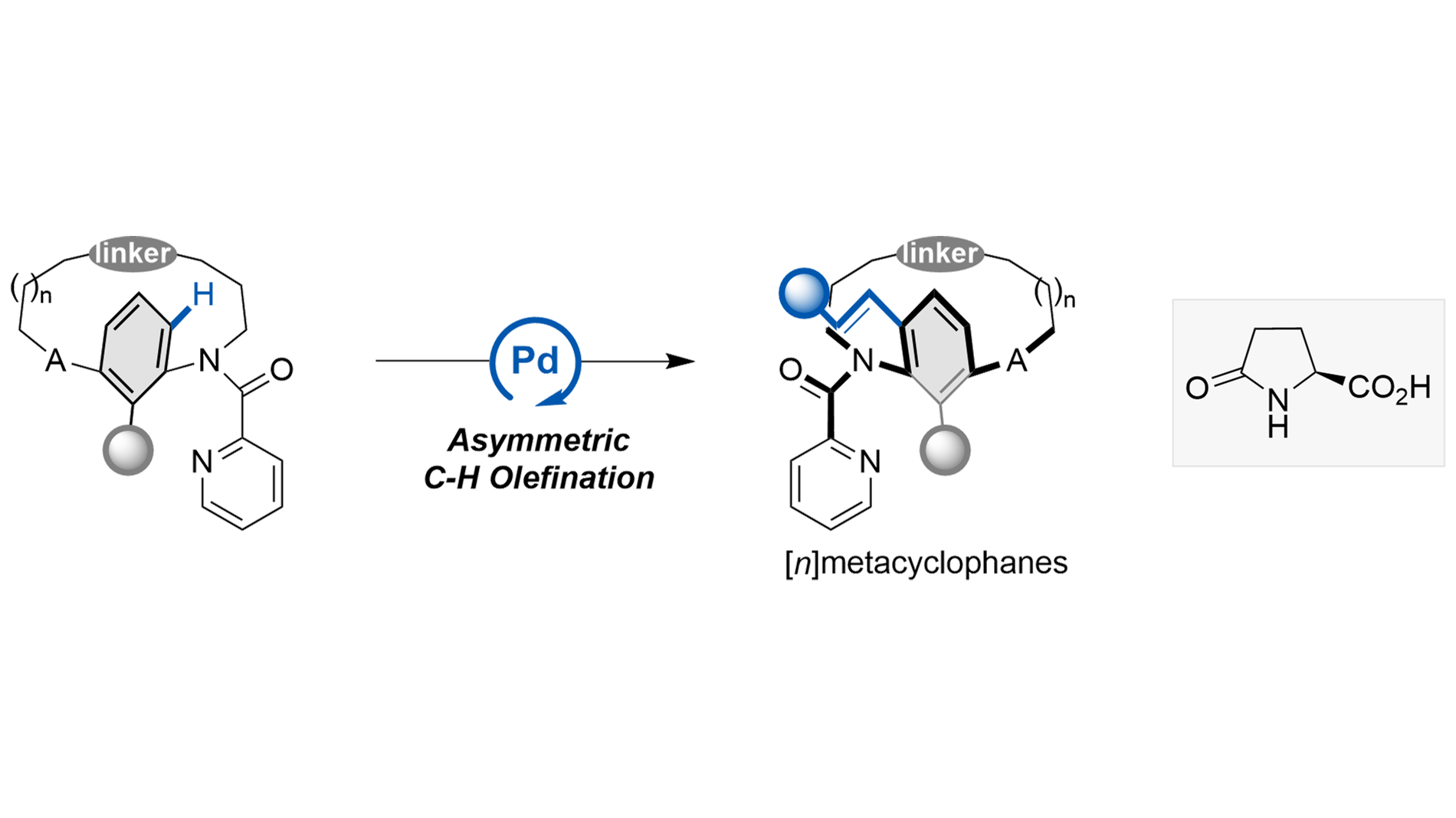
Keywords
Metacyclophanes, C–H olefination, asymmetric catalysis, palladium
1. Introduction
[n]Cyclophanes are molecules comprising aromatic rings bridged by alkyl chains, which are classified into ortho-, meta-, and para-subtypes according to the substitution pattern linking the arene to the ansa chain[1-4]. In meta- and paracyclophanes, the presence of sterically demanding substituents on the aromatic ring, combined with an optimally sized alkyl tether, can impart planar chirality by restricting the rotation of the arene around the bridge[5-7]. Conformationally defined metacyclophanes constitute the core structure of numerous bioactive natural products, such as complestatin[8], which exhibits inhibitory activity against HIV-1 integrase (Scheme 1A). Furthermore, optically pure metacyclophanes represent privileged scaffolds in pharmaceutical research[9-12], asymmetric catalysis[13,14], and supramolecular chemistry[15-20]. Recently, atroposelective and enantioselective synthesis of planar-chiral cyclophanes has garnered growing interest, with most efforts focusing on paracyclophanes[21-46]. In contrast, catalytic asymmetric synthesis of metacyclophanes remains less developed, primarily due to challenges associated with their conformational flexibility and significant ring strain[47-50].
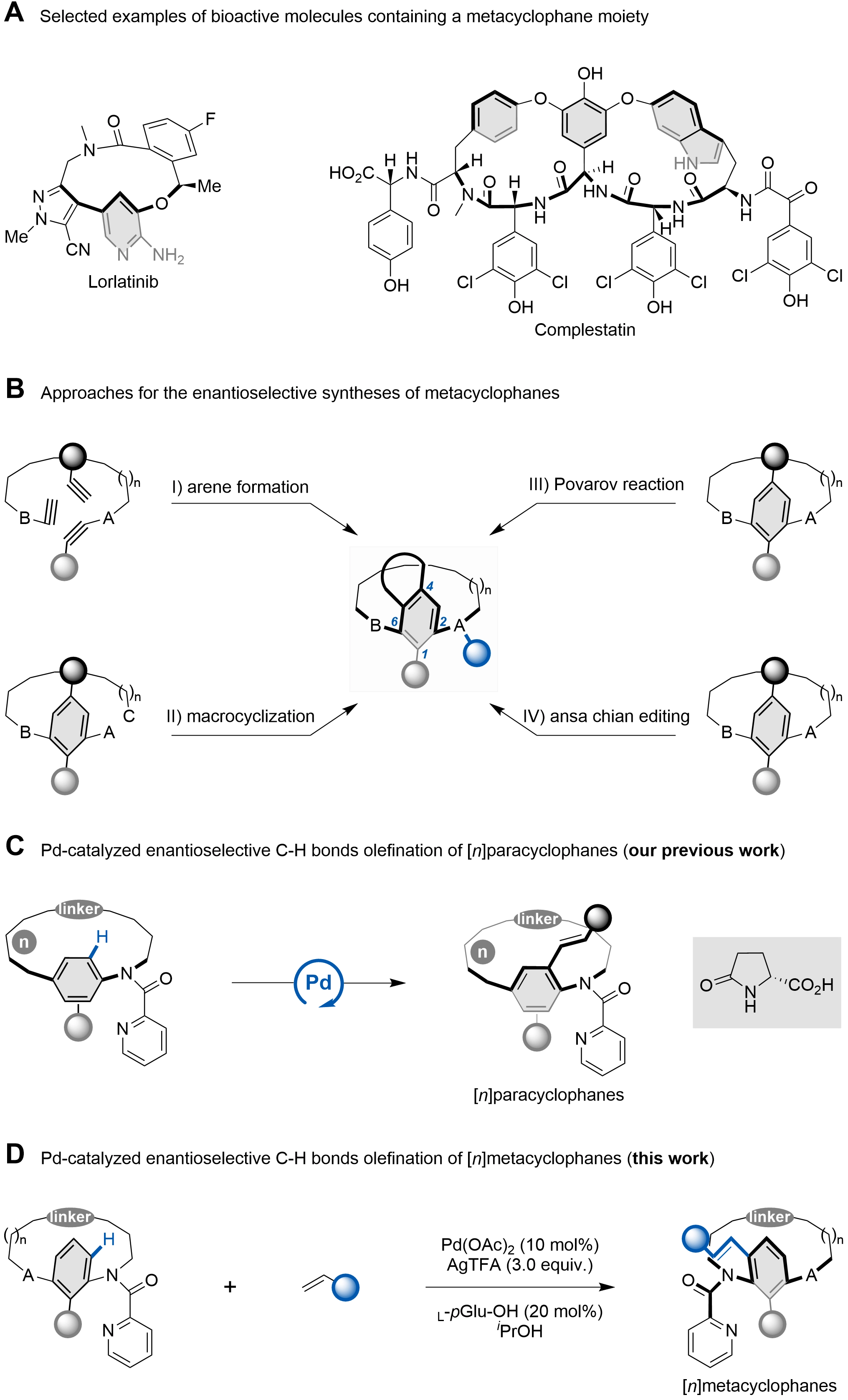
Scheme 1. Overview of metacyclophanes and design of a Pd(II)-catalyzed C–H olefination strategy for the atroposelective synthesis of [n]metacyclophanes. Created in ChemDraw.
In 2007, Tanaka and colleagues pioneered a rhodium-catalyzed atroposelective arene formation to afford [7]–[10]metacyclophanes in moderate yields (Scheme 1B, I)[51]. Subsequently, asymmetric macrocyclization strategies have emerged as an alternative route to metacyclophanes. In this context, Li and co-workers developed a Pd-catalyzed C–O bond-forming macrocyclization for the synthesis of enantioenriched planar-chiral metacyclophanes (Scheme 1B, II)[52]. Independently, Wang and Zhou reported N-heterocyclic carbene- or chiral phosphoric acid (CPA)-catalyzed asymmetric macrolactonizations to access indole-based metacyclophanes[13,53]. Beyond these methods, the Povarov reaction has also proven attractive for constructing such scaffolds. More recently, Li’s group described a CPA-catalyzed enantioselective desymmetric Povarov reaction, coupled with oxidative aromatization, to access planar-chiral [n]metacyclophanes (Scheme 1B, III)[14]. In these systems, conformational stability is maintained through bulky substituents on the benzene ring and a short ansa chain. Most recently, our group reported a CPA-catalyzed asymmetric ansa chain editing strategy for the synthesis of metacyclophanes (Scheme 1B, IV)[54]. This approach leverages the combined effects of ansa chain length, substituent bulk on the arene ring, and restricted rotation of the tertiary amide bond to cooperatively stabilize the resulting conformation.
The Pd/pGlu-catalyzed atroposelective C–H olefination has been well established for constructing enantioenriched scaffolds[55-67]. For example, Shi and co-workers developed an elegant picolinamide-directed asymmetric C–H olefination to access axially chiral anilides[59]. This catalytic system was subsequently extended to the construction of N–aryl peptide atropisomers and axially chiral styrenes[60-62]. More recently, we employed the Pd/pGlu platform in the late-stage C–H functionalization of cyclophanes, providing efficient access to diverse planar-chiral paracyclophanes (Scheme 1C)[40-42]. We hypothesized that the same strategy could be applicable to the atroposelective synthesis of metacyclophanes. Herein, we report a Pd-catalyzed late-stage C–H olefination of metacyclophanes enabled by a monoprotected amino acid ligand (Scheme 1D). Furthermore, our study revealed that introducing a picolinamide group onto the ansa chain offers additional conformational stabilization for the resulting metacyclophanes.
2. Experimental
Representative Synthesis of [n]metacyclophanes 3. A 10 mL-reaction tube equipped with a magnetic stirring bar was charged with metacyclophanes 1 (0.1 mmol, 1.0 equiv.), alkene 2 (0.2 mmol, 2.0 equiv.), Pd(OAc)2 (2.2 mg, 0.01 mmol, 0.1 equiv.), L-pGlu-OH (2.6 mg, 0.02 mmol, 0.2 equiv.) and AgTFA (66.3 mg, 0.3 mmol, 3.0 equiv.). The tube was sealed and iPrOH (2.0 mL) was injected into the tube via a syringe. The reaction mixture was allowed to stir at 60 °C (pre-heated metal module) under air for 12 h. Then, the reaction mixture was immediately cooled to room temperature. The solvent was evaporated and the residue was purified by alkaline alumina column chromatography (PE/EA = 2:1, v/v) affording the metacyclophanes 3.
3. Results and Discussion
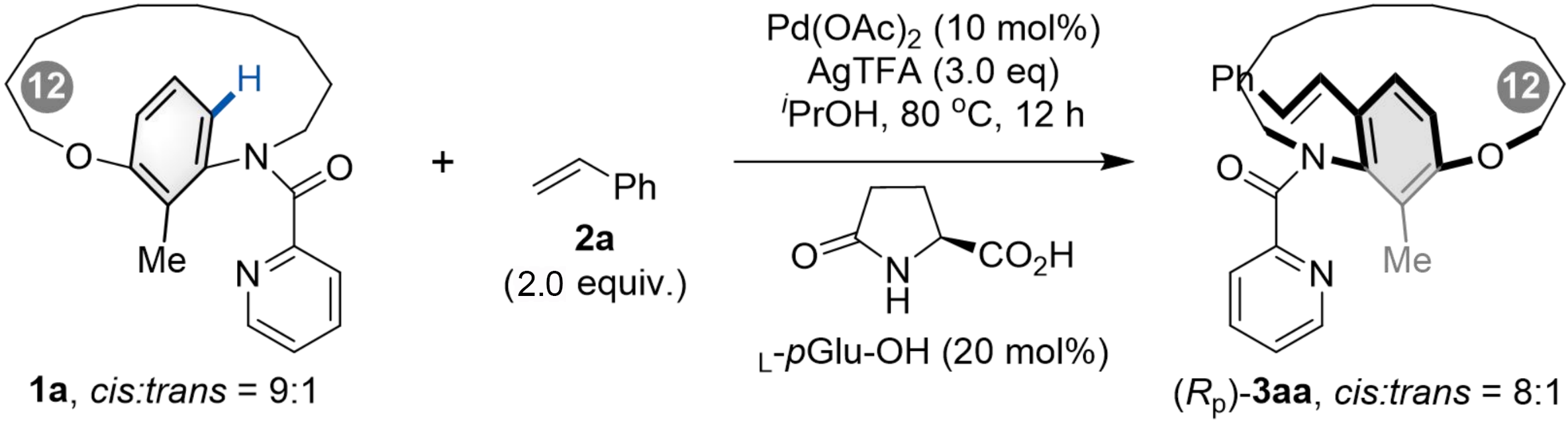
The Pd-catalyzed late-stage atroposelective C–H olefination of metacyclophane was investigated using picolinamide-derived [12]metacyclophane 1a and styrene 2a as model substrates. As summarized in Table 1, a series of palladium catalysts was evaluated with AgTFA (3.0 equiv.) as the oxidant, L-pGlu-OH (20 mol%) as the chiral ligand, and iPrOH as the solvent. When Pd(OAc)2 was used as the catalyst, product 3aa was obtained in high yield with 88% ee (entry 1). Catalysts such as Pd(PPh3)Cl2, Pd(COD)Cl2, and Pd(MeCN)2Cl2 also afforded excellent conversion, with ee values ranging from 69% to 71% (entries 3-4, 6). In contrast, PdCl2 and PdBr2 led to slightly lower reaction efficiencies (entries 2, 5). Screening of other oxidants, including Ag2O, AgOAc, Ag2SO4, AgNO3, and Ag3PO4, did not improve the reaction outcomes (entries 7-11). Further investigation of solvents revealed that protic solvents generally afforded the product in high yield and ee (entries 12-13), whereas MeCN and toluene gave 3aa in moderate yield (entries 14-15). Although aprotic solvents such as chlorobenzene and 1,2-dimethoxyethane (DME) resulted in excellent conversion, the product was obtained with low ee (entries 16-17). Interestingly, lowering the reaction temperature led to an increase in enantiomeric excess (entries 18-19), which is attributed to the suppression of racemization of 3aa under milder conditions. Conformational stability studies indicated a rotational barrier of 124.37 kJ·mol-1 for 3aa, corresponding to a half-life of 65.2 minutes for the rotation reaction at 110 °C. Both substrate 1a and product 3aa exist as inseparable mixtures of cis and trans rotamers, which originate from rotation about the N–CO bond of the 2-pyridyl formyl group. 1H NMR and X-ray diffraction analysis indicated that the cis isomer was found to be predominant[40-42,59-61].
Table 1. Optimization of the reaction conditions.
| Entrya | Variation from above conditions | Yield (%)b | ee (%)c |
| 1 | None | 99 | 88 |
| 2 | PdCl2 (10 mol%) | 72 | 69 |
| 3 | Pd(PPh3)Cl2 (10 mol%) | 99 | 71 |
| 4 | Pd(COD)Cl2 (10 mol%) | 99 | 69 |
| 5 | PdBr2 (10 mol%) | 84 | 69 |
| 6 | Pd(MeCN)2Cl2 (10 mol%) | 99 | 70 |
| 7 | Ag2O (3.0 equiv.) | 70 | 69 |
| 8 | AgOAc (3.0 equiv.) | 85 | 72 |
| 9 | Ag2SO4 (3.0 equiv.) | 68 | 72 |
| 10 | AgNO3 (3.0 equiv.) | 99 | 84 |
| 11 | Ag3PO4 (3.0 equiv.) | 75 | 73 |
| 12 | t-AmylOH as the solvent | 99 | 82 |
| 13 | TFE as the solvent | 99 | 84 |
| 14 | MeCN as the solvent | 64 | 83 |
| 15 | toluene as the solvent | 64 | 53 |
| 16 | PhCl as the solvent | 99 | 27 |
| 17 | DME as the solvent | 99 | 56 |
| 18 | 70 °C | 96 | 90 |
| 19 | 60 °C | 90 (80)d | 95 |
a: Reaction conditions: 1a (0.1 mmol), 2a (0.2 mmol, 2.0 equiv.), Pd(OAc)₂ (0.01 mmol, 10 mol%), L-pGlu-OH (0.02 mmol, 20 mol%), and oxidant (0.3 mmol, 3.0 equiv.) in iPrOH (2.0 mL). The mixture was stirred at 80 °C for 12 h under air. cis/trans ratio was determined by ¹H NMR analysis; b: The yields were determined by 1H NMR with 1,3,5-trimethoxybenzene (1/3 equiv.) as an internal standard; c: Enantiomeric excess (ee) was determined by HPLC on a chiral stationary phase; d: Yield refers to isolated yield. Numbering in grey circles indicates ansa chain length. ee: enantiomeric excess; TFE: trifluoroethanol; DME: 1,2-dimethoxyethane.
After establishing the optimal reaction conditions, we evaluated the substrate scope of alkenes in the Pd-catalyzed C–H olefination for the synthesis of [n]metacyclophanes (Scheme 2). Substituted vinylarenes with electron-neutral, electron-donating, and electron-withdrawing groups all proved compatible (3aa-3ai), affording [12]metacyclophanes in moderate to good yields with high to excellent enantioselectivities. Notably, substitution at the 3-position of the benzene ring further improved enantiocontrol (3ah, > 99% ee). A range of acrylates, including sterically hindered derivatives, also reacted smoothly to deliver the corresponding products (3aj-3an) in good yields with high enantioselectivities. Moreover, substrates bearing other functional groups, such as phosphate acrylate, dimethyl(phenyl)silane, and phenyl sulfone, were also suitable, providing [12]metacyclophanes (3ao-3aq) in moderate to good yields with high enantioselectivities. Additionally, vinylarenes including 2-vinylnaphthalene, 1-vinylpyrene, and vinylferrocene were efficiently transformed into the target metacyclophanes (3ar-3at).
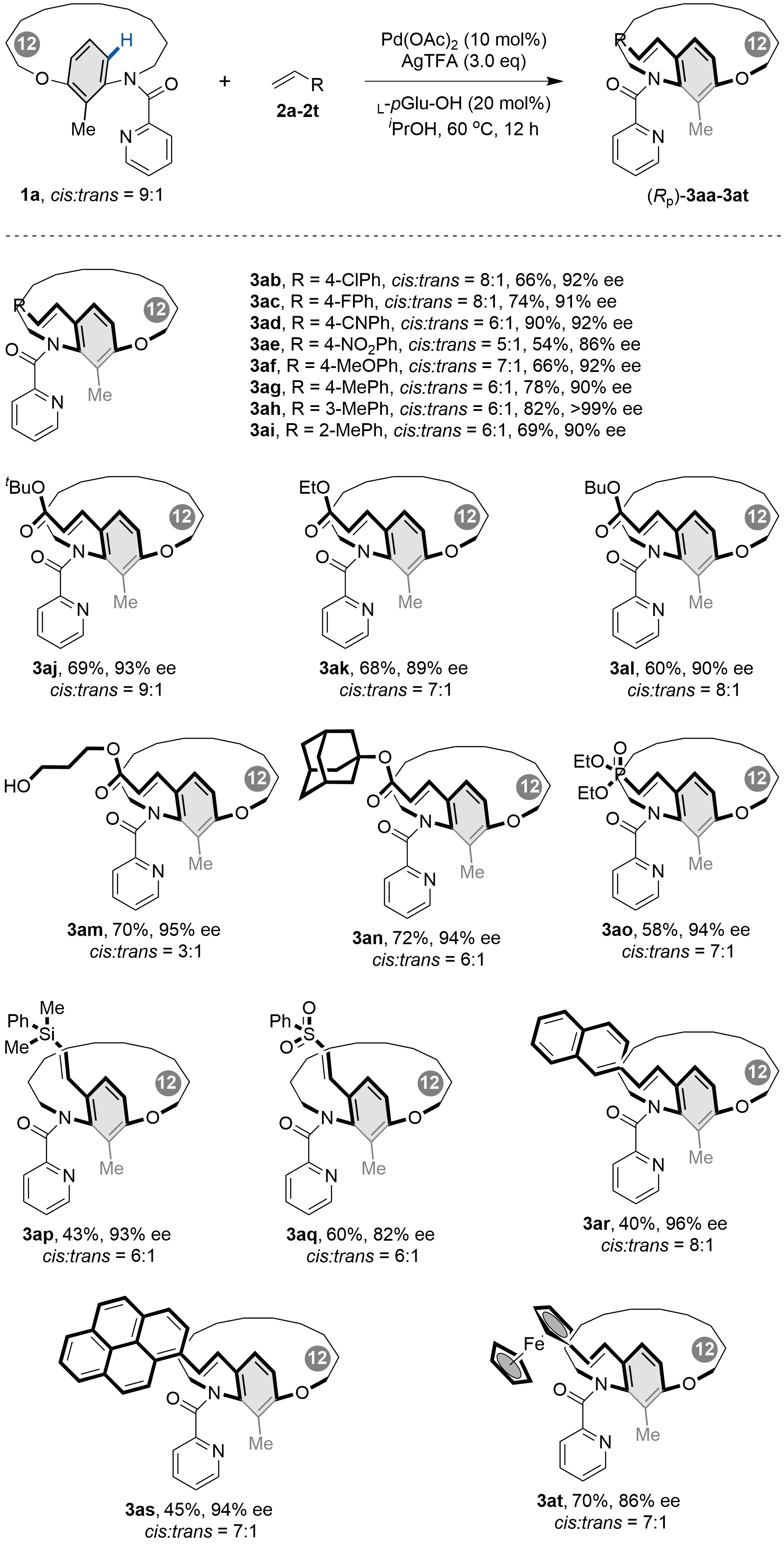
Scheme 2. Scope of alkenes. Reaction conditions: 1a (0.1 mmol, 1.0 equiv.), 2 (0.2 mmol, 2.0 equiv.), Pd(OAc)2 (0.01 mmol, 0.1 equiv.), L-pGlu-OH (0.02 mmol, 0.2 equiv.) and AgTFA (0.3 mmol, 3.0 equiv.) in iPrOH (2.0 mL) was stirred at 60 °C for 12 h under air. Created in ChemDraw.
We next examined the effect of ansa chain length and benzene ring substituents on the reaction (Scheme 3). For [9]-[11]metacyclophanes, the C–H olefination proceeded via a kinetic resolution pathway. Under standard conditions, using 2.0 equivalents of alkene 2a, [11]metacyclophane 3da was obtained in 68% yield with 0% ee, as both isomers were fully consumed. When the loading of 2a was reduced to 0.5 equivalents, product 3da was obtained in 37% yield with 81% ee, while the remaining starting material 1d was recovered in 35% yield and > 99% ee. A similar trend was observed for the [9]-[10]metacyclophanes (3ba–3ca), which also exhibited high selectivity under kinetic resolution conditions. In contrast, the longer-chain [13]-[16]- and [20]metacyclophanes underwent dynamic kinetic resolution, furnishing the corresponding products (3ea–3ia) in moderate to high yields with high enantioselectivities. Furthermore, [12]metacyclophane comprising heteroatom variations in the ansa chain were obtained in good yields with high enantioselectivity (3ka). More importantly, the reaction demonstrated broad tolerance for diverse aromatic substituents at the 2-position of the benzene ring, affording products 3la–3pa in synthetically useful yields and with high enantioselectivities.
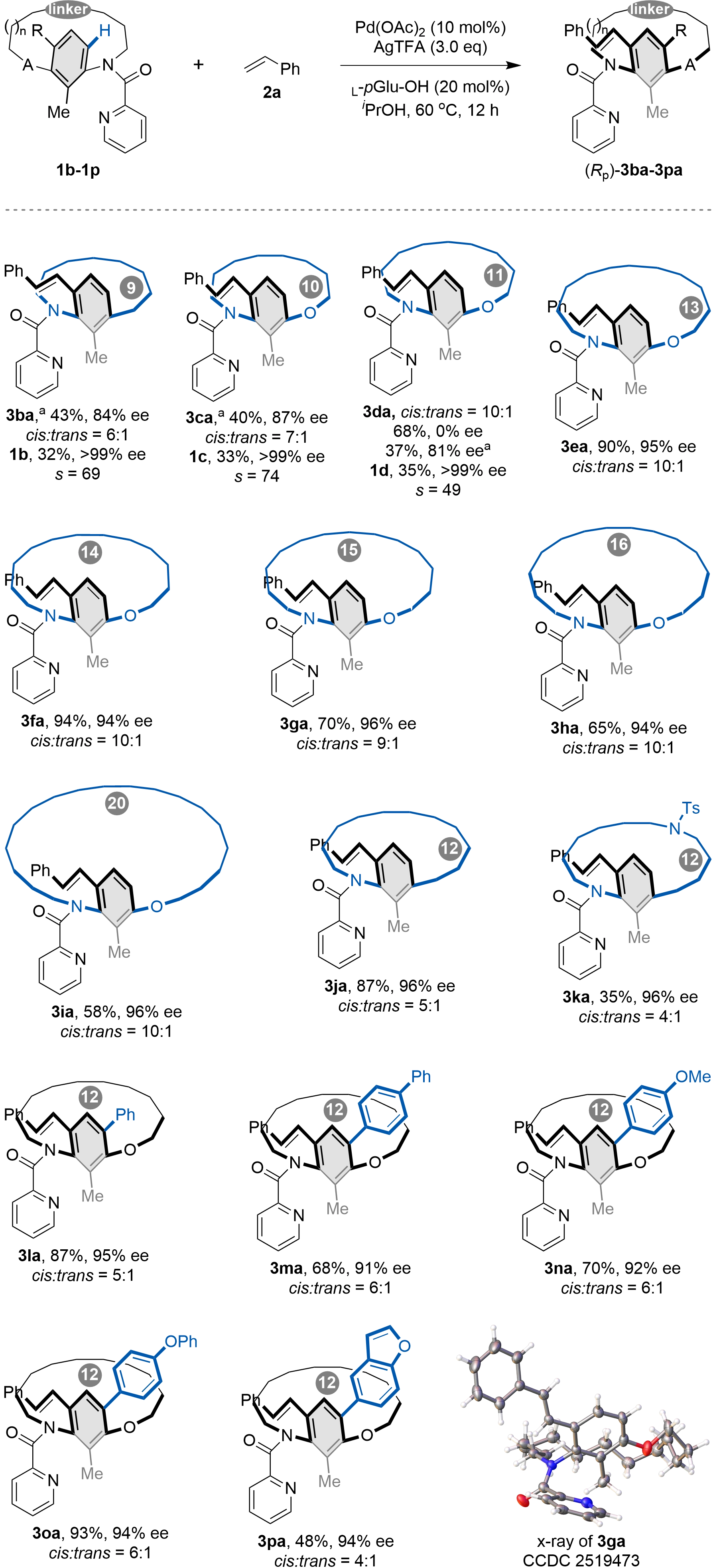
Scheme 3. Scope of the ansa chain and the benzene ring substituents. Reaction conditions: 1 (0.1 mmol, 1.0 equiv.), 2a (0.2 mmol, 2.0 equiv.), Pd(OAc)2 (0.01 mmol, 0.1 equiv.), L-pGlu-OH (0.02 mmol, 0.2 equiv.) and AgTFA (0.3 mmol, 3.0 equiv.) in iPrOH (2.0 mL) was stirred at 60 °C for 12 h under air. a2a (0.5 equiv.) was used, the reaction was performed for 8 h. Created in ChemDraw.
To further investigate the conformational stability of [n]metacyclophanes, racemization experiments were conducted on derivatives with varying ansa chain lengths (Scheme 4A). The results demonstrate that as the number of atoms in the ansa chain increased from 12 to 14 (3aa, 3ea to 3fa), the racemization energy barrier gradually rose. Additionally, the barriers for compounds 3ga, 3ha, and 3ia were determined to be 125.18, 124.93, and 124.39 kJ·mol-1, respectively. These observations contrast with the previously established trend[41,42,68], which suggested that increasing the length of the ansa chain reduces steric hindrance to aromatic ring rotation and thereby lowers conformational stability. Furthermore, the introduction of a picolinamide group onto the ansa chain enables concurrent rotation of the aromatic ring about both the ansa chain and the C–N axis. The overall conformational stability is governed by the combined steric hindrance arising from these two rotational modes.
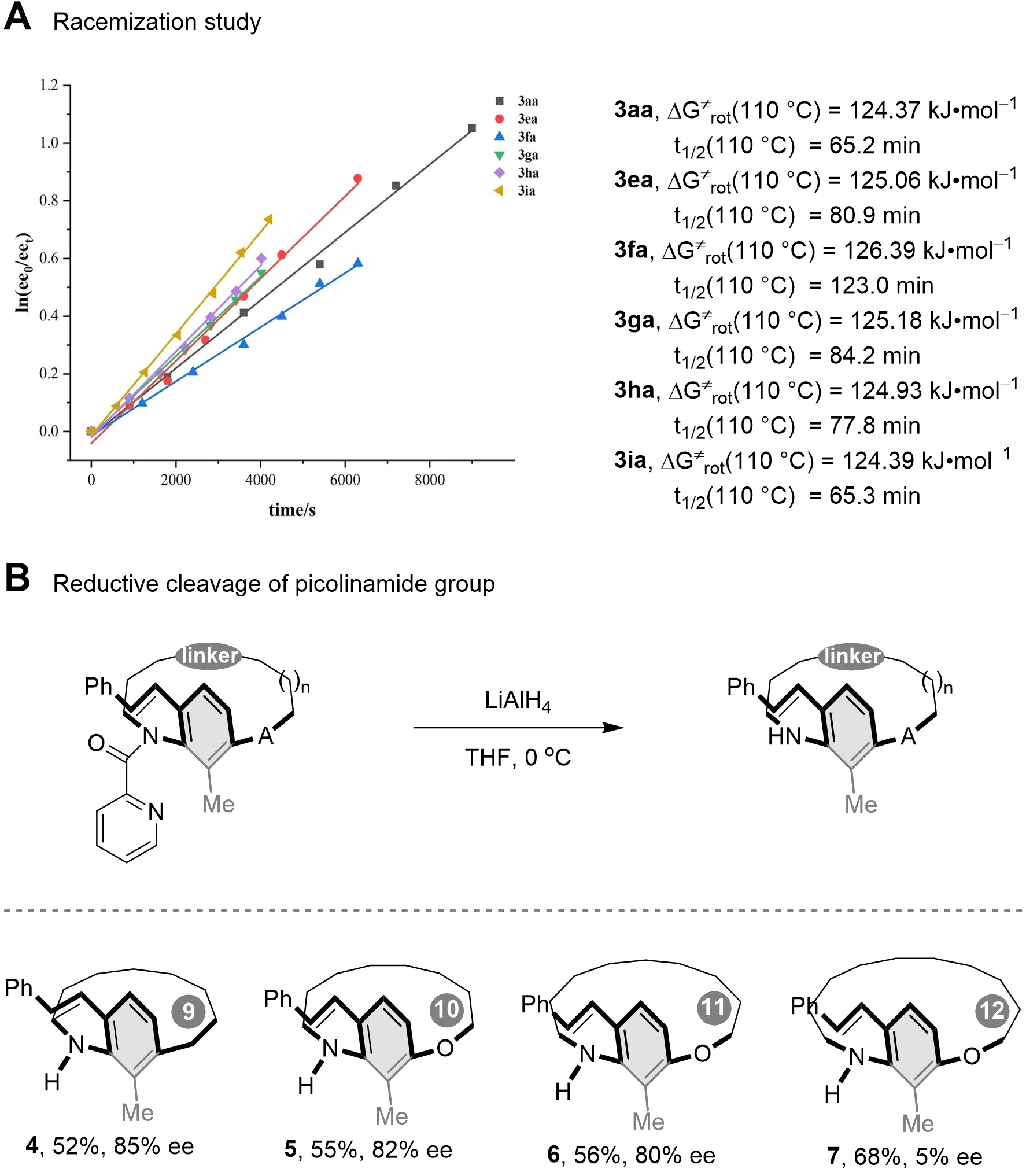
We also performed reduction experiments to further confirm the auxiliary role of the picolinamide group in stabilizing the conformation of the metacyclophanes (Scheme 4B). Compounds 3aa–3da were treated with lithium aluminium hydride (LiAlH4) to afford products 4-7. In the case of product 7, derived from [12]metacyclophane, the removal of the C–N rotational hindrance meant that the 12-atom ansa chain alone could not effectively restrict rotation of the benzene ring. Consequently, enantioselective control was poor (5% ee). In contrast, when the ansa chain contained ≤ 11 atoms, the chain itself was sufficiently short to restrict benzene-ring inversion without additional steric assistance. Thus, reduced products 4-6 exhibited almost no loss of enantiomeric excess, maintaining 80%-85% ee. These outcomes clearly demonstrate the auxiliary contribution of the picolinamide group around the C–N axis to conformational stability[41,42,69].
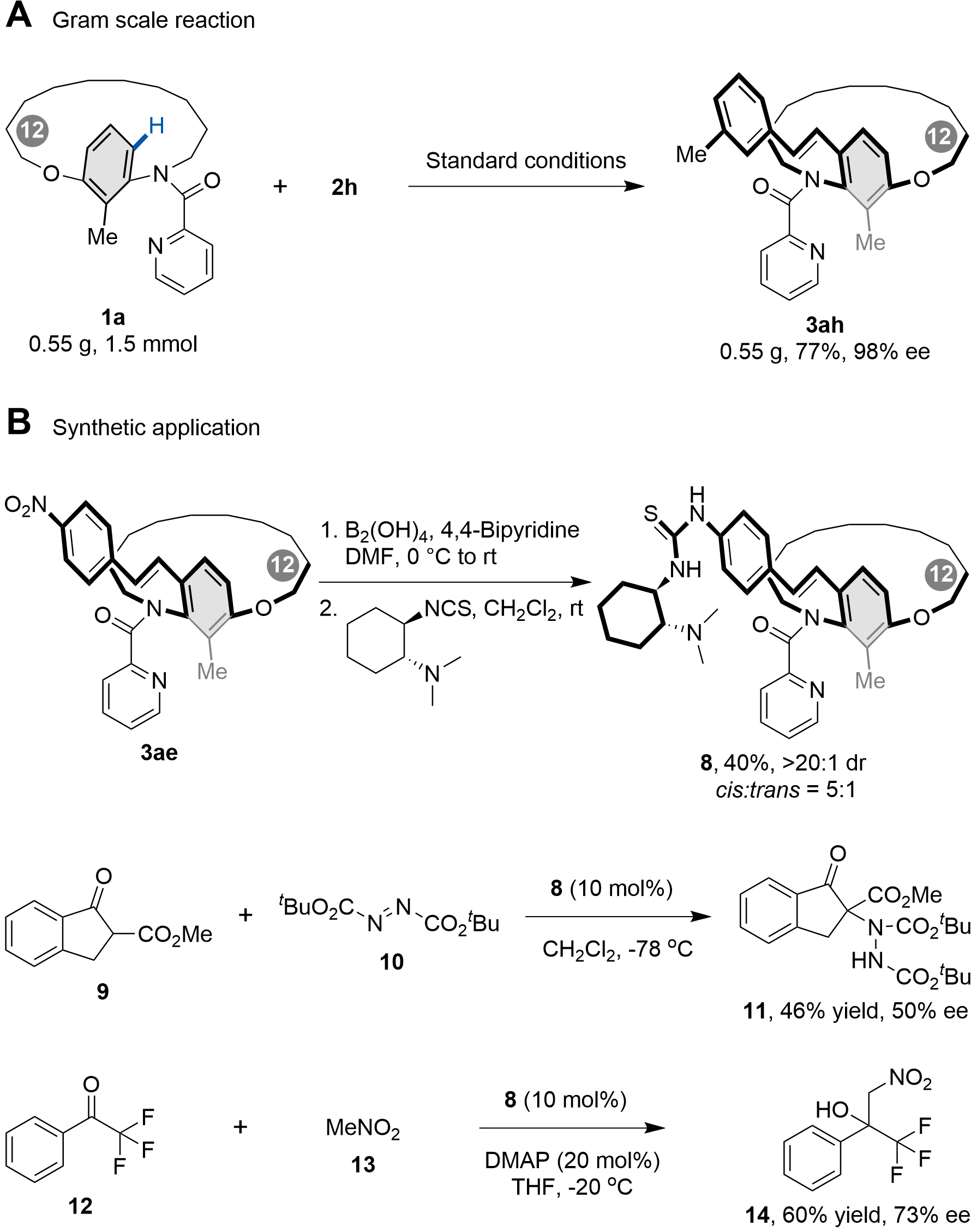
To further demonstrate the synthetic utility of this methodology, a gram-scale reaction of 1a with 2h was carried out (Scheme 5). The C–H olefination proceeded smoothly under the standard conditions, affording 0.55 g of metacyclophane 3ah in 98% ee. Subsequently, compound 3ae was transformed into a thiourea-based bifunctional catalyst. The resulting catalyst 8 was employed in the Michael addition of β-ketoester 9 to di-tert-butyl azodicarboxylate 10, furnishing product 11 in 46% yield and 50% ee. Additionally, catalyst 8 proved effective in catalyzing the Henry reaction between trifluoromethyl acetophenone 12 and nitromethane 13, affording the centrally chiral product 14 in 60% yield with 73% ee.
4. Conclusion
In summary, we have developed a Pd(II)-catalyzed enantioselective C–H olefination strategy for constructing [n]metacyclophanes. The protocol affords a range of metacyclophanes in high yields with excellent enantioselectivities. Studies on racemization indicate that the conformational stability of these macrocycles depends on the rotation of the aromatic ring along both the ansa chain and the C–N axis. The practicality of this approach is underscored by its scalability and the effective use of resulting products as catalysts in asymmetric transformations.
Supplementary materials
The supplementary material for this article is available at: Supplementary materials.
Authors contribution
Tang Q, Zhao C: Conceptualization, writing-original draft, writing-review & editing, data curation, formal analysis.
Yan XE, Li J, Dong Z, Zhai H: Methodology, investigation, data curation, formal analysis.
Conflicts of interest
The authors declare no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
The data and materials could be obtained from the corresponding author upon request.
Funding
This research was supported by the National Natural Science Foundation of China Foundation of China (Grant No. 22571022).
Copyright
© The Author(s) 2026.
References
-
1. Griffin RW. Meta-bridged aromatic compounds. Chem Rev. 1963;63(1):45-54.[DOI]
-
2. Cram DJ, Cram JM. Cyclophane chemistry: Bent and battered benzene rings. Acc Chem Res. 1971;4(6):204-213.[DOI]
-
3. Gleiter R, Hopf H, editors. Modern cyclophane chemistry. Weinheim: Wiley-VCH Verlag; 2004.[DOI]
-
4. Kotha S, Shirbhate ME, Waghule GT. Selected synthetic strategies to cyclophanes. Beilstein J Org Chem. 2015;11:1274-1331.[DOI]
-
6. Marshall JA. Trans-cycloalkenes and [a.b]betweenanenes, molecular jump ropes and double bond sandwiches. Acc Chem Res. 1980;13(7):213-218.[DOI]
-
7. Tanaka K. Catalytic enantioselective synthesis of planar chiral cyclophanes. Bull Chem Soc Jpn. 2018;91(2):187-194.[DOI]
-
8. Culp EJ, Waglechner N, Wang W, Fiebig-Comyn AA, Hsu YP, Koteva K, et al. Evolution-guided discovery of antibiotics that inhibit peptidoglycan remodelling. Nature. 2020;578(7796):582-587.[DOI]
-
10. Glunz PW, Mueller L, Cheney DL, Ladziata V, Zou Y, Wurtz NR, et al. Atropisomer control in macrocyclic factor VIIa inhibitors. J Med Chem. 2016;59(8):4007-4018.[DOI]
-
14. Wang Z, Lin J, Shao YB, Wang B, Li X. Enantioselective synthesis of macrocyclic planar chiral metacyclophanes via desymmetrization. ACS Catal. 2025;15(21):17938-17949.[DOI]
-
15. Han XN, Han Y, Chen CF. Supramolecular tessellations by the exo-wall interactions of pagoda[4]arene. Nat Commun. 2021;12:6378.[DOI]
-
16. Han XN, Han Y, Chen CF. Recent advances in the synthesis and applications of macrocyclic arenes. Chem Soc Rev. 2023;52(9):3265-3298.[DOI]
-
17. Niu S, Xiao H, Yang XD, Cong H. Chloride anion-induced dimer capsule based on a polyfluorinated macrocycle meta-WreathArene. Chin Chem Lett. 2023;34(7):108042.[DOI]
-
18. Song YH, Bian Q, Wang F, Liu J, Yang YH, Zhang YM, et al. Water-soluble stimuli-responsive supramolecular nanoagrochemicals based on macrocycle compounds. Coord Chem Rev. 2025;524:216299.[DOI]
-
20. Hirano S, Hara H, Hiyama T, Fujita S, Nozaki H. Synthetic and structural studies of [6]-, [7]- and [10]metacyclophanes. Tetrahedron. 1975;31(18):2219-2227.[DOI]
-
22. Dong Z, Li J, Zhao C. Catalytic enantioselective macrocyclization for the synthesis of planar-chiral cyclophanes: Recent updates. Eur J Org Chem. 2024;27(47):e202400841.[DOI]
-
24. Zhai H, Lv K, Li J, Wang J, Liu T, Zhao C. Rhodium(III)-catalyzed atroposelective indolization to access planar-chiral macrocycles. J Am Chem Soc. 2024;146(42):29214-29223.[DOI]
-
25. Zhao YH, Zhu D, Chen ZM. Enantioselective construction of planar-chiral molecules by catalytic asymmetric late-stage functionalizations. ChemCatChem. 2024;16(23):e202401312.[DOI]
-
27. Zhu K, Yang L, Yang Y, Wu Y, Zhang F. Recent advances toward the catalytic enantioselective synthesis of planar chiral cyclophanes. Chin Chem Lett. 2025;36(7):110678.[DOI]
-
31. Li J, Zhao C. Highly enantioselective synthesis of planar-chiral cyclophanes through a Brønsted acid-catalyzed asymmetric transfer hydrogenation. ACS Catal. 2023;13(21):14155-14162.[DOI]
-
32. Lv X, Su F, Long H, Lu F, Zeng Y, Liao M, et al. Carbene organic catalytic planar enantioselective macrolactonization. Nat Commun. 2024;15:958.[DOI]
-
34. Chen Z, Yang W, Jia M, Shi J, Li J, Chen H, et al. Synthesis of highly strained para-cyclophanes via ring-expansion [5,5]-sigmatropic rearrangement reaction. Nat Chem. 2025;17(8):1169-1178.[DOI]
-
36. Wang Z, Zhang XX, Sun Y, Zheng H, Li X. Enantioselective synthesis of planar/multiple chiral [n]cyclophanes through asymmetric allylation. Chin J Chem. 2025;43(11):1263-1270.[DOI]
-
38. Chen D, Zhou Y, Tung CH, Yu ZX, Xu Z. A desymmetric dearomatization cyclopropanation of [2.2]paracyclophane. CCS Chem. 2025;7(5):1509-1521.[DOI]
-
39. Li J, Dong Z, Chen Y, Yang Z, Yan X, Wang M, et al. N-Heterocyclic carbene-catalyzed enantioselective synthesis of planar-chiral cyclophanes via dynamic kinetic resolution. Nat Commun. 2024;15:2338.[DOI]
-
44. Wang J, Lv K, Wen Y, Liu T, Zhao C. Enantio-, atrop-, and diastereoselective macrolactonization to access type III cyclophanes. Nat Commun. 2025;16:3170.[DOI]
-
45. Yao T, Yuan C, Wang X, Zhai H, Zhao C. Organocatalytic enantio-, atrop-, and diastereoselective macrocyclization of quinone methides. CCS Chem. 2025.[DOI]
-
48. Kane VV, De Wolf WH, Bickelhaupt F. Synthesis of small cyclophanes. Tetrahedron. 1994;50(16):4575-4622.[DOI]
-
49. Piątek P, Kalisiak J, Jurczak J. Synthesis and chiroptical properties of two new planar-chiral macrocycles. Tetrahedron Lett. 2004;45(16):3309-3311.[DOI]
-
50. Ishida N, Sawano S, Murakami M. Stereospecific ring expansion from orthocyclophanes with central chirality to metacyclophanes with planar chirality. Nat Commun. 2014;5:3111.[DOI]
-
52. Wei S, Chen LY, Li J. Enantioselective synthesis of planar chiral macrocyclic metacyclophanes by Pd-catalyzed C–O cross-coupling. ACS Catal. 2023;13(11):7450-7456.[DOI]
-
54. Dong Z, Lv K, Yuan C, Wang L, Wang H, Li J, et al. Syntheses, mechanism insights, and anti-inflammatory activities of conformationally defined [n]metacyclophanes. ACS Catal. 2026;16(3):2182-2193.[DOI]
-
62. Jiang TY, Ke YT, Wu YJ, Yao QJ, Shi BF. Pd(II)-Catalyzed atroposelective C−H olefination: Synthesis of enantioenriched N-aryl peptoid atropisomers. Chem Commun. 2023;59(90):13518-13521.[DOI]
-
63. Jin L, Yao QJ, Xie PP, Li Y, Zhan BB, Han YQ, et al. Atroposelective synthesis of axially chiral styrenes via an asymmetric C–H functionalization strategy. Chem. 2020;6(2):497-511.[DOI]
-
65. Jiang BY, Zhou G, Jiang AL, Zhou T, Shi BF. Synthesis of axially chiral biaryl-2-carboxamides through Pd(II)-catalyzed atroposelective C−H olefination. Org Chem Front. 2024;11(13):3710-3716.[DOI]
-
67. Fan L, Zhou T, Yang X, Jiang M, Hu X, Shi B. Pd(II)-catalyzed enantioselective C−H olefination of 2-(arylsulfinyl)pyridines through kinetic resolution. Chin J Org Chem. 2022;42(10):3405-3418.[DOI]
-
68. Zhao G, Li J, Tang Q, Zhao C. Pd(II)-catalyzed highly atroposelective C–H alkynylation of [n]paracyclophanes. Synthesis. 2026.[DOI]
-
69. Wang X, Li J, Tang Q, Zhao C. Pd(II)-catalyzed atroposelective C–H olefination to access [n]naphthalenophanes. Org Chem Front. 2026;13(5):1523-1530.[DOI]
Copyright
© The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Publisher’s Note
Science Exploration remains a neutral stance on jurisdictional claims in published
maps
and institutional affiliations. The views expressed in this article are solely those
of
the author(s) and do not reflect the opinions of the Editors or the publisher.
Share And Cite



Science Exploration Style
Yan XE, Li J, Dong Z, Zhai H, Tang Q, Zhao C. Pd(II)-catalyzed atroposelective and enantioselective C−H olefination of [n]metacyclophanes. Chiral Chem. 2026;2:202606. https://doi.org/10.70401/cc.2026.0016
Tips
Copy completed.
Submit a Manuscript
Author Instructions
Cite this Article
Article Metrics
0
View
0
Download
Cited
Article Updates

Science Exploration Style
Yan XE, Li J, Dong Z, Zhai H, Tang Q, Zhao C. Pd(II)-catalyzed atroposelective and enantioselective C−H olefination of [n]metacyclophanes. Chiral Chem. 2026;2:202606. https://doi.org/10.70401/cc.2026.0016
copy
Share Link
copy