Enantio- and diastereoselective NHC/Cu-catalyzed intermolecular dearomative cyclopropanation of indoles with diazo esters
Jiawei Lu
Sheng Liu
Lin-Xin Ruan
*

,
Shu-Li You
*
,
Shi-Liang Shi
*
*Correspondence to:
Lin-Xin Ruan, State Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, University of Chinese Academy of Sciences, Shanghai 200032, China.
E-mail: ruanlinxin@sioc.ac, cn
Shu-Li You, State Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, University of Chinese Academy of Sciences, Shanghai 200032, China. E-mail: slyou@sioc.ac.cn.
Shi-Liang Shi, State Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, University of Chinese Academy of Sciences, Shanghai 200032, China. E-mail: shiliangshi@sioc.ac.cn.
Shu-Li You, State Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, University of Chinese Academy of Sciences, Shanghai 200032, China. E-mail: slyou@sioc.ac.cn.
Shi-Liang Shi, State Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, University of Chinese Academy of Sciences, Shanghai 200032, China. E-mail: shiliangshi@sioc.ac.cn.
Chiral Chem. 2026;2:202608. 10.70401/cc.2026.0017
Received: February 10, 2026Accepted: March 20, 2026Published: March 24, 2026
Abstract
We report a highly enantioselective N-heterocyclic carbene (NHC)/copper-catalyzed intermolecular dearomative cyclopropanation of indoles with diazo esters. This protocol enables the efficient construction of cyclopropane-fused indolines featuring quaternary stereogenic centers under mild conditions. A broad range of substituted indoles and diazo esters is tolerated, delivering the corresponding products in high yields with enantioselectivities of up to 90% ee. Notably, the employment of a bulky chiral NHC ligand is crucial for achieving effective stereocontrol over the challenging quasi-linear copper–carbenoid intermediate. Furthermore, gram-scale reactions and downstream transformations demonstrate the synthetic utility of this methodology. Mechanistic investigations, including kinetic isotope effect and Hammett studies, support a reaction pathway involving nucleophilic attack of the indole on a copper–carbenoid species. This earth-abundant copper catalytic system serves as a sustainable alternative to traditional noble-metal-based asymmetric carbene-transfer reactions.
Graphical Abstract
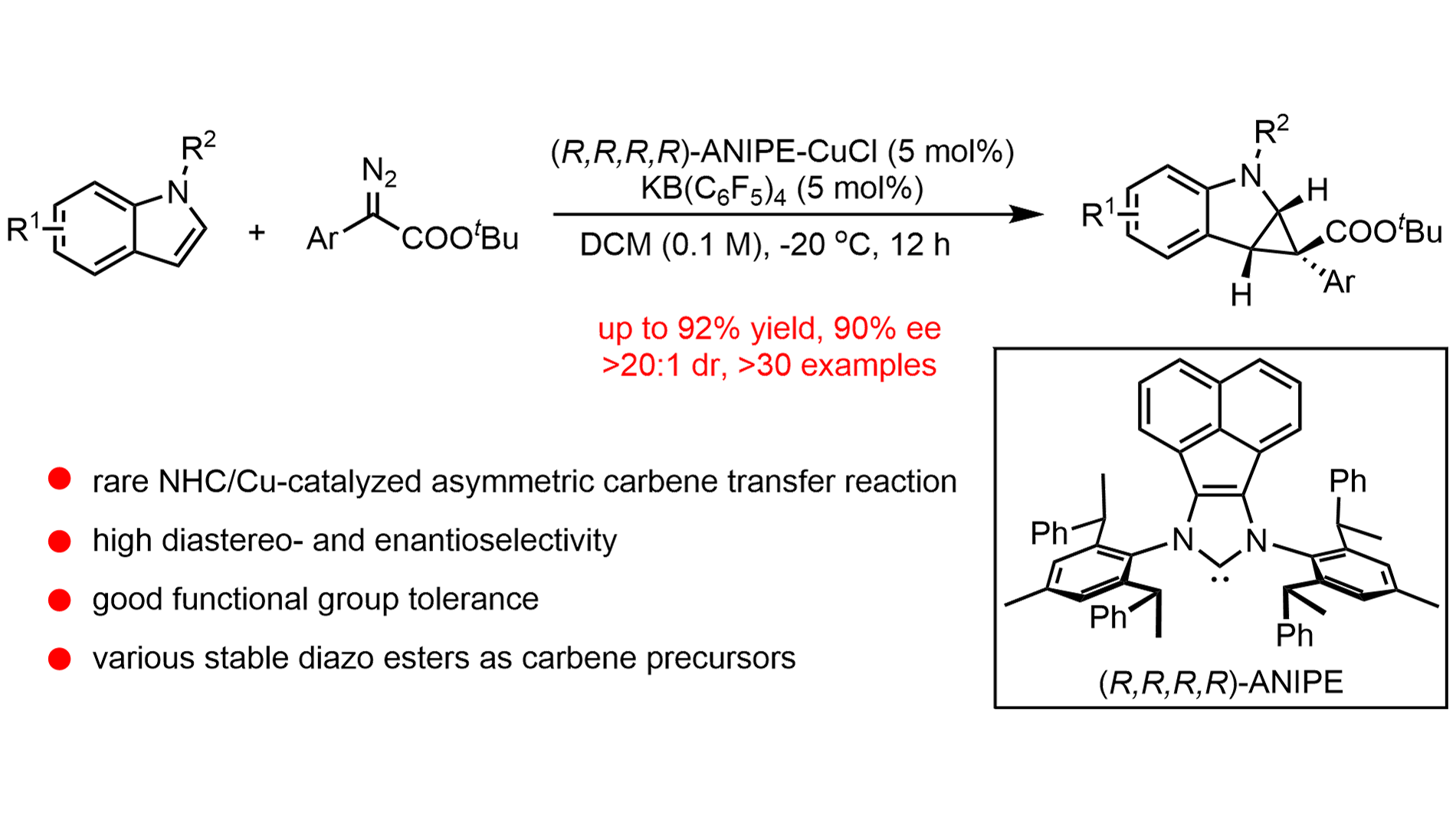
Keywords
Asymmetric cyclopropanation, chiral NHC ligand, copper catalysis, dearomatization, indole
1. Introduction
Dearomatization represents a fundamental and powerful strategy in organic synthesis, enabling the transformation of simple aromatic compounds into partially saturated cyclic frameworks characterized by enhanced structural complexity and stereochemical diversity[1-3]. In particular, catalytic asymmetric dearomatization (CADA) reactions have garnered significant attention, as they offer efficient and streamlined access to chiral molecules bearing multiple stereocenters from readily available aromatic precursors[4]. Among various aromatic substrates[5-8], indoles have emerged as exceptionally attractive platforms for dearomatization. Given that polycyclic indole derivatives are prevalent in alkaloid natural products and biologically active compounds[9-11], their asymmetric synthesis has stimulated sustained interest[12,13]. From a synthetic perspective, the dearomative functionalization of indole derivatives represents one of the most straightforward and versatile approaches for constructing these privileged molecular scaffolds[14,15].
Asymmetric dearomatization of indoles via carbene insertion has recently emerged as an effective strategy for the construction of fused indoline frameworks. Although asymmetric carbene-transfer reactions have been extensively investigated over the past decades[16-21], enantioselective dearomative cyclopropanation of indoles remains relatively underexplored. In 2024, Bi and co-workers[22] reported a rhodium-catalyzed intermolecular asymmetric dearomative cyclopropanation of indoles using trifluoromethyl N-triftosylhydrazones as carbene precursors, achieving excellent enantioselectivities of up to 99% ee (Figure 1a). The utilization of chiral dirhodium(II) paddlewheel complexes proved crucial, as their unique electronic and coordination properties facilitate the efficient generation and stabilization of metal–carbene intermediates, thereby enhancing both reactivity and stereocontrol[23,24]. Given the economic advantages and greater availability of copper compared to rhodium, the development of Cu-catalyzed asymmetric carbene-transfer reactions is of substantial practical interest. Boysen[25] and Pirovano[26] independently reported Cu-catalyzed intermolecular cyclopropanation of indoles with ethyl diazoacetate. However, these two protocols generally afforded only moderate enantioselectivity or unsatisfactory reaction yields (Figure 1b). In 2017, Zhou and co-workers[27] developed the first highly enantioselective Cu-catalyzed intramolecular cyclopropanation of indoles using a chiral spiro bisoxazoline ligand (Figure 1b, up to > 99% ee).
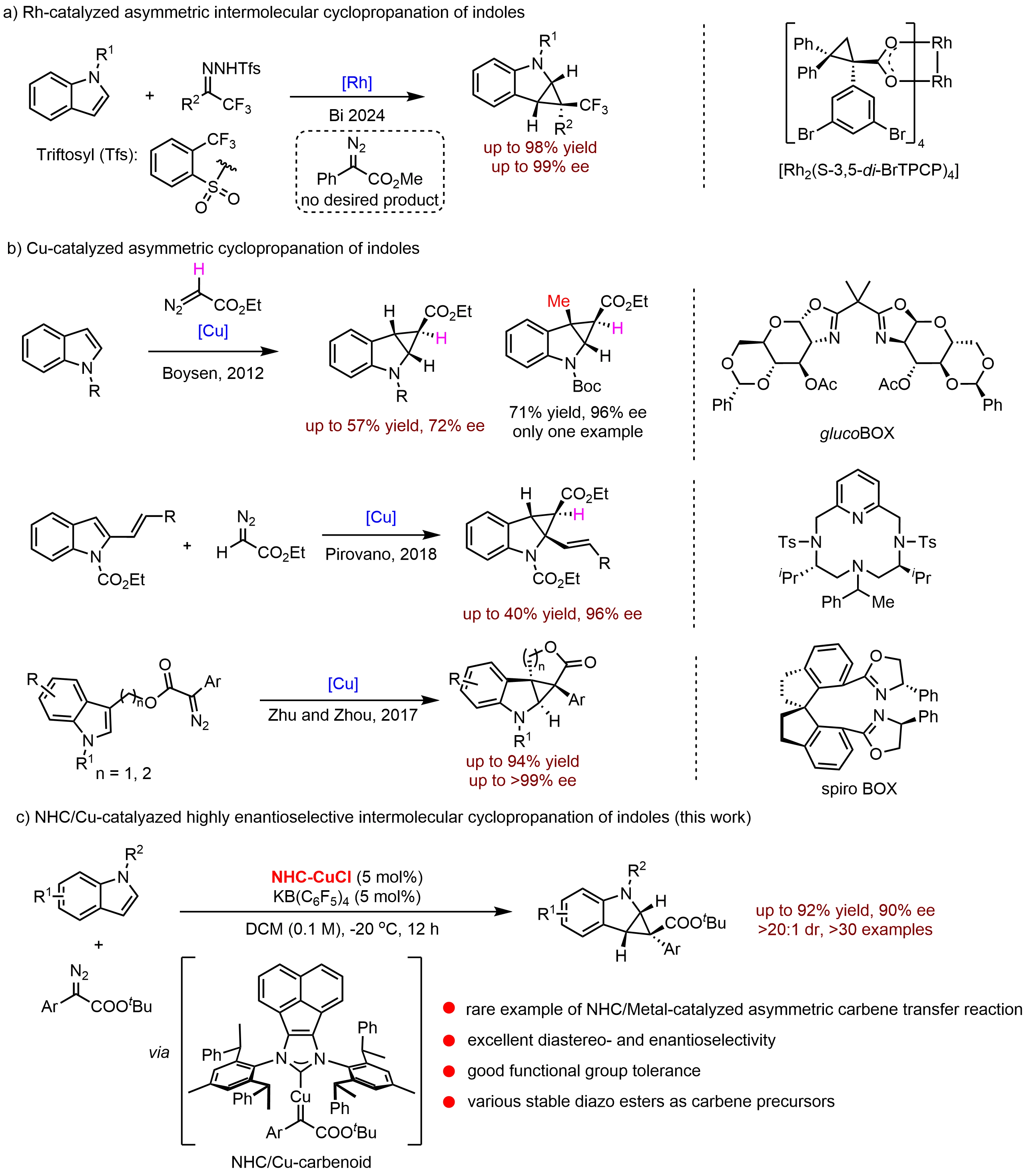
Despite their potential advantages, asymmetric dearomative cyclopropanation of indoles, particularly intermolecular processes, remains largely underexplored. This limitation primarily stems from the relative stability of indoles compared to simple alkenes[17]. Furthermore, several competing side reactions, such as the ring-opening of strained cyclopropanes to form quinoline derivatives[28], competitive C3–H insertion of indoles[22,29], and dimerization of carbene intermediates[30], often compromise reaction efficiency and stereochemical control. Consequently, highly efficient asymmetric intermolecular cyclopropanation of indoles based on earth-abundant metals remains rare. A major challenge lies in the scarcity of suitable chiral ligands capable of simultaneously promoting high reactivity and excellent stereocontrol[31,32].
In recent years, our group has developed a series of chiral N-heterocyclic carbene (NHC) ligands[33-37]. Characterized by their strong σ-donor ability and bulky, flexible steric profiles, these ligands have consistently delivered high catalytic efficiency and outstanding stereoselectivity across a diverse array of asymmetric transformations[38-45]. Motivated by these findings, we hypothesized that an NHC/Cu catalyst system could effectively facilitate asymmetric carbene transfer. Furthermore, we postulated that the deep chiral pocket created by our NHC ligand might address the challenging stereocontrol associated with the quasi-linear NHC/Cu(I)–carbenoid intermediate[46-49]. To the best of our knowledge, enantioselective carbene transfer reactions enabled by chiral NHC/metal catalysis have not been reported[49-53]. Herein, we report a rare example of enantioselective NHC/Cu-catalyzed intermolecular cyclopropanation of indoles with diazo esters (Figure 1c). This protocol provides efficient access to cyclopropane-fused indolines with broad functional group tolerance.
2. Experimental Section
General Considerations: Chemicals were commercially purchased from Adamas-beta, Bide Pharmatech Ltd., Aladdin, and J & K Chemical Co. Ltd., and were used directly without further purification unless otherwise stated. Copper chloride was purchased from Strem Chemicals Inc. and used as received. All the reactions were conducted under a N2 atmosphere using a glovebox or vacuum-line techniques, and the glassware was dried in an oven (140 °C) or flame-dried. 1H NMR, 13C NMR, and 19F NMR spectra were recorded at room temperature on an Agilent 400 MHz or 600MHz, Varian 400 MHz, or Bruker 400 MHz spectrometers and were calibrated using residual solvent as an internal reference (CDCl3: 7.26 ppm for 1H NMR and 77.16 ppm for 13C NMR). Enantiomeric excess was determined by high-performance liquid chromatography (HPLC) analysis using the chiral column described below in detail.
2.1 Typical procedure for asymmetric NHC/Cu catalyzed intermolecular cyclopropanation of indoles with diazo esters
In a nitrogen-filled glovebox, an oven-dried screw-cap reaction tube equipped with a magnetic stir bar was charged with L1-CuCl (8.8 mg, 0.01 mmol, 5 mol%) and KB(C6F5)4 (7.2 mg, 0.01 mmol, 5 mol%), the indole substrate (0.2 mmol, 1.0 equiv.), and dichloromethane (1 mL). The reaction tube was sealed with a screw-cap septum and stirred at -20 °C for 1 hour. The diazo compound (0.4 mmol, 2.0 equiv.) was dissolved in dichloromethane (1 mL) and frozen at -20 °C for 1 hour before being slowly added dropwise to the reaction mixture. The reaction mixture was stirred within the sealed tube at -20 oC for 12 h. After the reaction was complete, the mixture was concentrated under reduced pressure to give the crude product. The diastereomeric ratio (d.r.) value of the crude products was determined by 1H NMR. The crude residue was purified via column chromatography to afford the desired product. The enantiomeric excess (ee) value was determined by chiral HPLC analysis of the isolated product.
3. Results and Discussion
Our investigation began by screening various metal catalysts (Rh, Ag, Pd) with L1 for the reaction of N-protected indole 1 and diazo ester 2; however, these systems failed to promote the desired transformation or yielded racemic product 3 (Table 1, entries 1-3). Gratifyingly, employing ANIPE–CuCl afforded product 3 in 90% yield with 19% ee (entry 4). We observed that the steric bulk of the diazo ester significantly influenced enantioselectivity, with ee values improving as steric demand increased (entries 5-8). Notably, tert-butyl 2-diazo-2-phenylacetate provided an enhanced enantioselectivity of 49% ee (entry 8), a finding we attribute to improved enantioface differentiation of the NHC–copper carbenoid intermediate. Further optimization of the N-protecting group identified N-Piv as optimal, delivering the product in 87% yield and 72% ee (entry 11). While other NHC/Cu complexes showed slightly inferior results, common phosphine and bisoxazoline ligands were ineffective (entries 12-16). Lowering the temperature to -20 °C significantly boosted enantioselectivity but reduced the yield to 57% (entry 17), likely due to competitive diazo dimerization. Increasing the diazo ester loading to 2.0 equivalents restored the yield to 89% (entry 18). Finally, replacing NaBArF with KB(C6F5)4 marginally improved both yield and enantioselectivity (entry 19; see Tables S1-S6 for details).
Table 1. Optimization of the reaction conditions.
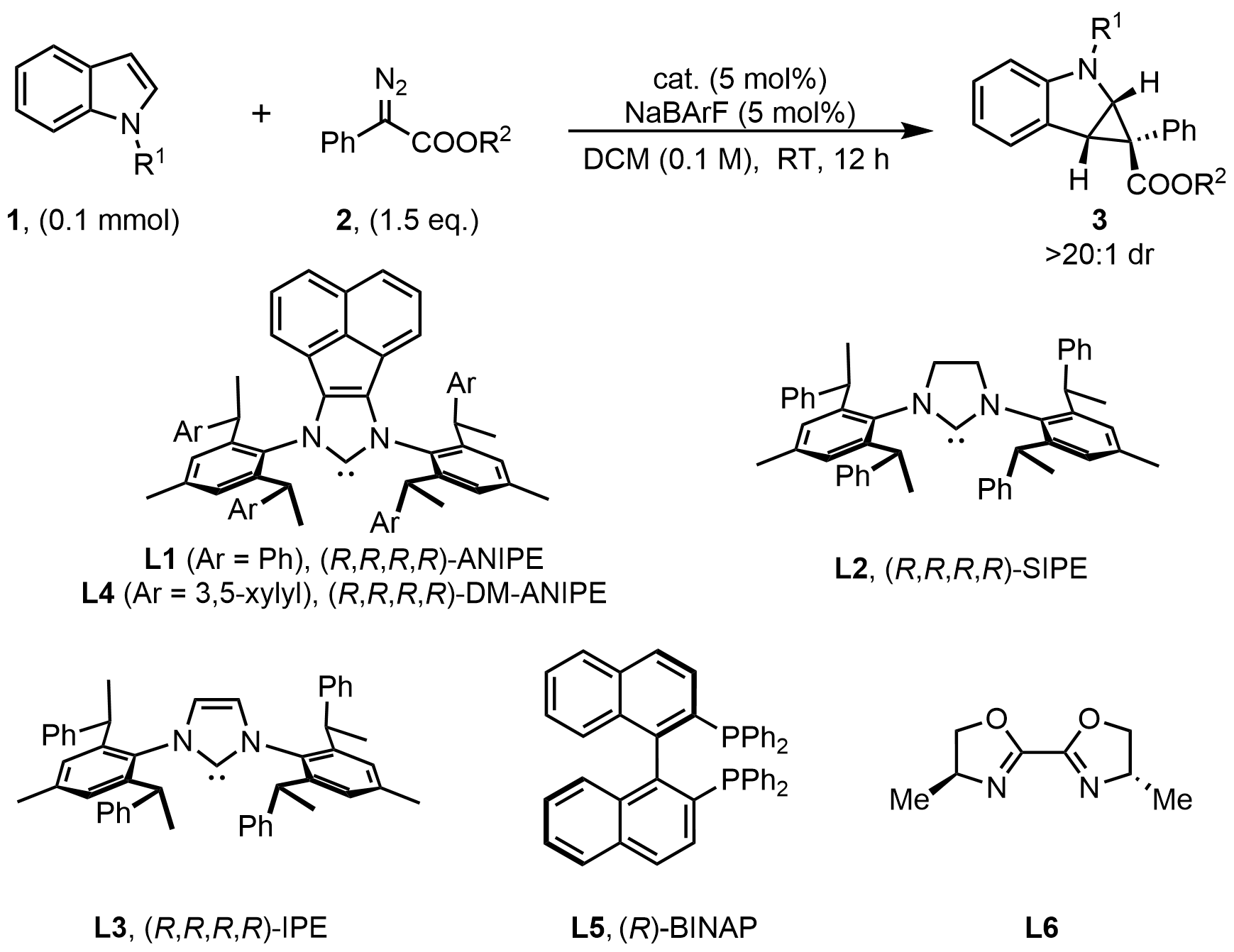
| Entry | R1 | R2 | cat. | Yield (%)a | ee (%)a |
| 1 | Boc | Et | L1/HCl + Rh2(OAc)4 | N.D. | N.D. |
| 2 | Boc | Et | L1-AgCl | N.D. | N.D. |
| 3 | Boc | Et | L1-Pd(cin)Cl | 82 | 0 |
| 4 | Boc | Et | L1-CuCl | 90 | 19 |
| 5 | Boc | Me | L1-CuCl | 99 | 14 |
| 6 | Boc | Bn | L1-CuCl | 99 | 22 |
| 7 | Boc | iPr | L1-CuCl | 99 | 33 |
| 8 | Boc | tBu | L1-CuCl | 95 | 49 |
| 9 | Ts | tBu | L1-CuCl | 32 | 69 |
| 10 | Ac | tBu | L1-CuCl | 39 | 61 |
| 11 | Piv | tBu | L1-CuCl | 87 | 72 |
| 12 | Piv | tBu | L2-CuCl | 82 | 56 |
| 13 | Piv | tBu | L3-CuCl | 84 | 41 |
| 14 | Piv | tBu | L4-CuCl | 94 | 37 |
| 15 | Piv | tBu | L5-CuCl | N.D | N.D. |
| 16 | Piv | tBu | L6-CuCl | N.D | N.D. |
| 17b | Piv | tBu | L1-CuCl | 57 | 85 |
| 18b,c | Piv | tBu | L1-CuCl | 86 | 85 |
| 19b,c,d | Piv | tBu | L1-CuCl | 89 | 89 |
Reaction condition: 1 (0.1 mmol), 2 (0.15 mmol), catalyst (5 mol%), NaBArF (5 mol%) and DCM (1.0 mL) under N2 atmosphere, 12 h. a: The yield and d.r. were determined by 1H NMR spectroscopy of crude sample with C2H2Cl4 as an internal standard; b: -20 ℃; c: Using 2.0 equiv. diazo ester; d: Using KB(C6F5)4 instead of NaBArF. DCM: dichloromethane; NMR: nuclear magnetic resonance.
With the optimized conditions in hand, we first examined the structural variation of the diazo esters. As presented in Figure 2, this cyclopropanation reaction proceeded smoothly utilizing a variety of (hetero)aromatic diazo esters, providing the corresponding products in good to excellent yields and good enantioselectivities (75-90% ee). Common functional groups, including fluoride (3d), chlorides (3e, 3f, 3g), bromide (3h), iodide (3i), ethers (3j-3l, 3n-3o), trifluoromethyl (3m) and ester (3p), were well tolerated. Heterocyclic substrates such as those derived from thiophene (3r) and benzodioxole (3l) proved compatible, providing fused indolines in synthetically useful yields. Additionally, the diazo ester derived from isoxazole-4-carboxylic acid afforded product 3s in moderate yield and enantioselectivity (58%, 71% ee). We next explored the scope of the indole component. The reaction proceeded in good yields (65-86%) and high enantioselectivity (82-88% ee), accommodating various electron-donating (e.g., methoxy, benzyloxy, silyloxy, piperonyl; 4a-d) and electron-withdrawing groups (e.g., fluorides (3v, 3w), chloride (3x), and bromides (3y, 3z)). Notably, the substitution position on the aromatic ring had no significant impact on enantioselectivity. The absolute configuration of the product was unambiguously confirmed by X-ray diffraction analysis of a single crystal of 3a (CCDC 2429515).
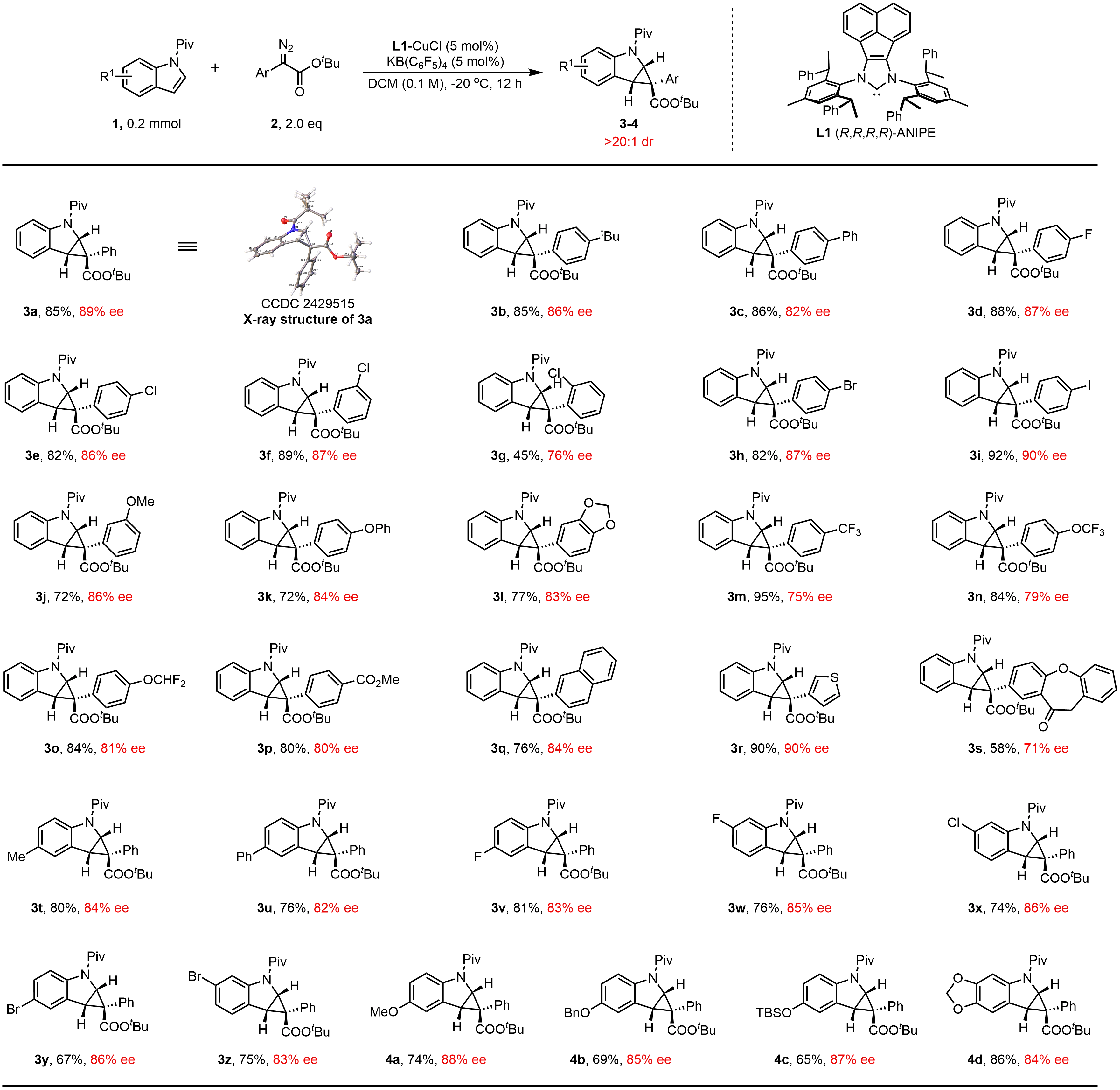
Figure 2. Substrate scopea. a: Standard conditions unless otherwise noted: 1 (0.2 mmol), 2 (0.4 mmol), L1-CuCl (0.01 mmol), KB(C6F5)4 (0.01 mmol), DCM (1mL), -20 °C, 12 h. Isolated yields are reported. Ee and dr were determined with HPLC and NMR analysis, respectively. DCM: dichloromethane; NMR: nuclear magnetic resonance; HPLC: high-performance liquid chromatography.
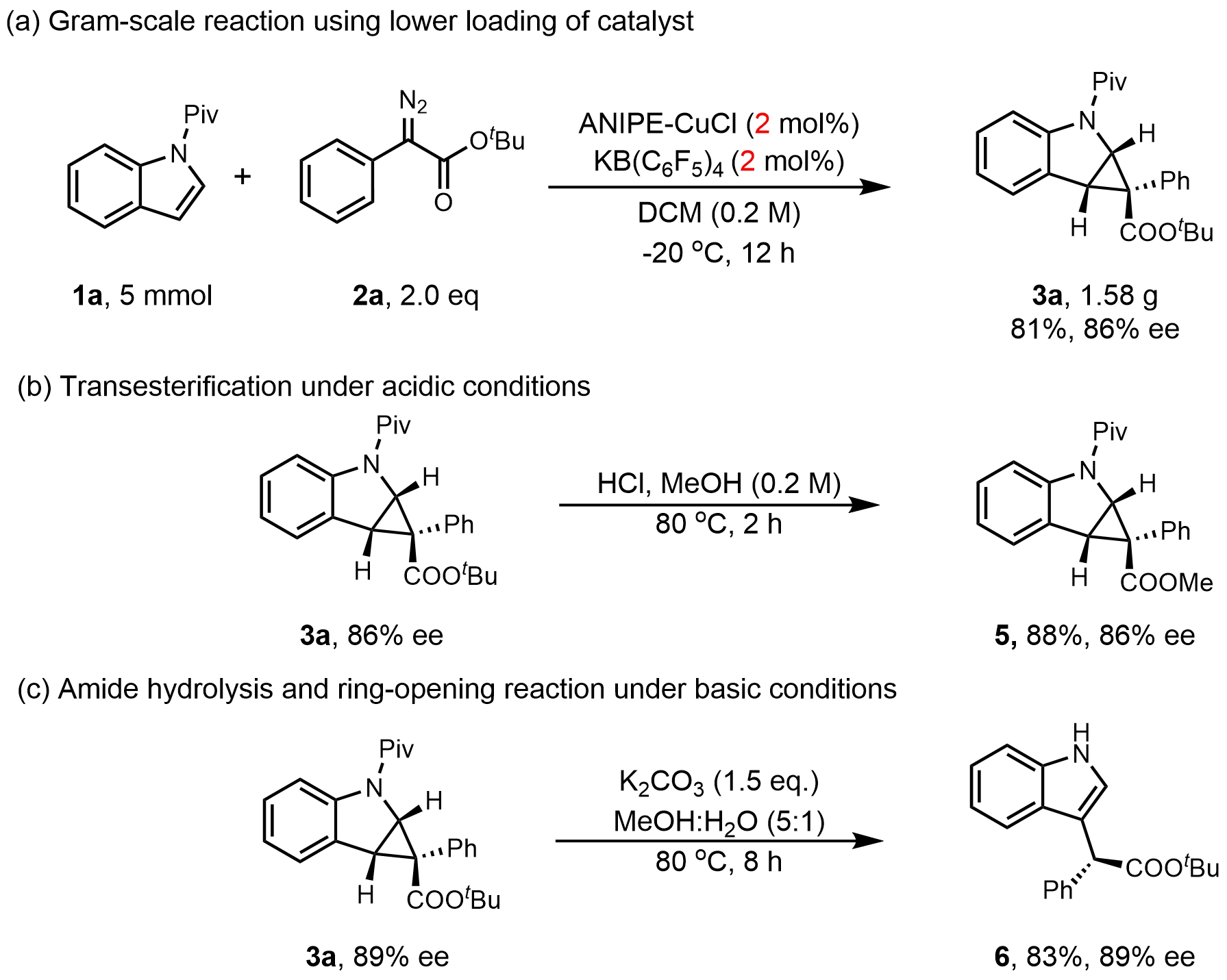
The operational robustness of this transformation was demonstrated on a gram-scale reaction. Notably, performing the reaction with 5 mmol of N-Piv indole 1a and 10 mmol of diazo ester 2a in dichloromethane, using just 2 mol% catalyst loading, afforded product 3a in 81% yield and 86% ee (Figure 3a). Further synthetic utility was illustrated by the transformation of 3a. Under acidic catalysis, transesterification of the ester moiety proceeded smoothly to furnish methyl ester 5 with complete retention of enantioselectivity (Figure 3b). Conversely, treatment with base promoted amide hydrolysis and ring-opening, yielding the C3-substituted indole derivative 6 while preserving stereochemical integrity (Figure 3c).
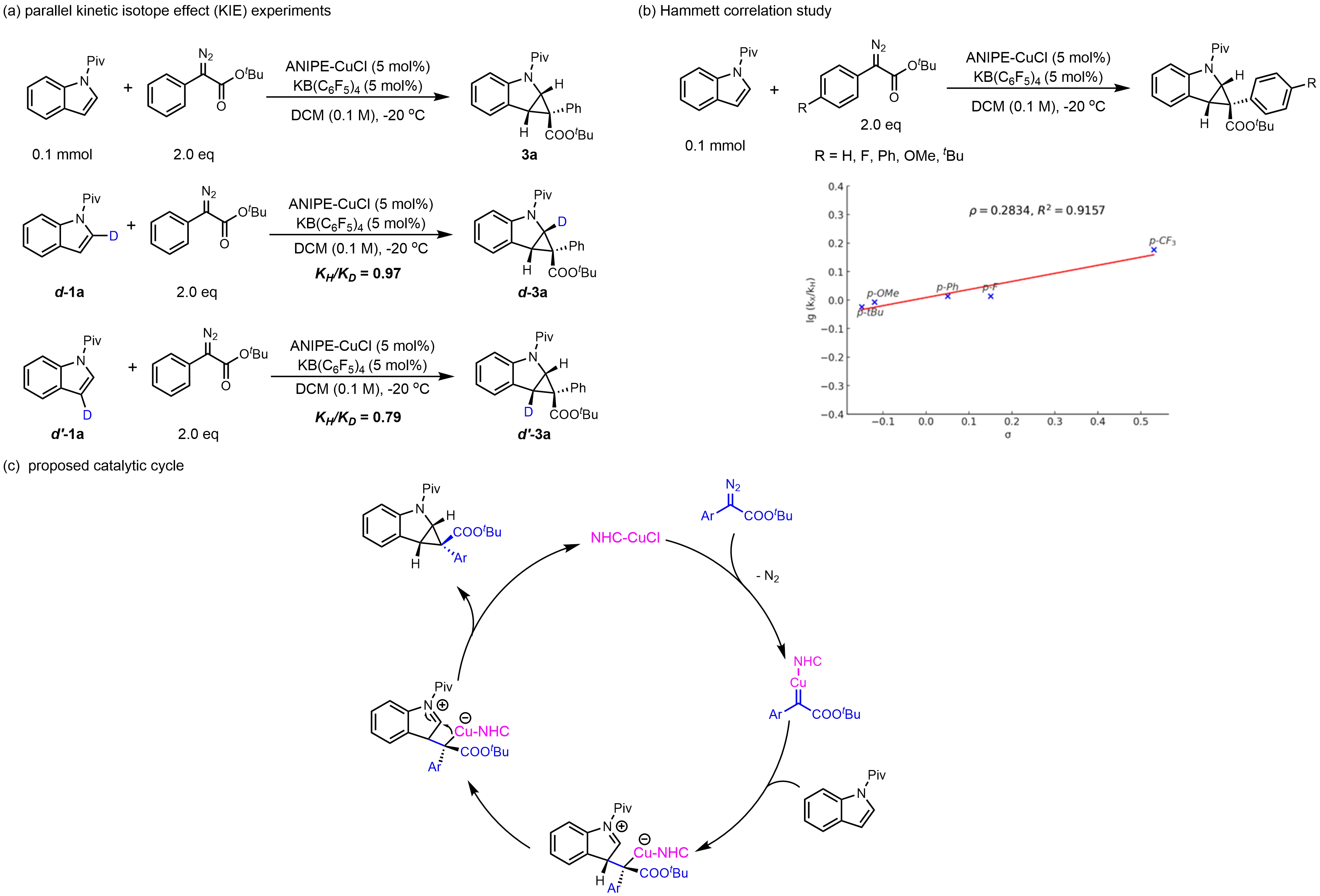
To gain insight into the reaction mechanism, a series of control experiments was conducted. Parallel kinetic isotope effect (KIE) studies using deuterated indoles revealed KIE values of 0.97 at the C2-position and 0.79 at the C3-position (Figure 4a). These observations suggest the presence of a secondary kinetic isotope effect, consistent with a change in hybridization from sp2 to sp3 at these positions during the reaction[54]. Subsequent Hammett analysis using para-substituted diazo esters established a linear free-energy relationship with a positive slope (ρ = 0.283, Figure 4b), indicating that the rate-determining step involves a transition state with partial negative charge accumulation[55]. Based on these experimental results and previous reports[22,56], a plausible catalytic cycle is proposed (Figure 4c). Initially, the NHC–Cu complex reacts with the diazo compound to extrude N2, generating a metal carbenoid intermediate. This is followed by nucleophilic attack of the indole at the carbene center to form a zwitterionic intermediate, which undergoes intramolecular cyclization to furnish the desired product. The use of our bulky yet flexible chiral NHC ligand is crucial for achieving effective control over both enantioselectivity and diastereoselectivity.
4. Conclusion
In summary, we have developed a highly efficient NHC/Cu-catalyzed asymmetric dearomative cyclopropanation of indoles using diazo esters as carbene precursors. This method provides straightforward access to structurally diverse cyclopropane-fused indolines bearing quaternary stereogenic centers in good yields with high levels of enantio- and diastereocontrol. The use of bulky chiral NHC ligands was found to be crucial for stabilizing the copper–carbenoid intermediate and enabling effective stereoinduction. Featuring broad substrate scope, excellent functional group tolerance, and scalability, this transformation highlights its practical utility in synthetic chemistry. Mechanistic investigations suggest a pathway involving nucleophilic attack of the indole on a copper–carbenoid species followed by intramolecular cyclization. This study demonstrates the substantial potential of chiral NHC ligands in metal-catalyzed asymmetric carbene-transfer chemistry.
Supplementary materials
The supplementary material for this article is available at: Supplementary materials.
Authors contribution
Shi SL, You SL: Conceptualization, supervision, writing-review & editing.
Ruan LX: Writing-original draft, writing-review & editing.
Liu S: Investigation, methodology.
Lu J: Investigation, methodology, writing-original draft.
Conflicts of interest
Shu-Li You is an Editorial Board Member of Chiral Chemistry. The other authors declare no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent to publication
Not applicable.
Availability of data and materials
The data reported in this paper are available in the main text or supplementary information, including methods, NMR data, HRMS data, HPLC spectra Crystal data and NMR spectra. Crystallographic data for the structures reported in this article have been deposited at the Cambridge Crystallographic Data Centre, under deposition numbers CCDC 2429515 (3a).
Funding
This work was financially supported by the National Natural Science Foundation of China (22325110 and 22501291), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB0610000), and the Shanghai Science and Technology Committee (25ZR1402566).
Copyright
© The Author(s) 2026.
References
-
1. López Ortiz F, Iglesias MJ, Fernández I, Andújar Sánchez CM, Ruiz Gómez G. Nucleophilic dearomatizing (DNAr) reactions of aromatic C,H-systems. a mature paradigm in organic synthesis. Chem Rev. 2007;107(5):1580-1691.[DOI]
-
2. Cheng YZ, Li MZ, Wang RX, Zhu LH, Shen WJ, Zou XX, et al. The development and perspective of dearomatization reaction. Prog Chem. 2024;36(12):1785-1829. Chinese.[DOI]
-
4. Zhuo CX, Zhang W, You SL. Catalytic asymmetric dearomatization reactions. Angew Chem Int Ed. 2012;51(51):12662-12686.[DOI]
-
5. Schinnerl M, Böhm C, Seitz M, Reiser O. New bis(oxazoline) ligands with secondary binding sites for the asymmetric cyclopropanation of furans. Tetrahedron Asymmetry. 2003;14(7):765-771.[DOI]
-
7. Pilsl LKA, Ertl T, Reiser O. Enantioselective three-step synthesis of homo-β-proline: A donor–acceptor cyclopropane as key intermediate. Org Lett. 2017;19(10):2754-2757.[DOI]
-
8. Fu J, Wurzer N, Lehner V, Reiser O, Davies HML. Rh(II)-catalyzed monocyclopropanation of pyrroles and its application to the synthesis pharmaceutically relevant compounds. Org Lett. 2019;21(15):6102-6106.[DOI]
-
11. Reisenbauer JC, Green O, Franchino A, Finkelstein P, Morandi B. Late-stage diversification of indole skeletons through nitrogen atom insertion. Science. 2022;377(6610):1104-1109.[DOI]
-
14. Zhu M, Zhang X, Zheng C, You SL. Energy-transfer-enabled dearomative cycloaddition reactions of indoles/pyrroles via excited-state aromatics. Acc Chem Res. 2022;55(17):2510-2525.[DOI]
-
16. Li ML, Yu JH, Li YH, Zhu SF, Zhou QL. Highly enantioselective carbene insertion into N–H bonds of aliphatic amines. Science. 2019;366(6468):990-994.[DOI]
-
17. Li YP, Li ZQ, Zhu SF. Recent advances in transition-metal-catalyzed asymmetric reactions of diazo compounds with electron-rich (hetero-) arenes. Tetrahedron Lett. 2018;59(24):2307-2316.[DOI]
-
20. Xu W, Xu MH. Rhodium(I)/chiral diene-catalyzed asymmetric carbene transformations. Acc Chem Res. 2026;59(1):179-194.[DOI]
-
21. Xia Y, Qiu D, Wang J. Transition-metal-catalyzed cross-couplings through carbene migratory insertion. Chem Rev. 2017;117(23):13810-13889.[DOI]
-
24. Chifotides HT, Dunbar KR. Rhodium compounds. In: Cotton FA, Murillo CA, Walton RA, editors. Multiple bonds between metal atoms. New York: Springer; 2006. p. 465-589.[DOI]
-
26. Pirovano V, Brambilla E, Tseberlidis G. Copper(I)(pyridine-containing ligand)] catalyzed regio- and steroselective synthesis of 2-vinylcyclopropa[b]indolines from 2-vinylindoles. Org Lett. 2018;20(2):405-408.[DOI]
-
29. Cai Y, Zhu SF, Wang GP, Zhou QL. Iron-catalyzed C–H fuctionalization of indoles. Adv Synth Catal. 2011;353(16):2939-2944.[DOI]
-
32. Brenna S, Ardizzoia GA. Carbene transfer and carbene insertion reactions catalyzed by a mixed-ligand copper(I) complex. Eur J Org Chem. 2018;2018(25):3336-3342.[DOI]
-
35. Wang ZC, Shi SL. Induced-fit chiral N-heterocyclic carbene ligands for asymmetric catalysis. Acc Chem Res. 2025;58(13):2157-2177.[DOI]
-
36. Jiang B, Shi SL. A chiral bifunctional NHC ligand promoted Ni/Al-catalyzed regio- and enantioselective C6–H alkylation of pyrimidines. ACS Catal. 2025;15(21):18824-18833.[DOI]
-
37. Zhang JW, Wu H, Shen D, Wu RK, Wang ZC, Hong X, et al. Enantioconvergent negishi cross-coupling of racemicsec-alkylzinc reagent with aryl halides enabled by BulkyN-heterocyclic carbene-Pd catalyst. CCS Chem. 2026;8(2):754-763.[DOI]
-
39. Liu X, Shi S. ANIPE-ligand-enabled copper-catalyzed asymmetric carboboronation of allenes with imines and diborons. Chin J Org Chem. 2024;44(6):1884-1896. Chinese.[DOI]
-
41. Sun B, Ruan LX, Zhao R, Zhang J, Niu R, Luo Q, et al. Dynamic kinetic asymmetric allylation, propargylation and crotylation of ketones using copper catalysis. Nat Synth. 2024;3(9):1091-1103.[DOI]
-
42. Chen G, Liu JM, Ruan LX, Shi SL. Selective dynamic kinetic asymmetric aldehyde–alkyne reductive coupling. Nat Synth. 2025;4(12):1630-1639.[DOI]
-
43. Wang ZC, Luo X, Zhang JW, Liu CF, Koh MJ, Shi SL. Enantioselective C–C cross-coupling of unactivated alkenes. Nat Catal. 2023;6(11):1087-1097.[DOI]
-
46. Ye X, Sun B, Shi SL. Contra-electronegativity transmetallation unlocks alkene carbomagnesiation to access quaternary stereocentres. Nat Chem. 2026.[DOI]
-
48. Raubenheimer HG, Cronje S, Olivier PJ. Synthesis and characterization of mono(carbene) complexes of copper and crystal structure of a linear thiazolinylidene compound. J Chem Soc Dalton Trans. 1995;2:313.[DOI]
-
49. Trose M, Nahra F, Cazin CSJ. Dinuclear N-heterocyclic carbene copper(I) complexes. Coord Chem Rev. 2018;355:380-403.[DOI]
-
50. Hölzel T, Belyaev A, Terzi M, Stenzel L, Gernert M, Marian CM, et al. Linear carbene pyridine copper complexes with sterically demanding N, N′-bis(trityl)imidazolylidene: Syntheses, molecular structures, and photophysical properties. Inorg Chem. 2021;60(23):18529-18543.[DOI]
-
52. Pérez PJ, Díaz-Requejo MM, Rivilla I. Gold-catalyzed naphthalene functionalization. Beilstein J Org Chem. 2011;7:653-657.[DOI]
-
53. Kastrati A, Jaquier V, Garbo M, Besnard C, Mazet C. Pd-catalyzed regioselective cyclopropanation of 2-substituted 1, 3-dienes. ACS Org Inorg Au. 2023;3(5):291-298.[DOI]
-
54. Rasmussen T, Jensen JF, Østergaard N, Tanner D, Ziegler T, Norrby PO. On the mechanism of the copper-catalyzed cyclopropanation reaction. Chem Eur J. 2002;8(1):177-184.[DOI]
-
55. Hansch C, Leo A, Taft RW. A survey of Hammett substituent constants and resonance and field parameters. Chem Rev. 1991;91(2):165-195.[DOI]
Copyright
© The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Publisher’s Note
Science Exploration remains a neutral stance on jurisdictional claims in published
maps
and institutional affiliations. The views expressed in this article are solely those
of
the author(s) and do not reflect the opinions of the Editors or the publisher.
Share And Cite



Science Exploration Style
Lu J, Liu S, Ruan LX, You SL, Shi SL. Enantio- and diastereoselective NHC/Cu-catalyzed intermolecular dearomative cyclopropanation of indoles with diazo esters. Chiral Chem. 2026;2:202608. https://doi.org/10.70401/cc.2026.0017
Tips
Copy completed.
Submit a Manuscript
Author Instructions
Cite this Article
Article Metrics
0
View
0
Download
Cited
Article Updates

Science Exploration Style
Lu J, Liu S, Ruan LX, You SL, Shi SL. Enantio- and diastereoselective NHC/Cu-catalyzed intermolecular dearomative cyclopropanation of indoles with diazo esters. Chiral Chem. 2026;2:202608. https://doi.org/10.70401/cc.2026.0017
copy
Share Link
copy