Catalytic enantioselective construction of saturated (hetero)cycles via radical cross-coupling
Xinlong Luo
#

,
Gan He
#
,
Xiao Lin
#
,
Kunyao Huang
Zhen Xu
Haohua Huo
*
*Correspondence to:
Haohua Huo, State Key Laboratory of Physical Chemistry of Solid Surfaces, Key Laboratory of Chemical Biology of Fujian Province, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, Fujian, China.
E-mail: hhuo@xmu.edu.cn
Chiral Chem. 2026;2:202614. 10.70401/cc.2026.0025
Received: March 13, 2026Accepted: May 06, 2026Published: May 07, 2026
This manuscript is made available in its unedited form to allow early access to the
reported findings. Further editing will be completed before final publication. As
such,
the content may include errors, and standard legal disclaimers are applicable.
Abstract
Chiral saturated (hetero)cyclic scaffolds constitute privileged structural motifs in natural products, pharmaceuticals, and bioactive molecules, where their three-dimensional architectures often confer superior physicochemical and pharmacological properties compared to planar aromatic systems. Despite their importance, general and modular strategies for the enantioselective synthesis of these C(sp3)-rich frameworks remain limited. In recent years, enantioselective radical cross-coupling (RCC) has emerged as a transformative strategy that addresses these limitations by enabling the direct coupling of abundant radical precursors, including alkenes, carboxylic acids, alkyl halides, and C(sp3)–H bonds, in a largely functional-group-agnostic manner. By leveraging transition-metal catalysis with chiral ligands, these methods achieve exquisite stereocontrol while providing modular access to structurally diverse saturated (hetero)cycles under mild conditions. This review comprehensively surveys recent advances in the catalytic enantioselective construction of saturated (hetero)cycles via RCC, organized into three major strategic classes: enantioconvergent cross-coupling of racemic substrates, enantioselective hydrofunctionalization of (hetero)cyclic alkenes, and direct enantioselective C(sp3)–H RCC. We critically analyze synthetic achievements, mechanistic insights, and substrate scope encompassing diverse ring sizes (from three-membered to bridged polycyclic systems) and heteroatom incorporation (N, O, S, P, Si, Ge), and discuss persistent challenges including macrocycle synthesis, C(sp3)–C(sp3) bond formation, multicomponent couplings, catalyst diversification beyond nickel and cobalt, expansion of C(sp3)–H functionalization scope, and mechanistic elucidation. This review provides a comprehensive understanding of the current state of the field and highlights promising directions for future development in accessing enantioenriched (hetero)cyclic architectures with broad applications in pharmaceutical discovery, materials science, and chemical biology.
Keywords
Saturated (hetero)cycles, enantioselective synthesis, radical cross-coupling
1. Introduction
Chiral saturated (hetero)cyclic skeletons represent one of the most prevalent structural motifs in natural products, pharmaceutical agents, and biologically active molecules. The strategic introduction of saturation into pharmaceutical candidates often leads to improvements in a variety of medicinal properties[1,2]. Saturated heterocyclic systems, in particular, exhibit enhanced target binding affinity, solubility, metabolic stability, and specificity, frequently correlating with improved clinical success rates compared to their planar aromatic counterparts[3]. Despite their desirability, modular access to these C(sp3)-rich molecular architectures, especially through enantioselective synthesis, remains underdeveloped. Stereochemistry plays a defining role in the chemical, physical, and biological properties of chiral molecules. Specific enantiomers and/or diastereomers frequently exhibit distinct interactions with chiral biological receptors (e.g., enzymes and proteins), leading to critical differences in pharmacokinetics, pharmacodynamics, and therapeutic efficacy[4-6]. Traditional approaches to chiral saturated (hetero)cycles typically rely on polar bond disconnections, including asymmetric dearomative hydrogenation[7-10], classic cycloaddition reactions[11-14], and intramolecular cyclization[15] strategies. However, these methods often depend on substrate-specific aromatic precursors or require de novo synthesis of bespoke cyclization substrates, thereby limiting access to diverse chemical space. Consequently, the development of general and modular strategies for the enantioselective construction of saturated (hetero)cycles from simple and widely available starting materials is of significant interest and synthetic utility.
Recent advances in radical cross-coupling (RCC)[16-18] reactions have provided a transformative solution to address this gap, offering great promise for extending fragment-coupling logic to the direct coupling of native functionalities. Unlike polar bond disconnection strategies, RCC mechanisms are largely functional-group-agnostic, allowing, in principle, for the coupling of abundant radical precursors, such as alkenes, carboxylic acids, alkyl halides, aldehydes, and even ubiquitous C(sp3)–H bonds, in virtually any desired combination to generate complex structures from simple feedstock chemicals. This approach greatly expands access to C(sp3)-rich chemical space and enables the formation of otherwise elusive saturated (hetero)cycles. Moreover, by leveraging the synthetic versatility of transition-metal catalysis, modular regulation of the coordination environment around the metal center via chiral ligands facilitates radical metalation and subsequent enantioselective cross-coupling, thereby enabling exquisite stereochemical control over the coupling process. As a result, enantioselective RCC has emerged as a powerful and increasingly versatile strategy for the construction of enantioenriched saturated (hetero)cycles.
This review comprehensively summarizes and discusses significant advances in the enantioselective construction of saturated (hetero)cycles via catalytic RCC, with a focus on synthetic achievements, mechanistic insights, unresolved challenges, and future opportunities. Through this analysis, we aim to provide a thorough understanding of the current state of the field and identify promising directions for future research and development in accessing enantioenriched (hetero)cycles. We categorize the methodological explorations into three main classes based on distinct reaction paradigms (Figure 1): (1) enantioconvergent
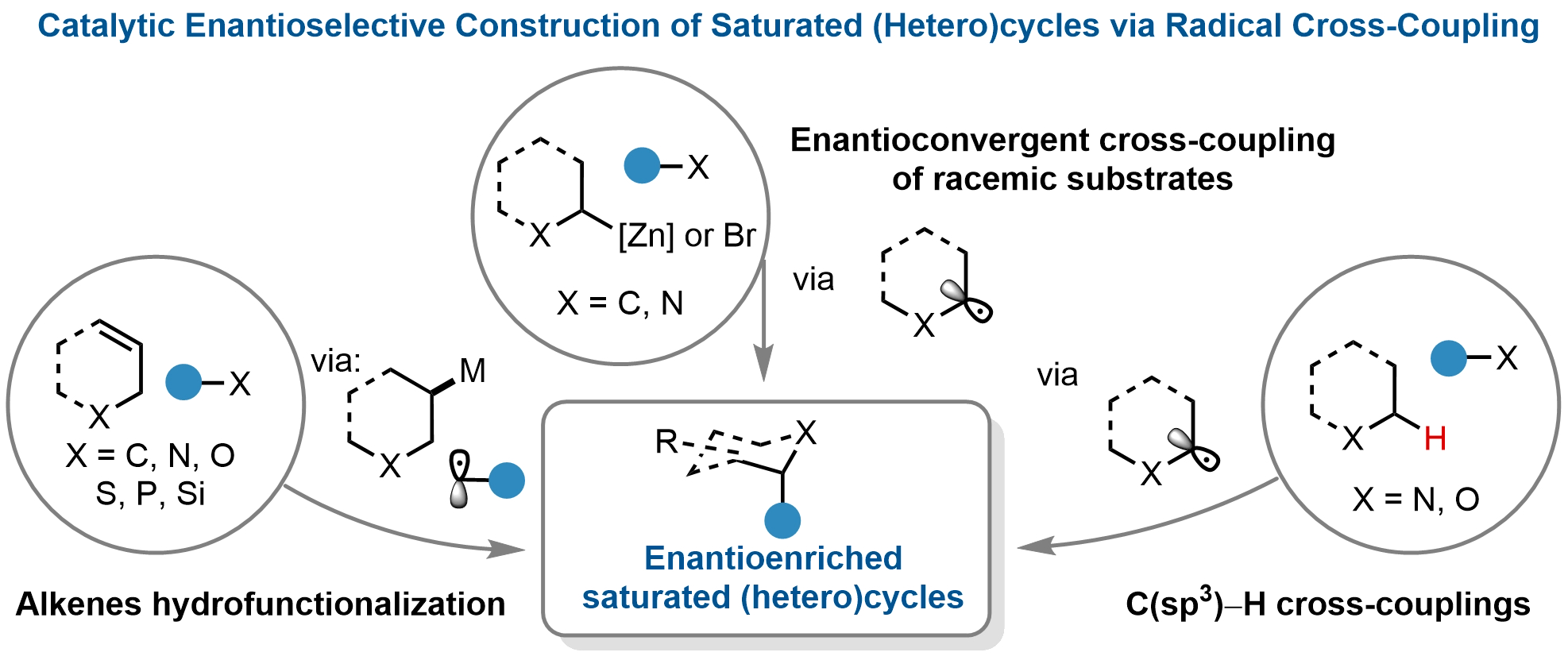
Figure 1. Catalytic enantioselective construction of saturated (hetero)cycles via radical cross-coupling.
2. Catalytic Enantioselective Construction of Saturated (Hetero)cycles via RCC
2.1 Enantioconvergent cross-coupling of racemic nucleophiles or electrophiles
In 2013, the Fu group reported a groundbreaking enantioconvergent Negishi cross-coupling that represents the first example of coupling a racemic alkyl nucleophile with an alkyl electrophile to generate enantioenriched products (Figure 2)[50]. Specifically, racemic α-zincated N-Boc-pyrrolidine, prepared in situ from commercially available N-Boc-pyrrolidine, undergoes cross-coupling with unactivated secondary alkyl halides in the presence of a chiral 1,2-diamine/nickel catalyst to afford 2-alkylpyrrolidines with high enantioselectivity (up to 94% ee) and good yields (up to 96%).
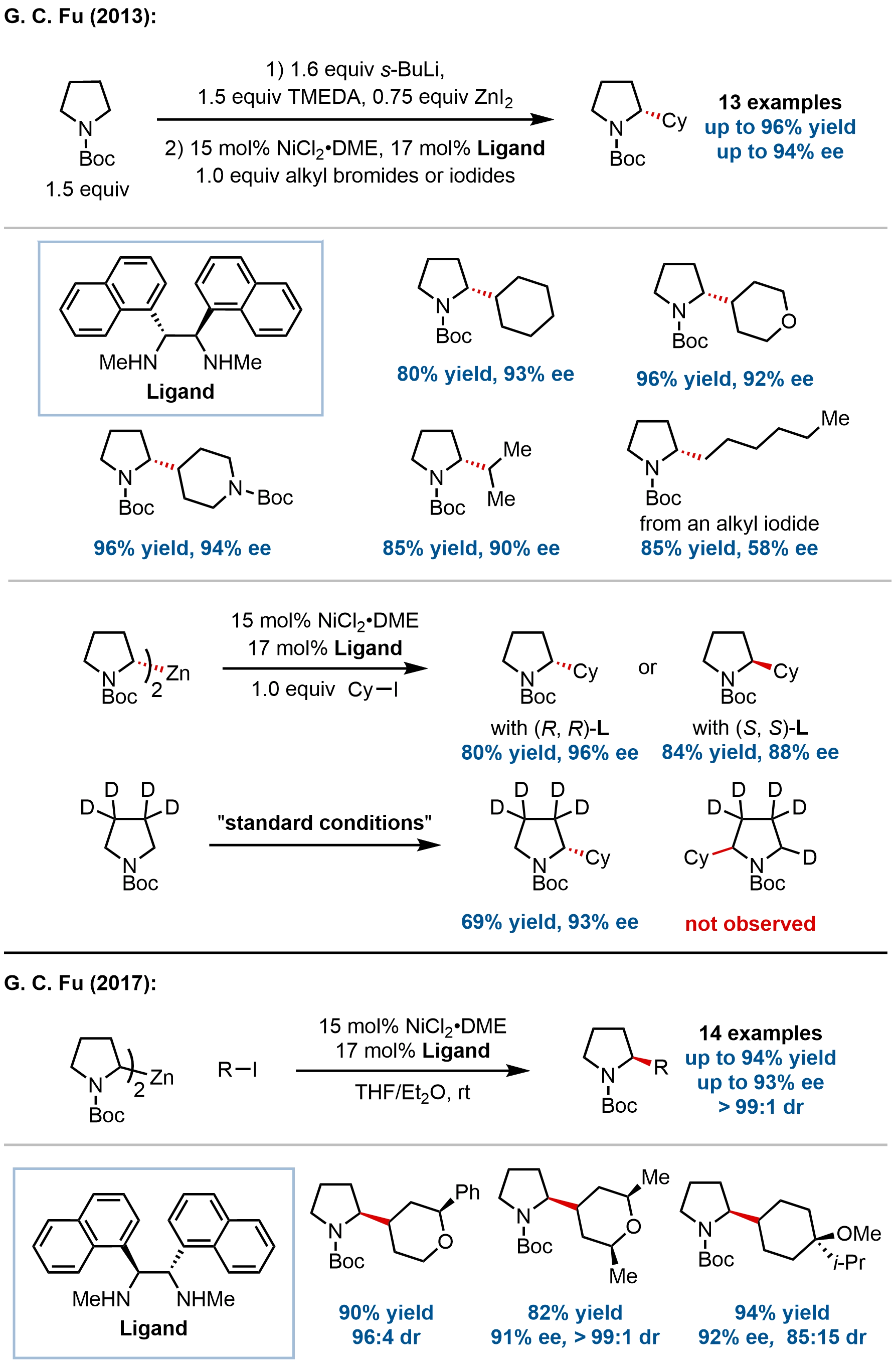
Figure 2. Enantioconvergent cross-coupling of racemic α-zincated N-Boc-pyrrolidine for the synthesis of enantioenriched α-alkylated N-Boc-pyrrolidines.
This method provides a one-pot, catalytic asymmetric route to an important class of nitrogen-containing heterocycles. The substrate scope encompasses various cyclic secondary alkyl iodides, heterocyclic electrophiles, and acyclic secondary alkyl halides, with particularly high enantioselectivities observed for secondary electrophiles. Both alkyl iodides and bromides serve as competent coupling partners, though bromides generally require elevated temperatures and provide more modest yields. In contrast, primary alkyl iodides and bromides provided products with moderate enantioselectivity (58% ee).
Mechanistic investigations revealed several key insights. First, the α-zincated N-Boc-pyrrolidine is configurationally stable at room temperature in the absence of a nickel catalyst. Second, cross-coupling experiments using an enantioenriched organozinc reagent
Building upon their earlier work, Fu and coworkers extended the enantioconvergent Negishi cross-coupling to achieve control of vicinal stereocenters, representing the first doubly stereoconvergent alkyl–alkyl bond formation[51]. Using a chiral 1,2-diamine/nickel catalyst, racemic α-zincated N-Boc-pyrrolidine couples with cyclic alkyl iodides (as mixtures of stereoisomers) to afford α-alkylated pyrrolidines with excellent stereoselectivity.
The substrate scope includes 4-monosubstituted cyclohexyl iodides with diverse substituents, providing products with high enantioselectivity and diastereoselectivity. The method also accommodates 4,4-disubstituted and 3,5-disubstituted cyclohexyl iodides, as well as oxygen heterocycles. Chiral electrophiles, including 3-substituted carbocyclic and heterocyclic iodides, react with high stereoselectivity to form cis-1,3-disubstituted products, while 3,3-disubstituted and bicyclic/polycyclic secondary alkyl iodides also couple with good stereoselectivity.
Mechanistic studies revealed that the electrophile undergoes isomerization during the reaction, with both diastereomers converging to the same product. Control experiments excluded SN2-type halogen exchange, suggesting stereoconvergence occurs through reversible iodine transfer between the alkyl radical and nickel. The major product consistently bears the new substituent in the equatorial position, potentially due to kinetic preference during radical capture or reversible nickel–carbon bond formation. The absolute configuration at the pyrrolidine α-position is controlled by the chiral catalyst, while the electrophile-derived stereocenter is substrate-controlled. Conformationally flexible secondary alkyl electrophiles showed modest stereoselectivity under standard conditions.
In addition to enantioconvergent cross-coupling of racemic nucleophiles, Liu and coworkers recently developed an orthogonal approach employing racemic cyclopropyl electrophiles with achiral terminal alkynes to access enantioenriched cyclopropanes, a transformation that remained underdeveloped due to severe chemoselectivity challenges posed by highly reactive cyclopropyl radicals (Figure 3)[52]. This fundamental challenge in enantioconvergent RCC arises from diminished chemoselectivity caused by side reactions of highly reactive alkyl radicals with closed-shell reactants. The issue is particularly acute for cyclopropyl radicals, which are more reactive than many unstrained alkyl radicals (C–H bond dissociation energy: PhC((CH2)2)–H, 93 kcal/mol;
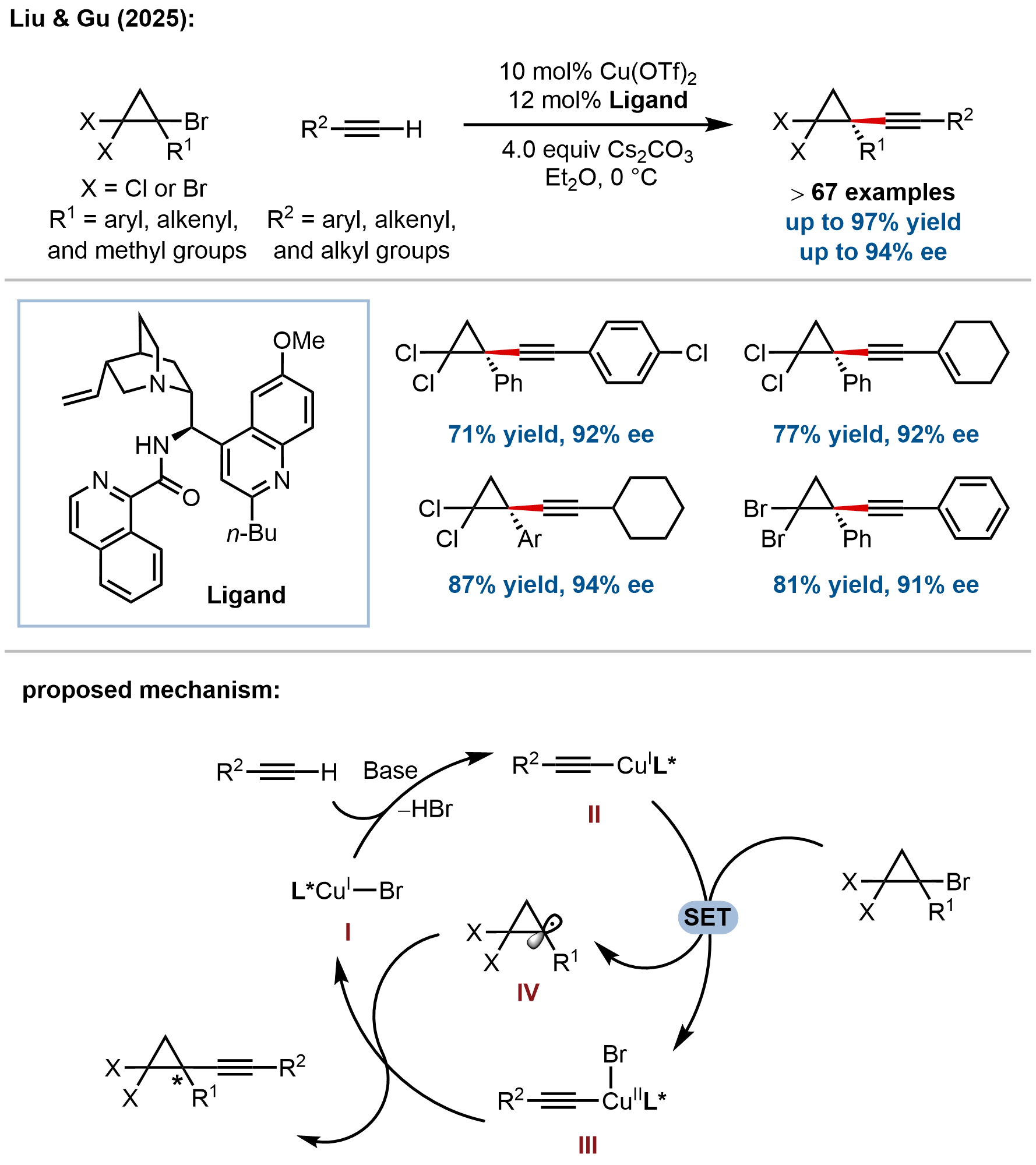
Figure 3. Enantioconvergent cross-coupling of cyclopropyl halides with terminal alkynes enabling modular access to enantioenriched cyclopropanes.
The proposed mechanism involves single-electron transfer (SET) between in situ-generated copper(I) acetylide complex II and racemic cyclopropyl halides to form Cu(II) acetylide III and cyclopropyl radical IV, followed by enantioselective C–C bond coupling. The key innovation lies in the use of Cu(II) salts with hard ligands/counterions [Cu(OTf)2] in combination with hard chiral
Very recently, Fu and coworkers extended this strategy to unactivated racemic cyclic electrophiles, reporting a nickel-catalyzed asymmetric arylation of racemic 3-iodopyrrolidines with arylzinc reagents to afford enantioenriched 3-arylpyrrolidines[53]. This method represents the first example of an enantioconvergent substitution of a cyclic alkyl electrophile lacking any stabilizing substituent, and it enables the streamlined synthesis of enantioenriched pyrrolidine derivatives.
2.2 Enantioselective hydrofunctionalization of (hetero)cyclic alkenes
Complementing enantioconvergent cross-coupling strategies, Rong and coworkers recently developed a catalyst-tuned approach for the divergent synthesis of chiral C2- and C3-alkylated pyrrolidines through desymmetrization of readily available 3-pyrrolines
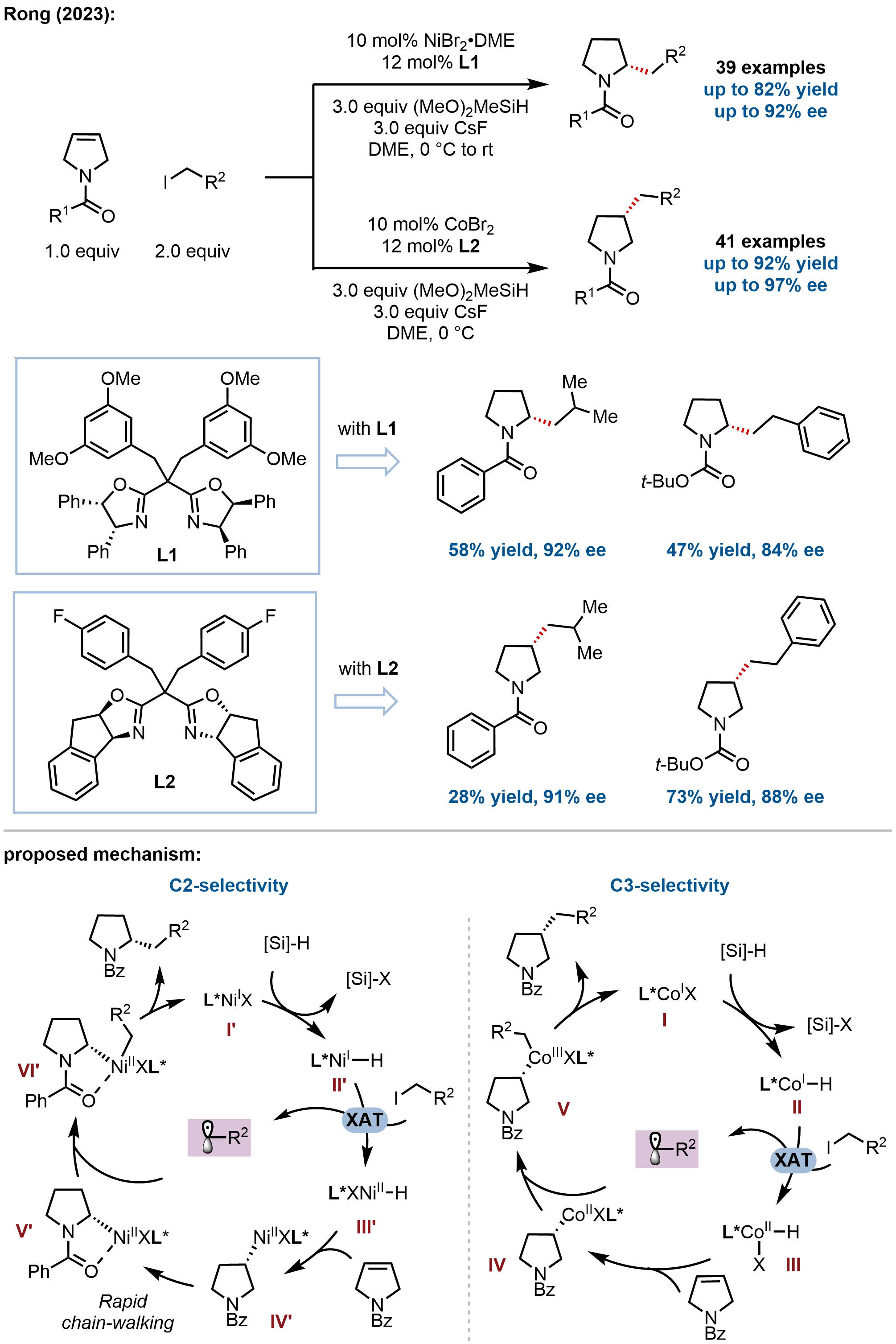
Figure 4. Divergent access to enantioenriched C2- and C3-alkylated pyrrolidines via alkene hydroalkylation.
Mechanistic studies revealed that the catalyst plays a decisive role in dictating the regioselectivity. In the cobalt-catalyzed system, the Co(I) catalyst reacts with silane and base to form a cobalt hydride Co(I)–H species. Subsequent halogen-atom abstraction (XAT) with the alkyl iodide generates a Co(II)–H species and an alkyl radical. The Co(II)–H species undergoes migratory insertion into the carbon–carbon double bond, affording an alkyl-Co(II) complex. This intermediate then captures the previously generated alkyl radical to yield a Co(III) intermediate, which finally undergoes reductive elimination to deliver the C3-selective hydroalkylation product and regenerate the Co(I) catalyst. In contrast, in the nickel-catalyzed system, the coordinated Ni(I)X precursor reacts with silane and base to form a Ni(I)–H species. After XAT with the alkyl iodide, a Ni(II)–H species is produced. Migratory insertion then furnishes an
Extending their catalyst-tuned hydroalkylation methodology, Rong and coworkers developed cobalt-catalyzed asymmetric hydromethylation of 3-pyrrolines and glycals, addressing the challenge of enantioselective methyl group installation, particularly demanding due to the minimal steric profile of methyl groups and propensity for β-H elimination leading to chain-walking byproducts (Figure 5)[55]. Using a chiral IndaBox/cobalt catalyst, diverse N-acyl-protected 3-pyrrolines undergo hydromethylation with CH3I to afford 3-methylpyrrolidines in up to 84% yield and with up to 99% ee (75 examples). The substrate scope encompasses aryl, heteroaryl, aliphatic, and cycloalkane acyls with excellent functional group tolerance. Notably, isotopically labeled methyl sources (CD3I, 13CH3I) provide deuterated and 13C-labeled products with comparable efficiency, enabling stable isotope tracer applications. The methodology extends to glycals, affording 2-deoxy-C-glycosides with outstanding β-selectivity (β/α > 20:1). This approach provides streamlined two-step access to (S)-3-methylpyrrolidine (82% overall yield), replacing traditional five- to six-step routes. The proposed mechanism follows their previously established Co(I)–H-mediated hydrometalation, XAT, methyl radical capture, and reductive elimination sequence.
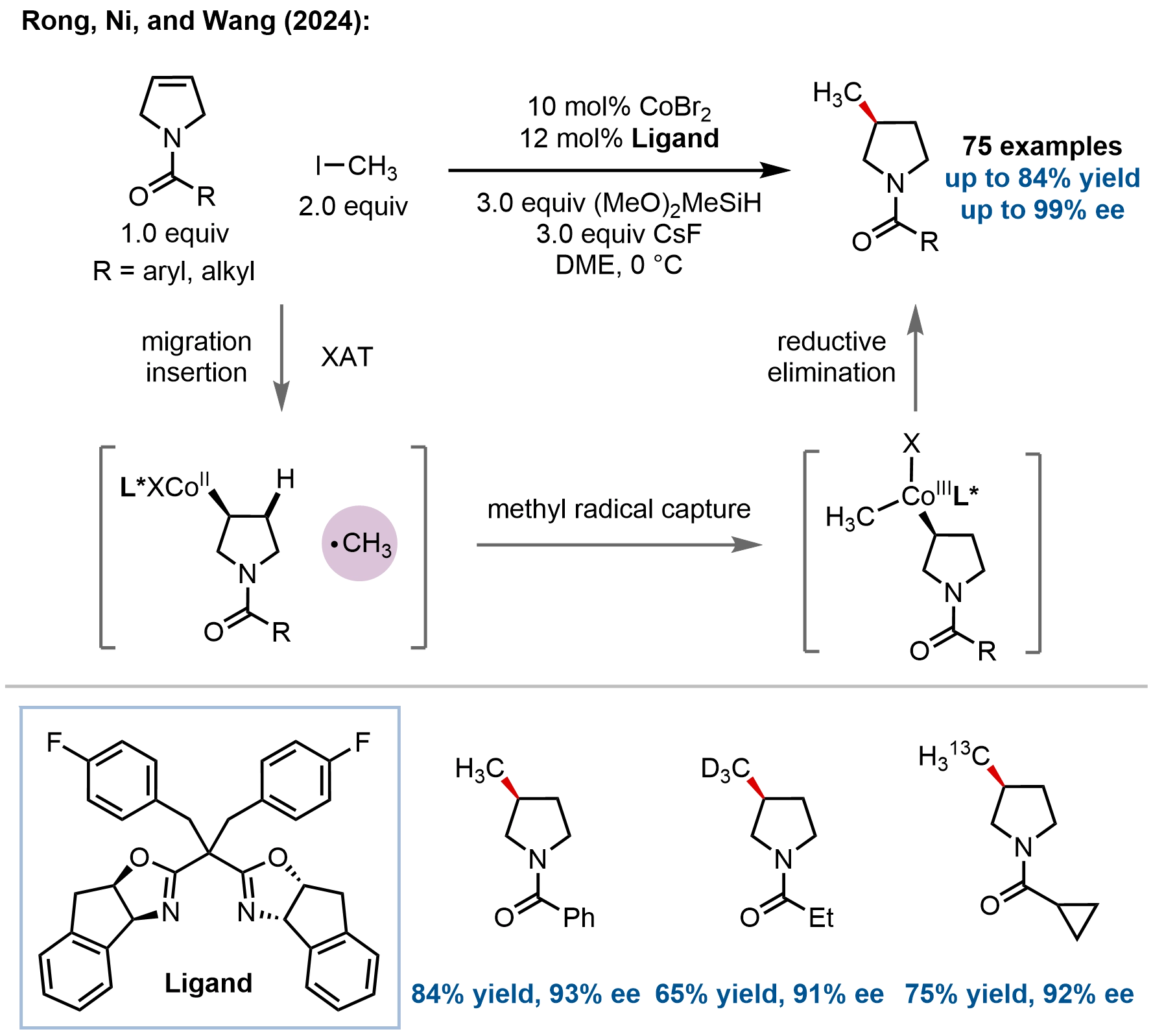
Figure 5. Cobalt-catalyzed enantioselective hydromethylation of 3-pyrrolines for the synthesis of N-protected 3-methylpyrrolidines.
In addition to saturated five-membered nitrogen heterocycles, Zhu and coworkers extended asymmetric migratory hydroalkylation to six-membered N-heterocycles, developing a nickel-catalyzed enantioselective synthesis of chiral α-alkylated azacycles from readily available N-heterocyclic alkenes (Figure 6)[56]. Using a chiral bisoxazoline/nickel catalyst, N-benzoyl-1,2,3,6-tetrahydropyridine and related substrates undergo migratory hydroalkylation with alkyl iodides to afford α-alkylated piperidines in up to 89% yield with up to 95% ee as single regioisomers (30 examples). The substrate scope encompasses diverse alkyl iodides bearing functional groups including trifluoromethyl, aryl/alkyl halides, ethers, silyl, ketone, esters, carbamate, and heterocycles (benzothiazole, thiophene). Various nitrogen protecting groups (amides, carbamates) are compatible. Notably, both remote olefins and proximal enamines undergo asymmetric hydroalkylation with comparable reactivity.
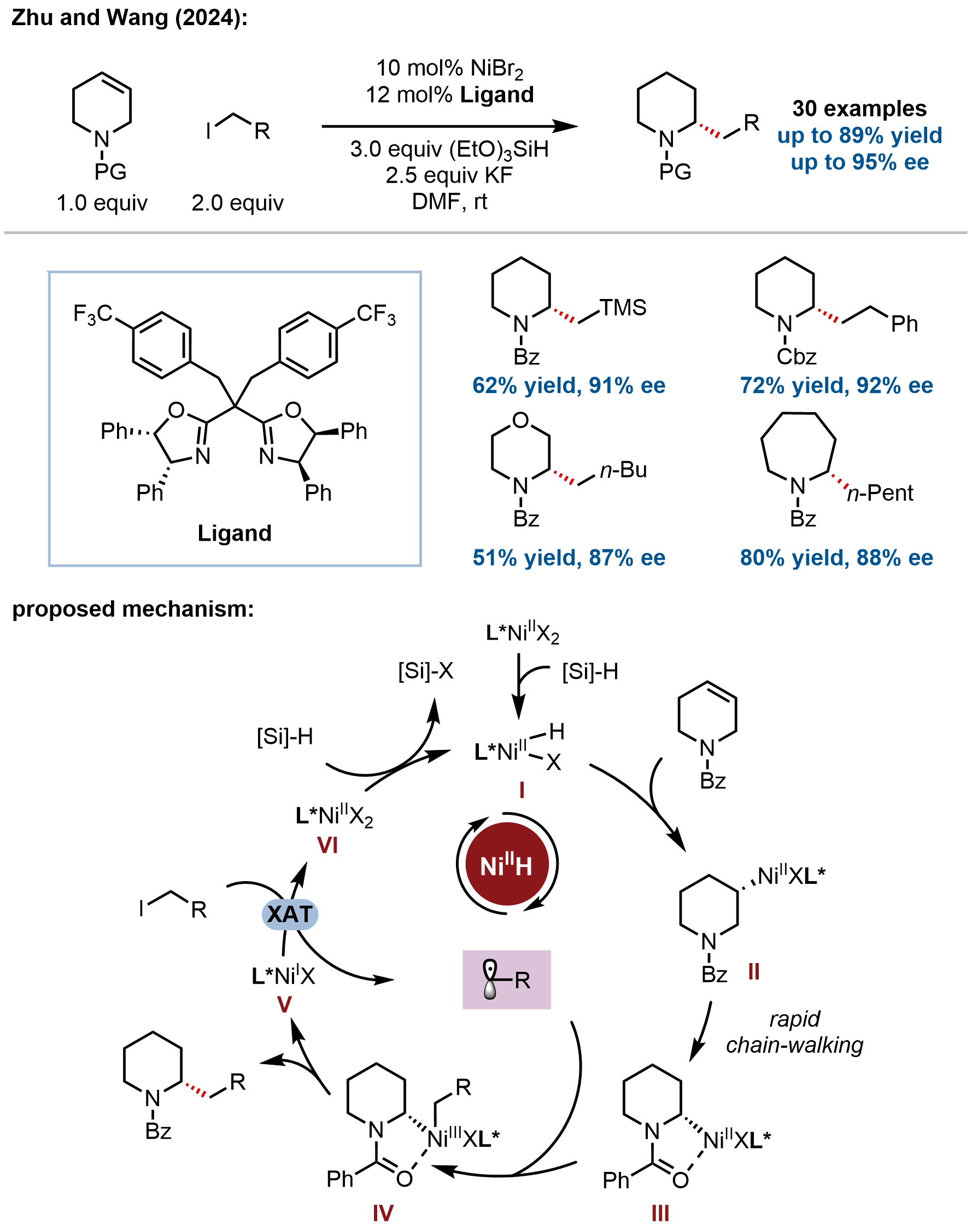
Figure 6. Asymmetric migratory hydroalkylation of six-membered N-heterocyclic alkenes enabling construction of enantioenriched α-alkylated piperidines.
The transformation was proposed to proceed through a directed hydronickelation–chain-walking mechanism. The
Extending enantioselective hydrofunctionalization beyond nitrogen heterocycles, Fu, Lu, Liu, and coworkers developed
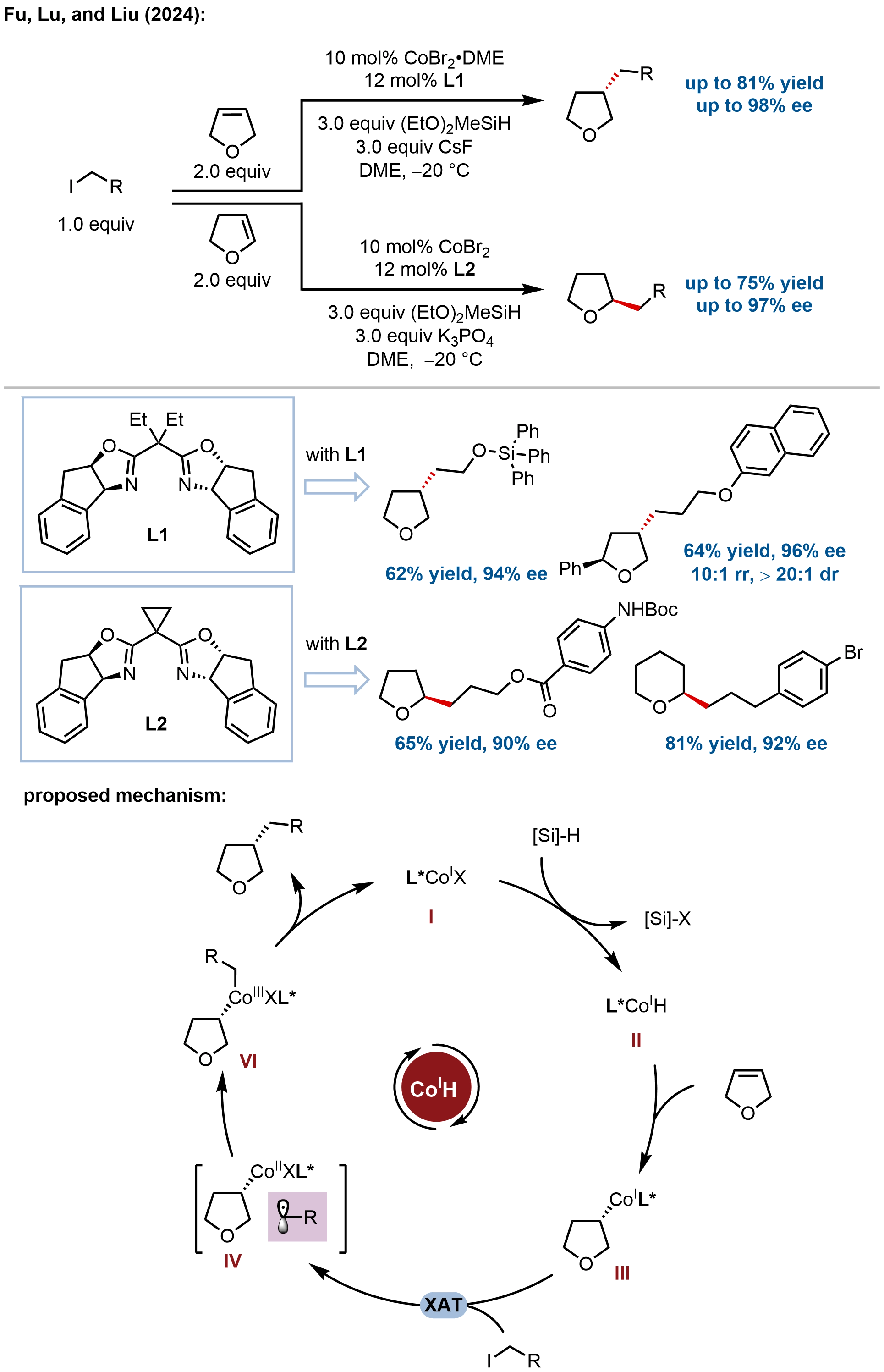
Figure 7. Ligand-controlled cobalt-catalyzed hydroalkylation of oxyheterocyclic alkenes for the synthesis of enantioenriched oxygen heterocycles.
Mechanistic investigations revealed a Co(I)-Co(II)-Co(III) catalytic cycle. Density functional theory (DFT) calculations confirmed that the high-spin Co(I)–H species undergoes alkene coordination and migratory insertion, with alkene hydrometalation as the stereodetermining step. Interaction region indicator analysis revealed that in a relevant transition state, the oxygen atom in alkene substrates exhibits substantial C–H···O interactions with ligand hydrogens, representing a noncanonical enantioselectivity control mode distinct from chelation-directing or conjugated group strategies. This oxygen-assisted weak interaction enables high enantioselectivity without canonical auxiliary groups, as evidenced by moderate enantioselectivity (32-58% ee) with norbornene substrates lacking oxygen atoms.
Addressing the challenge of synthesizing chiral cyclic sulfones, important bioisosteres of ketones and carboxylic acids with demonstrated biological activities, Hu and coworkers developed nickel-catalyzed regiodivergent and enantioselective hydroalkylation of 2-sulfolenes (Figure 8)[58]. The key innovation lies in achieving ligand-controlled regioselectivity within the same catalytic system: neutral PYROX ligand favors C3-alkylation, while anionic BOX ligand favors C2-alkylation. The substrate scope encompasses diverse functional groups including halo, boryl, nitrile, triflate, phenol, alcohol, ketone, protected amines, and heterocycles (phthalimide, furan, thiophene). Late-stage functionalization of drug-derived alkyl iodides (indomethacin, natural products) proceeds smoothly.
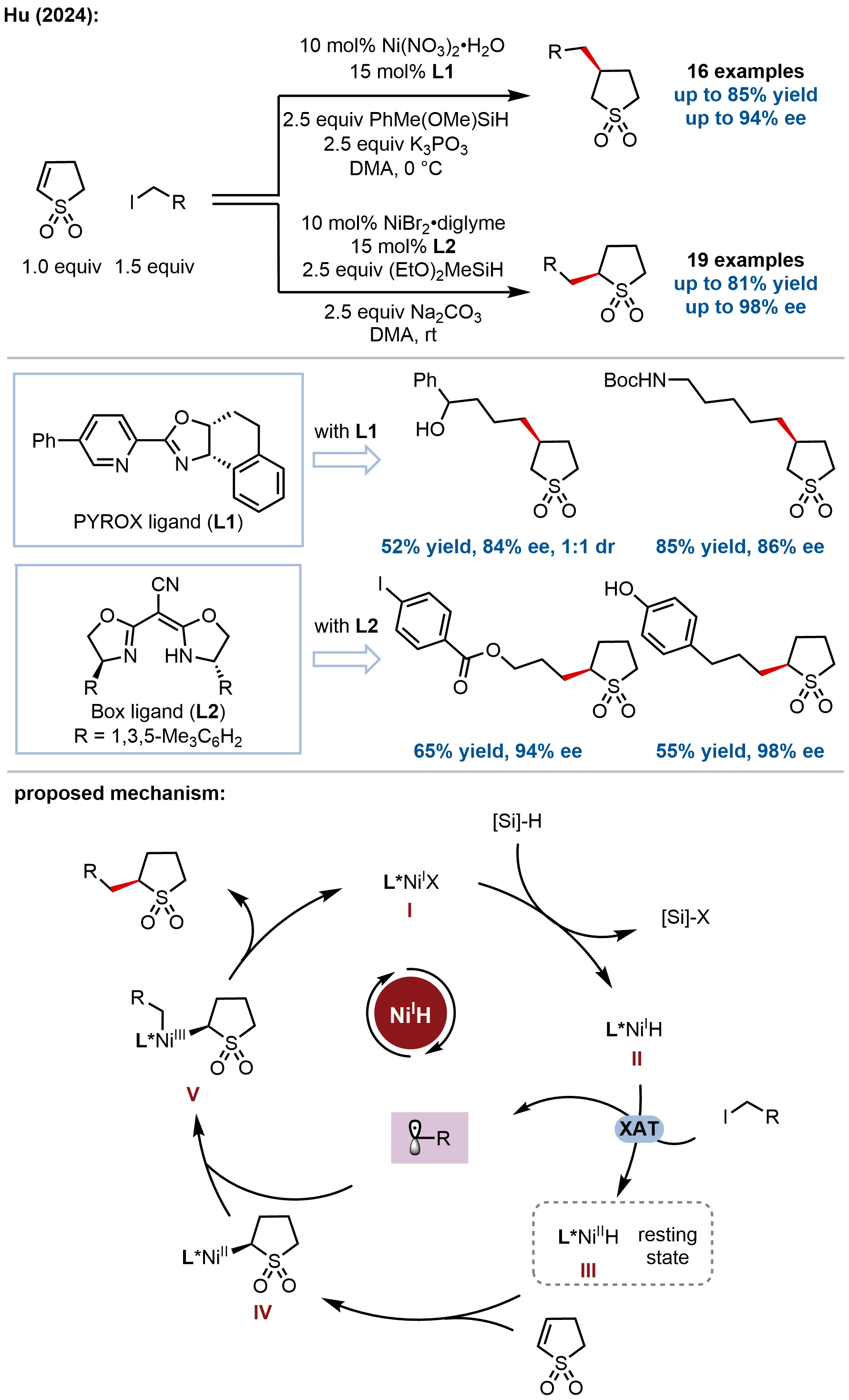
Figure 8. Regiodivergent and enantioselective hydroalkylation of sulfolenes enabling the synthesis of enantioenriched cyclic sulfones.
Mechanistic investigations revealed a Ni(I)/Ni(III) catalytic cycle with Ni(II)–H as the resting state. DFT calculations revealed that
In concurrent work with Hu’s nickel-catalyzed sulfolene hydroalkylation, Rong and coworkers developed cobalt-catalyzed asymmetric hydroalkylation of unactivated five-membered S- and O-heterocyclic alkenes through a desymmetrization strategy, addressing the challenge of constructing chiral centers at C3-positions (Figure 9)[59]. Using a chiral IndaBox/cobalt catalyst,
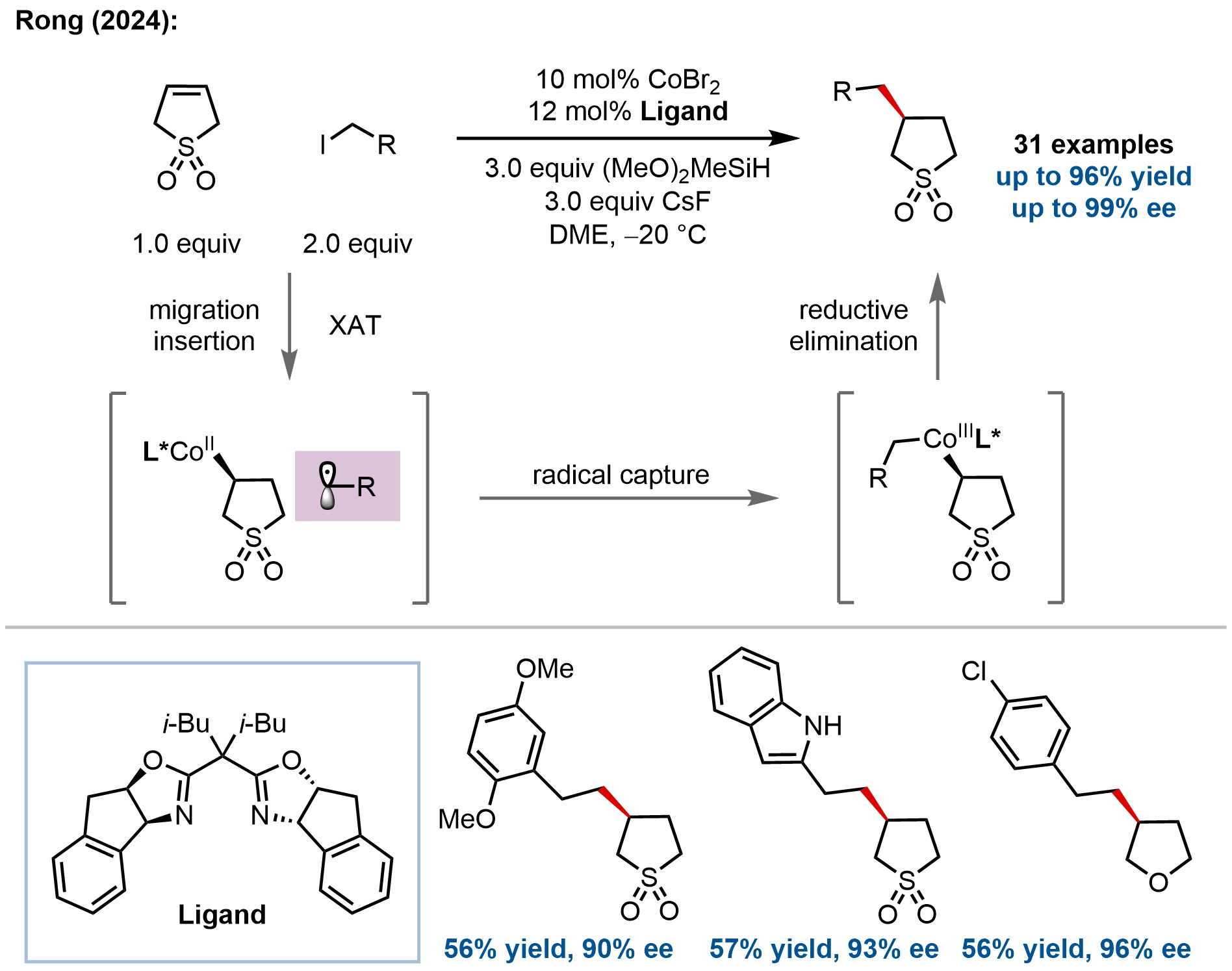
Figure 9. Cobalt-catalyzed asymmetric hydroalkylation of five-membered S- and O-heterocyclic alkenes for the synthesis of enantioenriched S- and O-heterocycles.
Mechanistic investigations confirmed a radical pathway. The proposed mechanism involves a Co(I)-Co(II)-Co(III) catalytic cycle: the
In a very recent contribution, Wang, Zhu, and coworkers developed an enantio- and diastereoselective synthesis of P-stereogenic phospholane oxides via a cobalt-catalyzed desymmetric hydroalkylation of prochiral 2,5-dihydro-1H-phosphole oxides with primary alkyl iodides (Figure 10)[60]. This work addresses a long-standing challenge in constructing chiral phospholane skeletons, privileged scaffolds in asymmetric catalysis, by simultaneously establishing two discrete stereocenters (C- and P-stereogenic) in a single operation. The reaction features a broad substrate scope, accommodating various aryl- and alkyl-substituted phosphole oxides and diverse functionalized alkyl iodides, including those derived from complex bioactive molecules, affording the corresponding phospholane oxides in good yields with excellent enantioselectivities (up to 93% yield, up to 96% ee) and diastereoselectivities
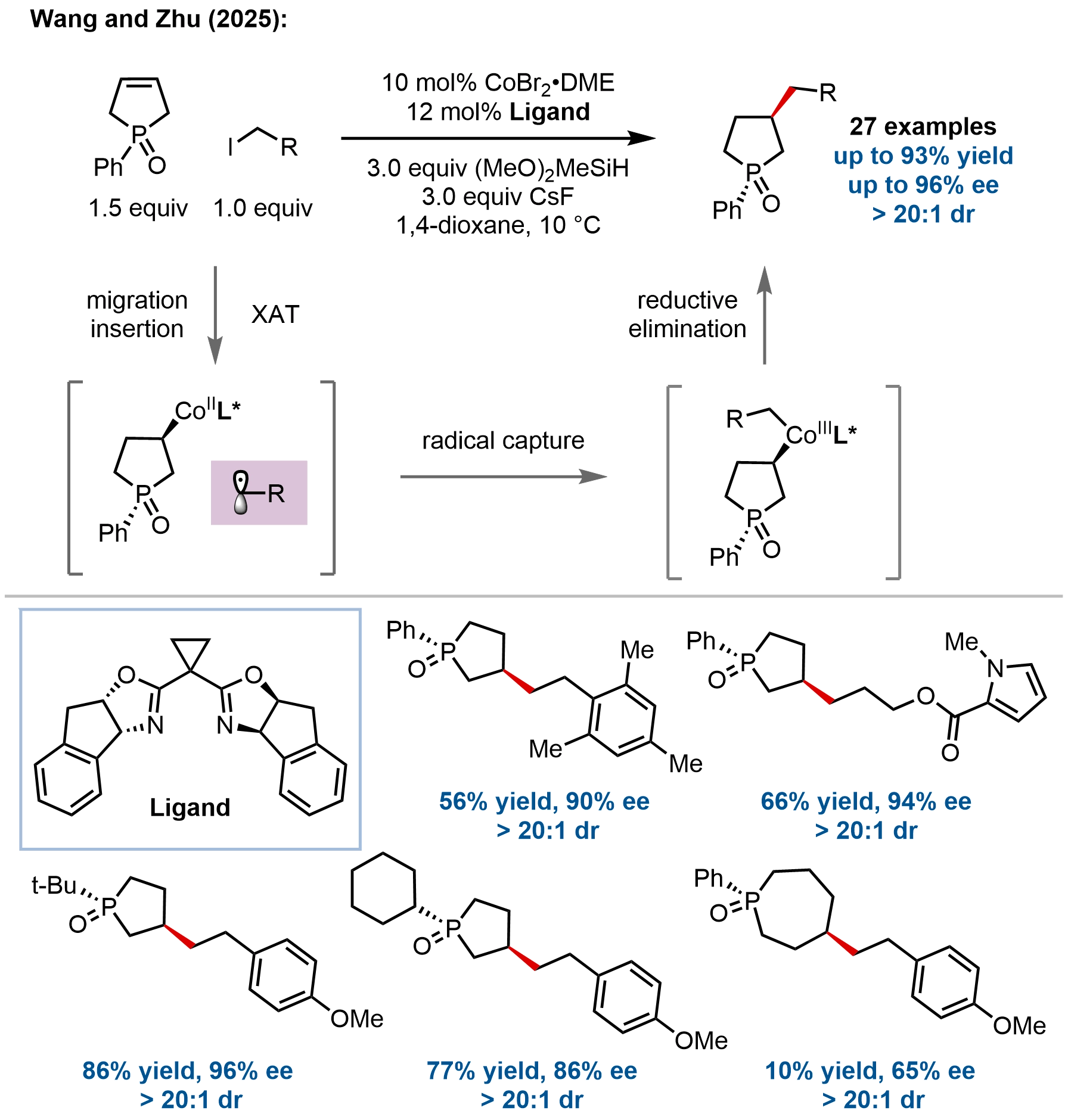
Figure 10. Cobalt-catalyzed desymmetric hydroalkylation for the synthesis of P-stereogenic phospholane oxides.
Mechanistic studies suggested a Co(I)-Co(II)-Co(III) catalytic cycle that aligns with the mechanisms established in related
Concurrently with the cobalt-catalyzed approach to P-heterocycles, Hu and coworkers reported a distinct nickel-catalyzed enantioselective hydroalkylation of unsaturated cyclic phosphinates, achieving the simultaneous construction of nonadjacent P- and C-stereocenters (Figure 11)[61]. This work represents the first application of nickel-hydride (Ni–H) catalysis to the formation of an enantioenriched non-carbon stereocenter. The protocol operates under mild conditions via a dynamic kinetic asymmetric transformation (DyKAT) of racemic six-membered phosphinate substrates. Key to the success is the use of a chiral bisoxazoline ligand with 2-naphthyl substituents, which, in conjunction with NiI2 and (EtO)3SiH, delivers 3-alkylated products with excellent regio-, diastereo-, and enantioselectivity.
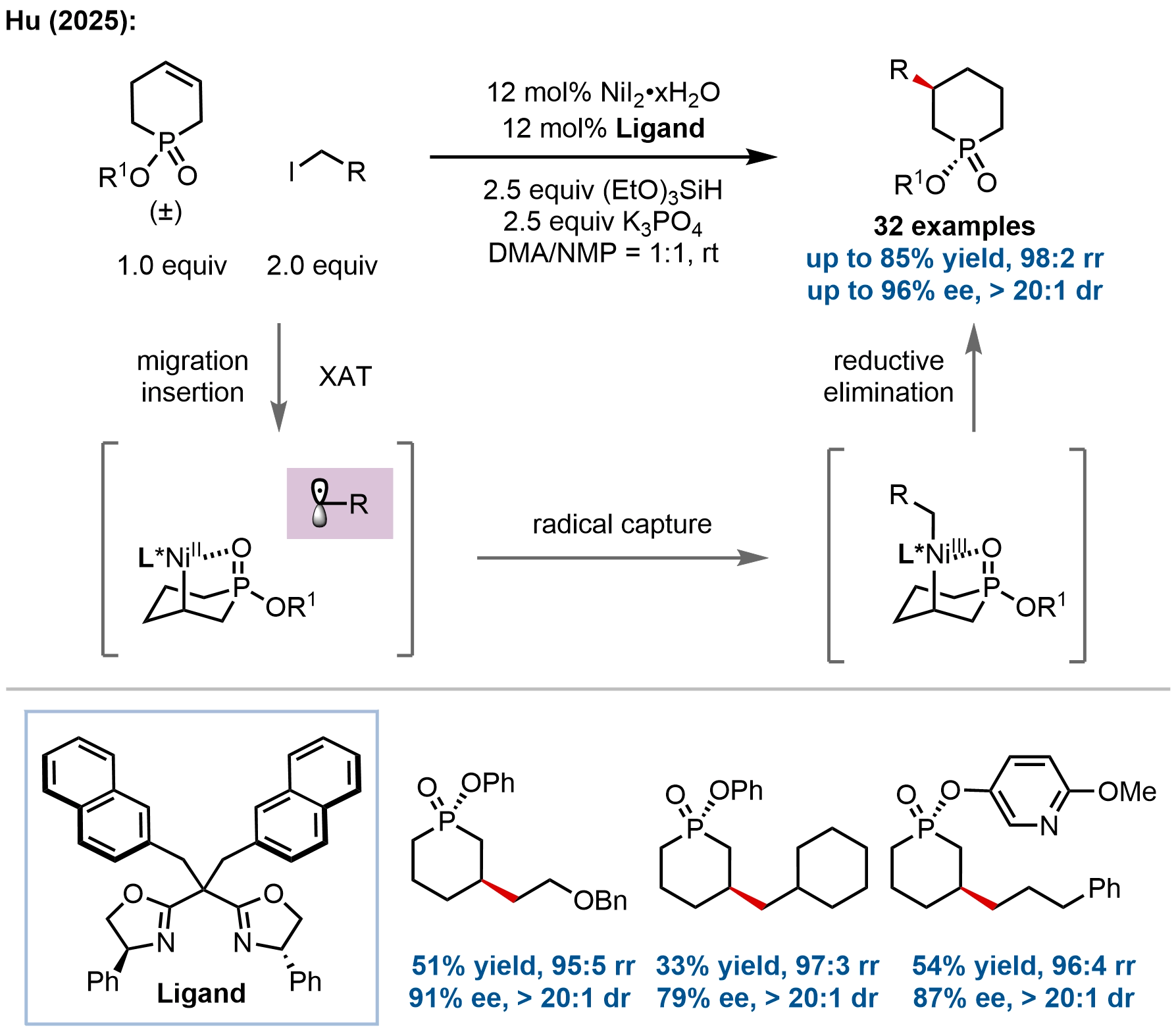
Figure 11. Nickel-catalyzed hydroalkylation of cyclic phosphinates for the synthesis of enantioenriched P-heterocycles
Mechanistic investigations confirmed a DyKAT pathway wherein both enantiomers of the racemic starting material are interconverted and converge to a single product stereoisomer. The proposed catalytic cycle involves Ni(II)–H formation, migratory insertion, and chain-walking to equilibrate isomeric alkyl-nickel intermediates. Selective capture of an alkyl radical by a key
In a significant expansion of their cobalt-catalyzed hydrofunctionalization platform, Fu, Lu, and coworkers developed a modular enantioselective hydroalkylation of vinylsilanes, allylsilanes, and their germanium congeners, enabling the divergent synthesis of
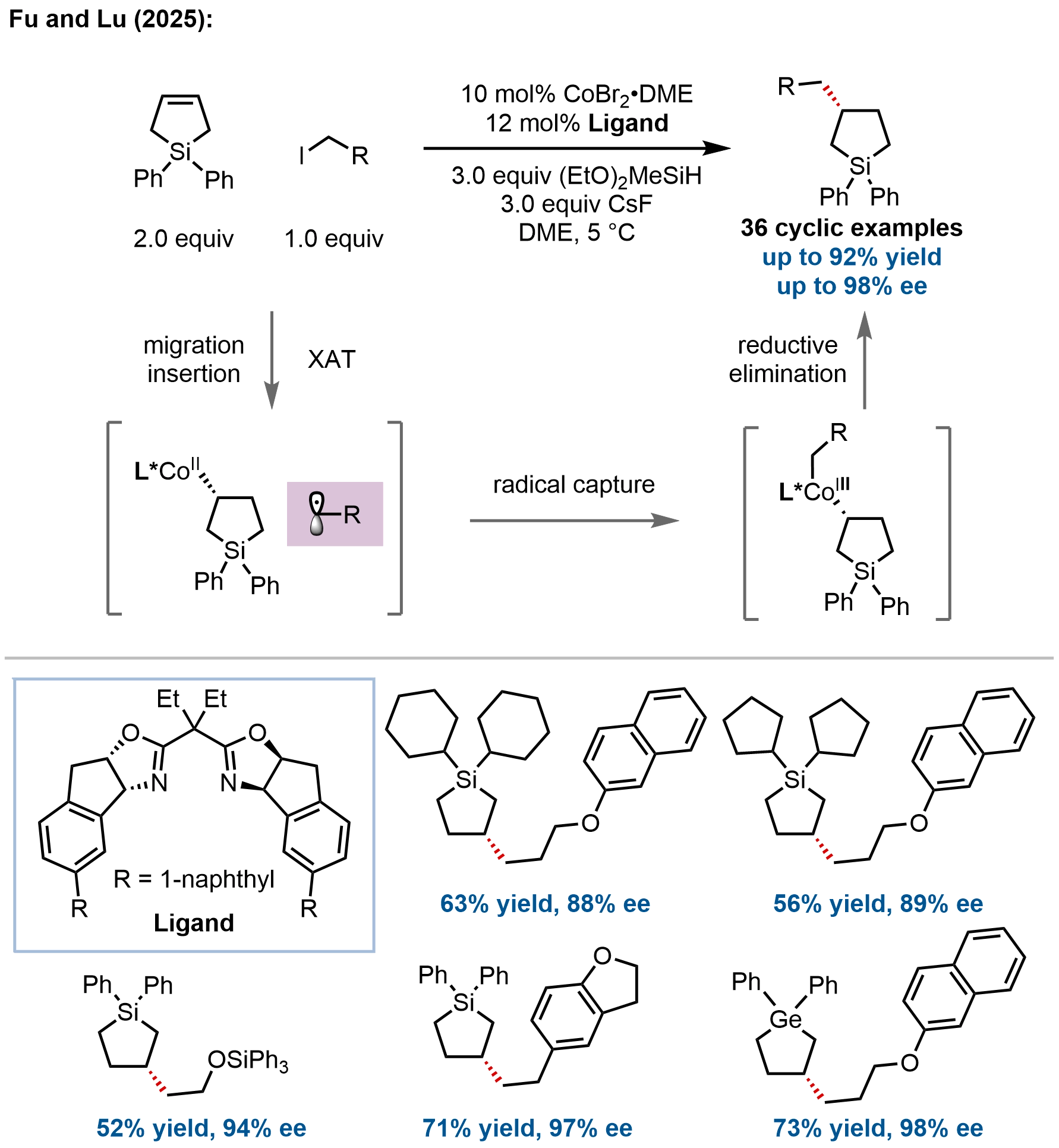
Figure 12. Cobalt-catalyzed enantioselective hydroalkylation of sila- and germacyclopent-3-enes for the synthesis of β-stereogenic cyclic organometalloids.
Mechanistic investigations revealed a Co(I)/Co(III) catalytic cycle wherein a Co(I)–H species undergoes enantioselective migratory insertion into the alkene as the stereodetermining step, followed by radical capture and reductive elimination. This is consistent with their previously established mechanism for the hydroalkylation of oxyheterocyclic alkenes (Figure 7). Distortion/interaction analysis and energy decomposition studies revealed that while the silicon atom enhances reaction activity, the dominant factor controlling enantioselectivity is steric interaction between the silyl substituent and the chiral catalyst, rather than electronic effects. This work not only provides a versatile platform for synthesizing chiral organosilanes and organogermanes but also establishes a new paradigm for leveraging main-group metalloid substituents as stereo-controlling auxiliaries in enantioselective radical catalysis.
In a significant advance beyond saturated heterocycles, Zhu and coworkers addressed the formidable challenge of enantioselective construction of carbocyclic scaffolds by developing a nickel-catalyzed asymmetric migratory hydroalkylation of readily available racemic cycloalkenes (Figure 13)[63]. This method enables the convergent synthesis of thermodynamically disfavored 1,2-cis disubstituted cycloalkanes bearing vicinal stereocenters with exceptional levels of regio-, diastereo-, and enantiocontrol. The key innovation lies in the design of a DyKAT that leverages a transformable ester substituent on the cycloalkene substrate to differentiate reactivity among rapidly interconverting regio- and stereo-isomeric alkylnickel intermediates. Using a chiral bisoxazoline/nickel catalyst, the reaction accommodates a broad range of racemic cycloalkenyl esters and primary alkyl iodides, delivering the desired cis-products in high yields (up to 75%) with excellent enantioselectivities (up to > 99% ee) as single regioisomers.
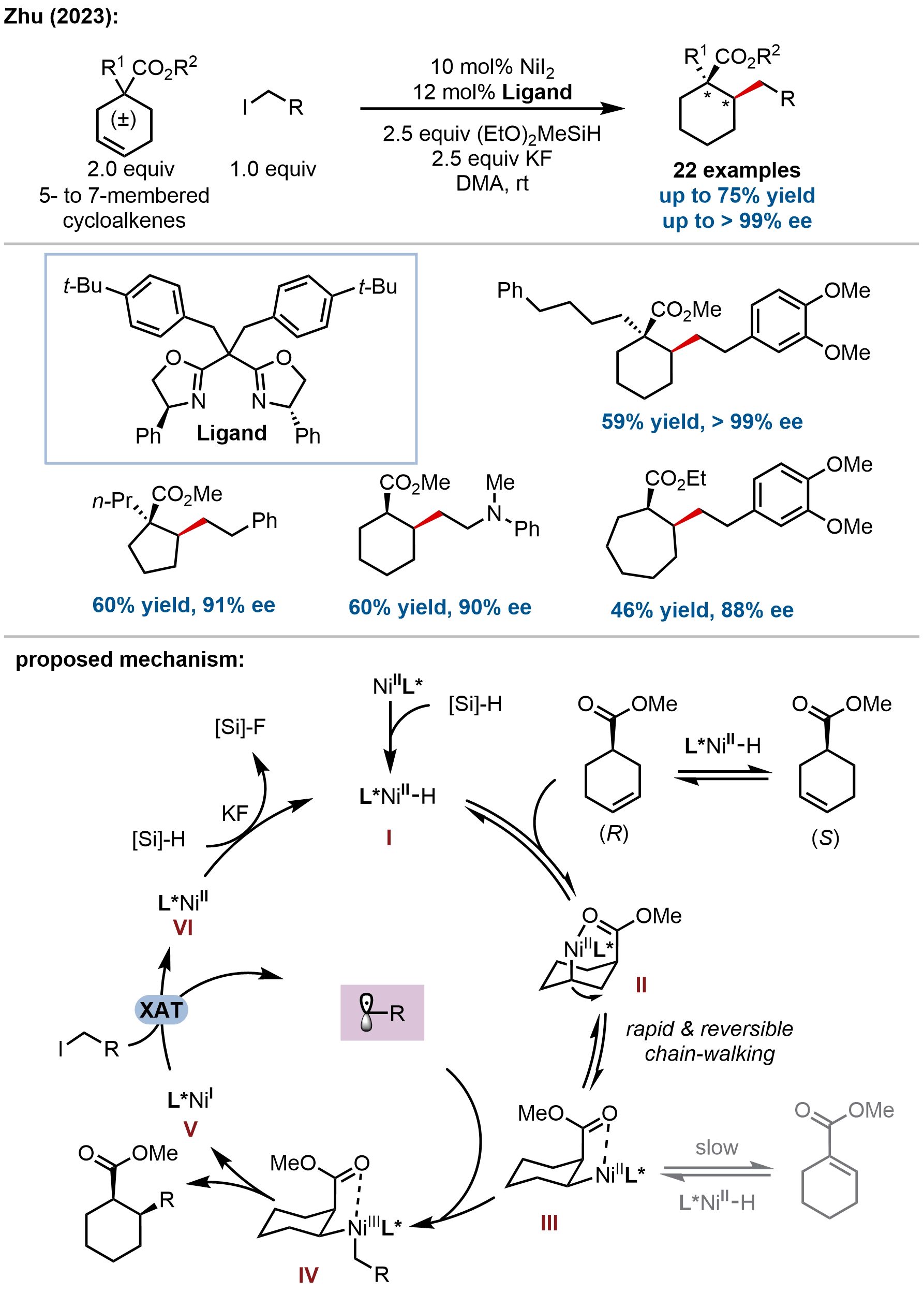
Figure 13. Nickel-catalyzed asymmetric migratory hydroalkylation of racemic cycloalkenes for the synthesis of enantioenriched 1,2-cis disubstituted cycloalkanes.
Mechanistic investigations elucidated a sophisticated Ni–H-catalyzed chain-walking cascade that underpins the DyKAT process. The in situ-generated L*Ni(II)–H species undergoes rapid and reversible migratory insertion into the cycloalkene, initiating a
In terms of bridged bicyclic structures, Fu, Lu, and coworkers recently developed an enantioselective ring-retentive hydroalkylation of strained oxa- and azabicyclo[2.2.1]alkenes, addressing the longstanding challenge of functionalizing bridged bicyclic scaffolds without disrupting their core architecture (Figure 14)[64]. While the thermodynamic driving force of strain energy release typically favors ring-opening pathways, this cobalt-catalyzed protocol enables the direct construction of tertiary carbon stereocenters within the intact bicyclic framework with exceptional enantiocontrol (up to 99% ee).
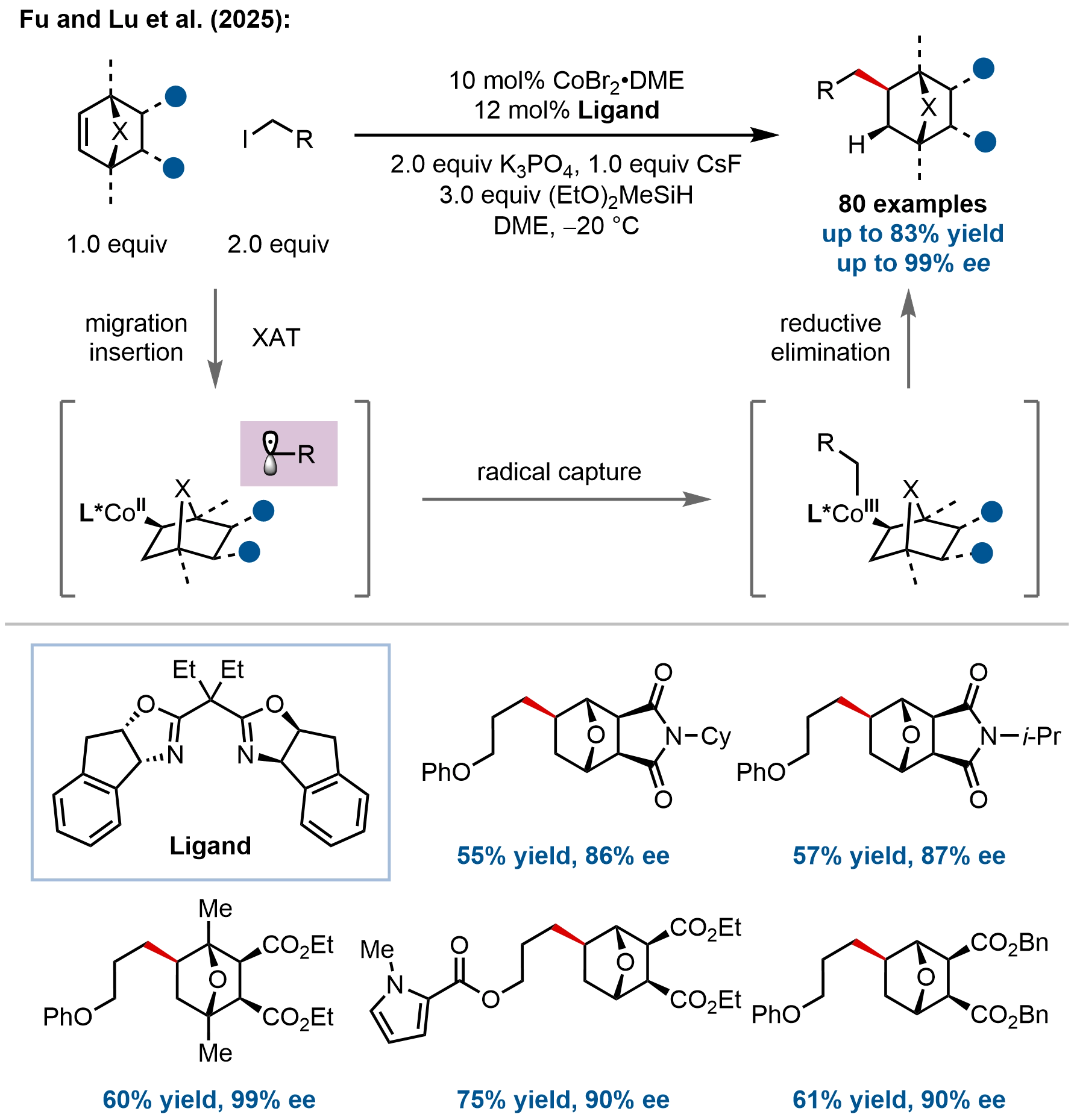
Figure 14. Cobalt-catalyzed enantioselective hydroalkylation of oxa- or azabicyclic alkenes enabling construction of enantioenriched bridged bicyclic frameworks.
Mechanistic studies, including deuterium labeling, radical clock experiments, and DFT calculations, support a Co(I)/Co(II)/Co(III) catalytic cycle wherein enantioselective alkene hydrometalation constitutes the stereodetermining step. This mechanism aligns with their previously established hydrofunctionalization manifolds (Figure 7 and Figure 12). This work establishes a powerful platform for the enantioselective synthesis of pharmacologically relevant bridged bicyclic architectures and expands the scope of cobalt-catalyzed hydroalkylation to strained, non-conjugated ring systems.
In a significant advance toward the synthesis of saturated carbocycles, Rong and coworkers developed a cobalt-catalyzed enantioselective desymmetrizing remote hydroalkylation of prochiral cyclohexene derivatives, enabling modular access to chiral multi-substituted cyclohexanes bearing three distinct C(sp3) stereocenters (Figure 15)[65]. This strategy addresses the formidable challenge of constructing densely functionalized six-membered carbocycles that are difficult to access via conventional methods. Through meticulous ligand optimization, a sterically tuned bisoxazoline ligand bearing geminal 3,5-di-tert-butylbenzyl substituents was identified as crucial for achieving excellent regio-, diastereo-, and enantiocontrol (up to > 20:1 dr, 96% ee) under mild conditions. The reaction exhibits a broad substrate scope, accommodating a diverse array of functionalized primary alkyl iodides, as well as various substituted cyclohexene esters and diol derivatives.
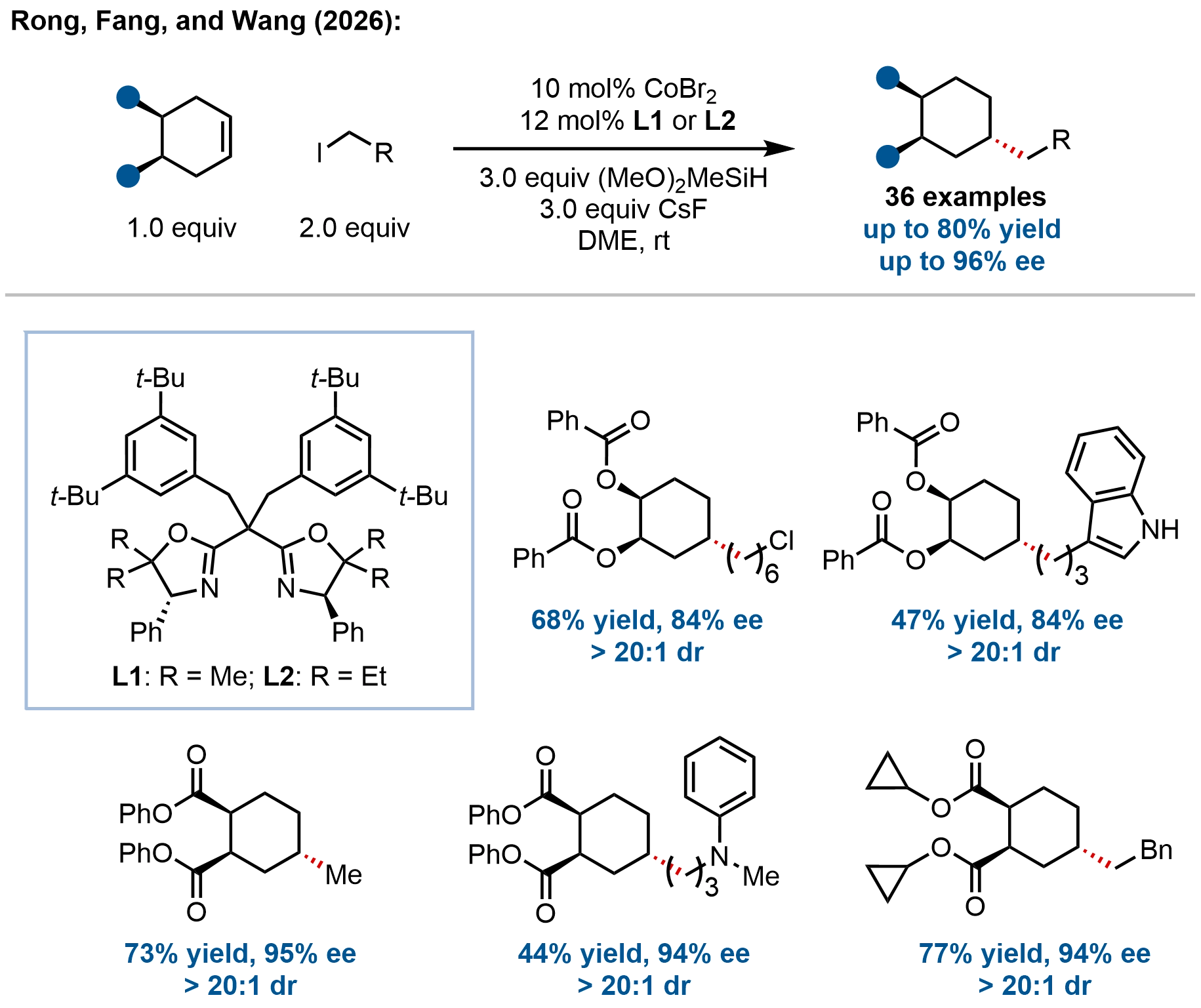
Figure 15. Cobalt-catalyzed enantioselective desymmetrizing hydroalkylation of prochiral cyclohexene derivatives for the synthesis of chiral multi-substituted cyclohexanes.
Mechanistic studies support a Co(I)/Co(III) catalytic cycle involving stereodetermining alkene hydrometalation followed by radical capture and reductive elimination, consistent with established cobalt-hydride manifolds (Figure 5, Figure 7, Figure 9, Figure 10,
Extending the low-valent cobalt-hydride catalytic platform, Fu, Lu, and coworkers developed a facial-selective hydroalkylation of cyclopropenes, enabling rapid and modular access to multi-substituted cyclopropanes (Figure 16A)[66]. This method addresses a gap in metal-hydride catalysis, where Cu–H systems typically require activated electrophiles and Ni–H catalysis depends on coordinating auxiliaries for high selectivity. Through judicious ligand optimization, the Co–H system demonstrated broad substrate scope, accommodating diverse primary and secondary alkyl iodides, various functional groups, and a range of substituted cyclopropenes, delivering the desired products in moderate to good yields with excellent facial-selectivities (up to > 20:1 dr). Preliminary asymmetric induction (up to 94% ee) was also demonstrated using a phosphinooxazoline (PHOX) ligand, suggesting future potential for enantioselective variants.
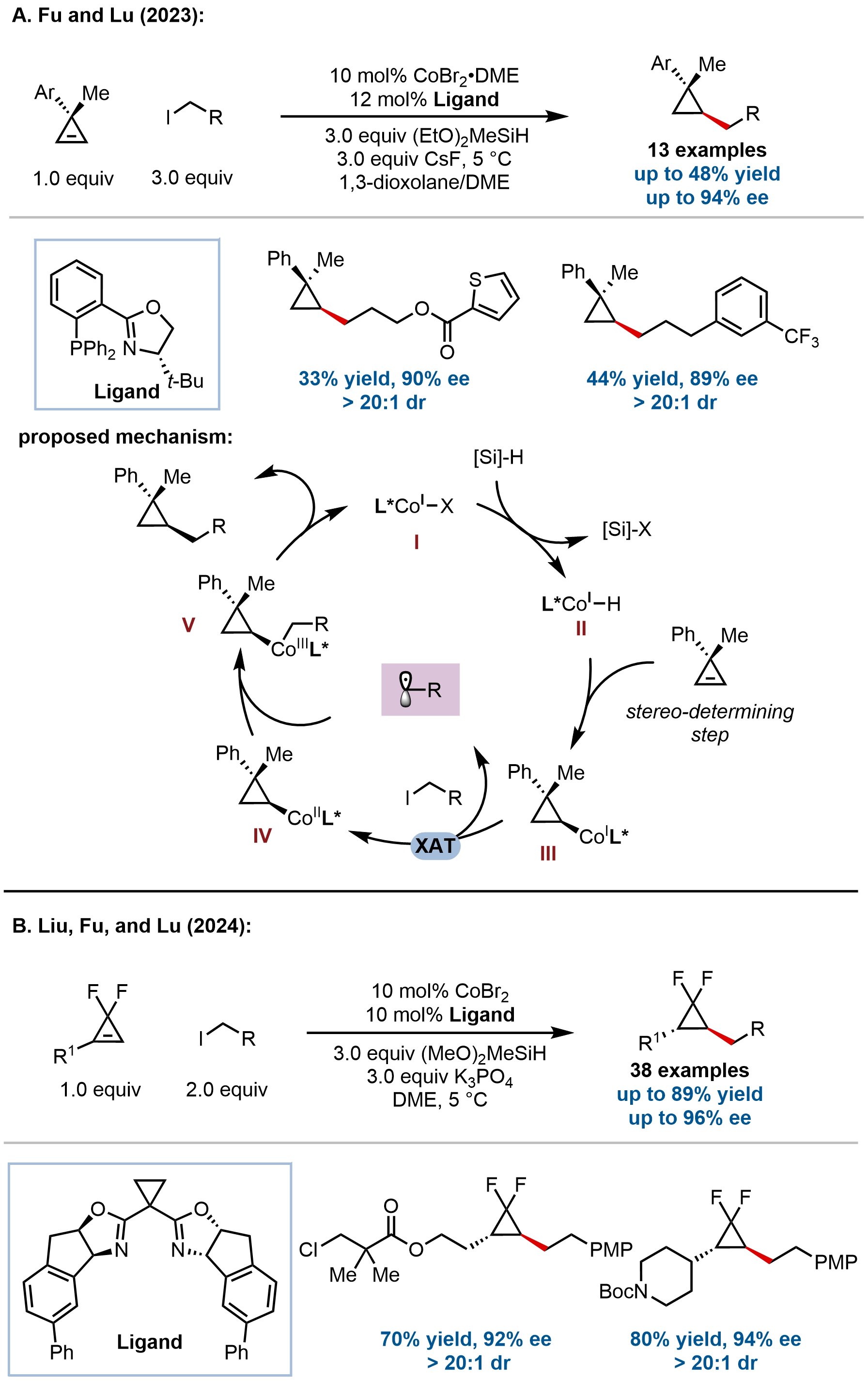
Figure 16. Cobalt-catalyzed hydroalkylation of cyclopropenes for the synthesis of enantioenriched cyclopropanes.
Mechanistic studies support a Co(I)/Co(III) catalytic cycle wherein stereodetermining syn-hydrometalation of the cyclopropene by a Co–H species is followed by radical capture and reductive elimination. Notably, comparative experiments revealed that the Co–H system exhibits superior facial-selectivity over Ni–H catalysis in the absence of directing groups, underscoring the unique sensitivity of cobalt to steric differentiation. This work further expands the scope of cobalt-catalyzed hydroalkylation to strained,
Building on their cobalt-catalyzed platform, Liu, Fu, Lu, and coworkers further extended this strategy to the stereoselective synthesis of chiral gem-difluorocyclopropanes bearing vicinal stereocenters via hydroalkylation of gem-difluorocyclopropenes (Figure 16B)[67]. This method achieves excellent enantio- and diastereocontrol (up to > 20:1 dr, 96% ee) without requiring chelation-directing groups, accommodating a broad range of functionalized alkyl iodides and enabling late-stage modification of bioactive molecules.
In a complementary approach to two-component hydrofunctionalizations, Yin and coworkers developed a nickel-catalyzed regio-, diastereo-, and enantioselective migratory alkylboration of endocyclic heterocyclic alkenes, enabling rapid and modular access to 2,4-disubstituted saturated oxygen heterocycles from readily available starting materials (Figure 17)[68]. This three-component coupling strategy addresses the long-standing challenge of constructing this specific substitution pattern by leveraging a
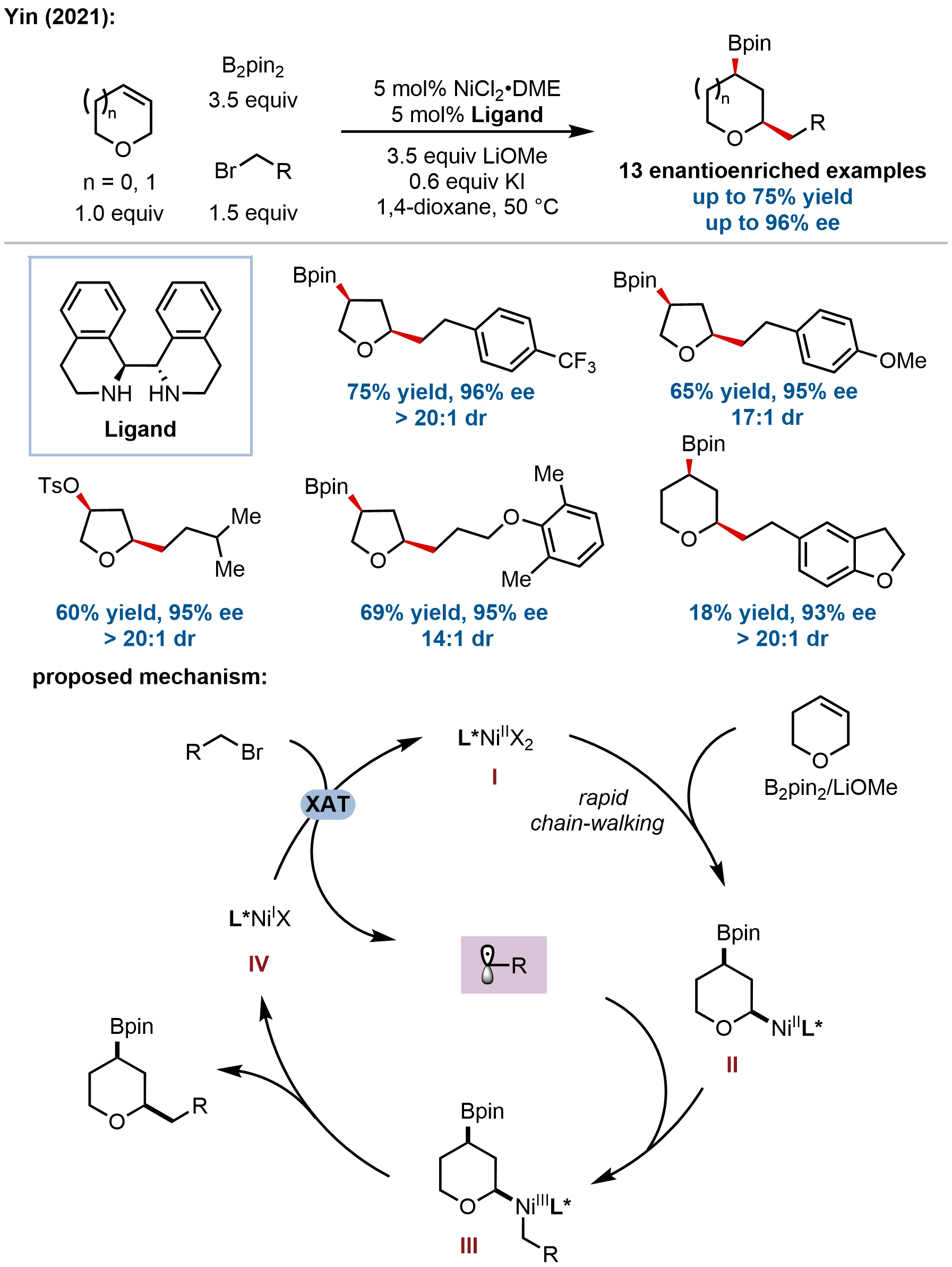
Figure 17. Nickel-catalyzed regio-, diastereo-, and enantioselective migratory alkylboration for the synthesis of 2,4-disubstituted saturated oxygen heterocycles.
Mechanistic investigations support a radical pathway wherein alkyl bromides undergo XAT to generate alkyl radicals. A proposed Ni(I)/Ni(III) catalytic cycle involves: (i) transmetalation of a Ni(II) species with the diboron reagent, (ii) regioselective migratory insertion guided by heteroatom coordination, (iii) chain-walking via iterative β-hydride elimination/reinsertion without dissociation of the nickel from the substrate, and (iv) radical capture by an alkylnickel(II) species to form a Ni(III) intermediate, followed by stereospecific reductive elimination to forge the C–C bond with high cis-diastereoselectivity. The stereochemical outcome suggests that the diastereoselectivity is likely controlled during reductive elimination from isomeric Ni(III) intermediates, with the
2.3 Direct enantioselective C(sp3)–H RCC
Enantioselective C(sp3)–H RCC represents one of the most straightforward approaches to access chiral saturated (hetero)cycles, as it bypasses the need for prefunctionalized starting materials by directly leveraging ubiquitous C–H bonds as latent nucleophiles. In 2022, our group reported the first direct enantioselective C(sp3)–H RCC of heterocycles, achieving the modular synthesis of enantioenriched α-(hetero)aryl saturated azacycles (Figure 18)[69]. This method leverages our previously established
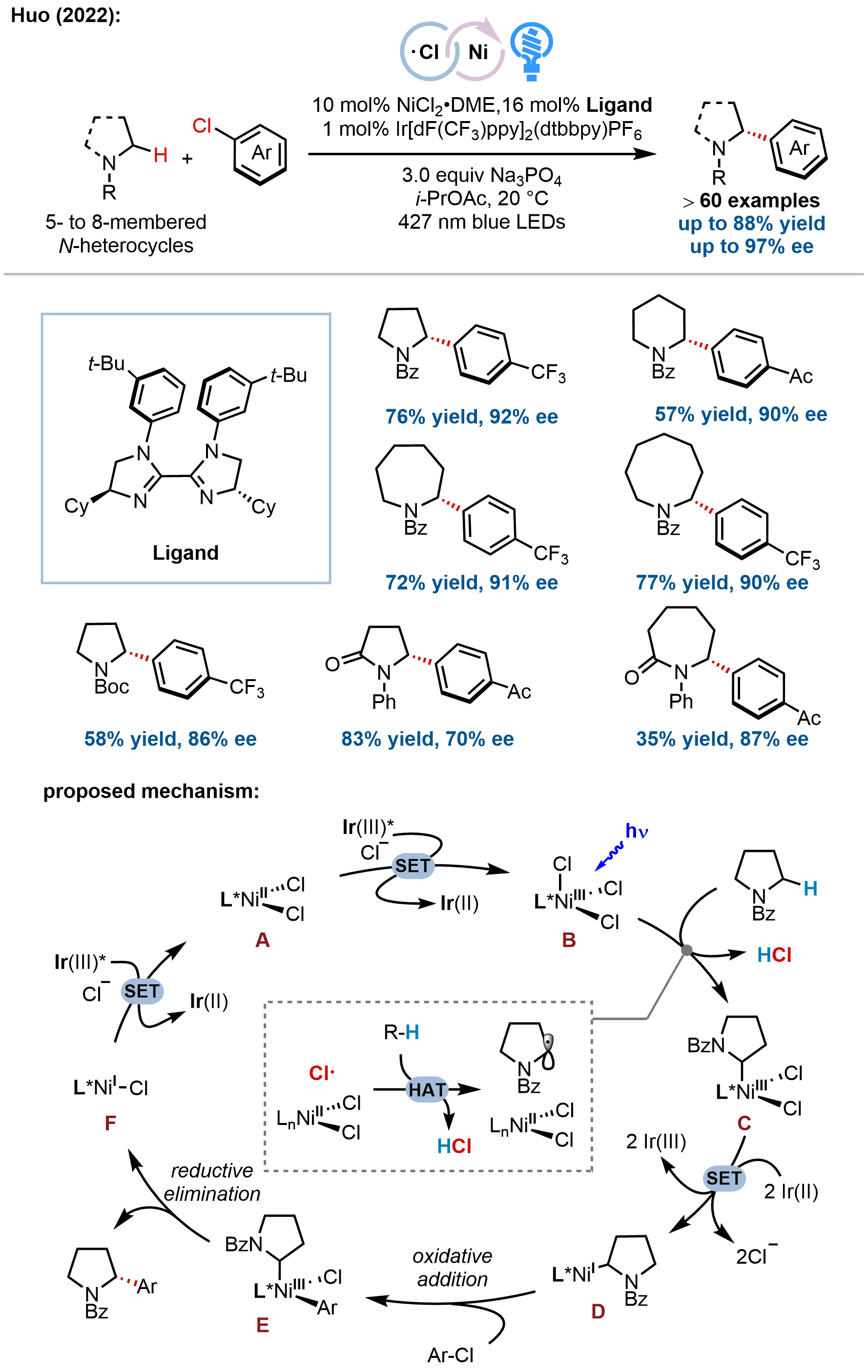
Figure 18. Direct enantioselective α-(hetero)arylation of saturated azacycles via metallaphotoredox catalysis.
Mechanistic studies elucidated a sophisticated Ni(I)/Ni(III) catalytic cycle wherein the nickel catalyst orchestrates multiple roles: chlorine radical generation, α-amino radical capture, cross-coupling, and asymmetric induction. Key evidence includes radical trapping experiments confirming α-amino radical intermediacy, luminescence quenching and cyclic voltammetry supporting the oxidation of a Ni(II) dichloride species to Ni(III) by the excited photocatalyst, and control experiments ruling out alternative pathways involving Ni(0) or preformed Ni(II) aryl chloride complexes. The proposed mechanism proceeds via: (i) photoredox-mediated oxidation of Ni(II) to Ni(III), (ii) LMCT-driven chlorine radical elimination and HAT to generate an α-amino radical, (iii) radical rebound to form a Ni(III) alkyl intermediate, (iv) photocatalytic reduction to Ni(I), (v) oxidative addition of aryl chloride, and (vi) reductive elimination to furnish the enantioenriched product.
Building on our chlorine-radical-promoted metallaphotoredox platform, our group further extended this strategy to achieve site- and enantioselective α-acylation of saturated N-heterocycles with abundant carboxylic acids (Figure 19)[77]. This method exploits the electrophilic chlorine radical generated via LMCT from a Ni(III)–Cl species as a highly selective hydrogen atom abstractor that preferentially activates cyclic α-amino C–H bonds even in the presence of intrinsically weaker yet polarity-mismatched C–H bonds, such as benzylic, allylic, acyclic α-amino, and α-oxy methylene groups. The protocol features broad substrate scope, accommodating diverse alkyl and aryl carboxylic acids, various ring sizes (4- to 8-membered N-heterocycles), and complex bioactive molecules, delivering α-acylated azacycles in good yields with excellent site- (typically > 20:1 rr) and enantioselectivity (up to 99% ee). The synthetic utility was compellingly demonstrated by the modular synthesis of eight previously inaccessible Taxol derivatives via
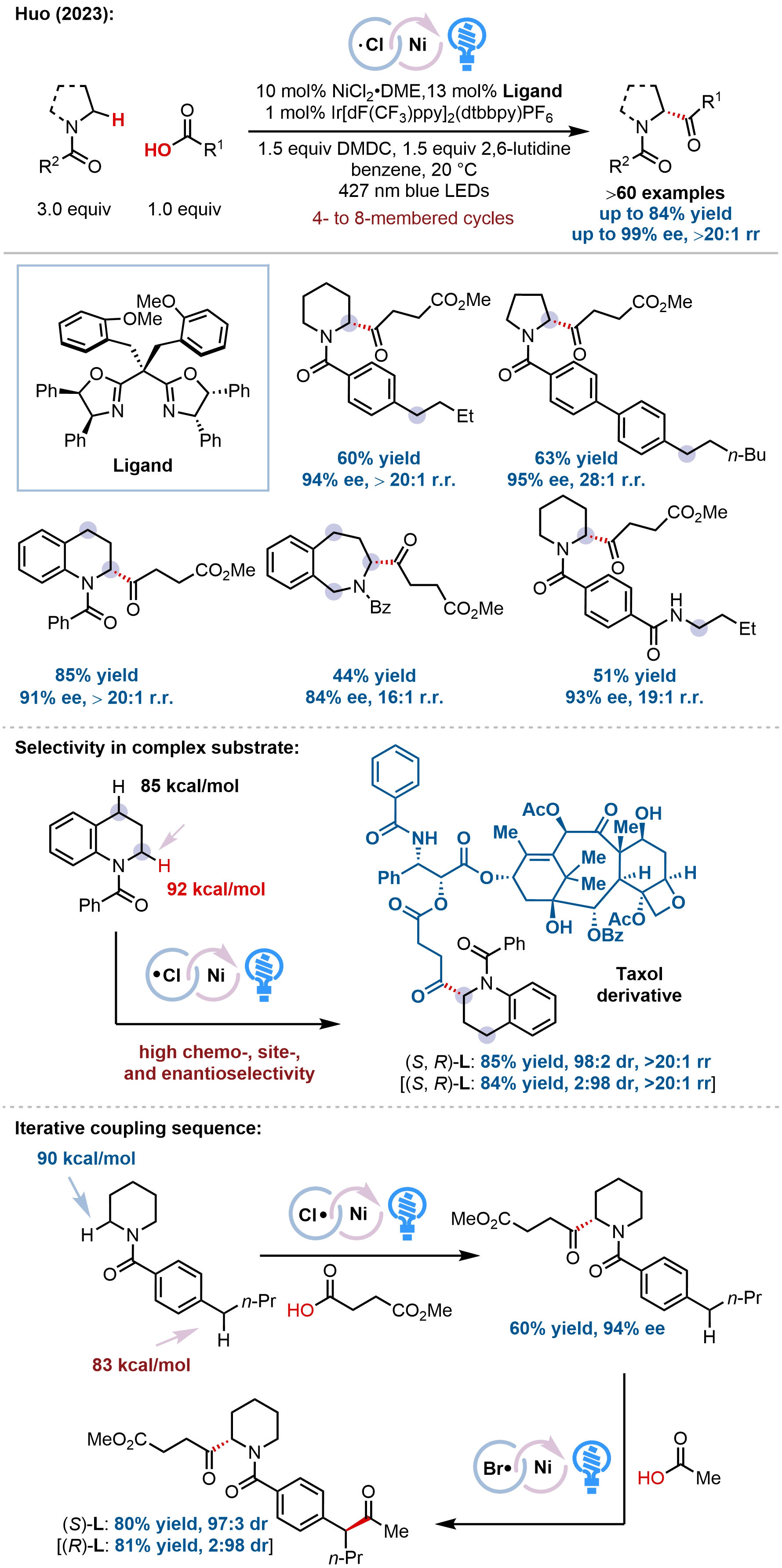
Figure 19. Site- and enantioselective α-acylation of saturated N-heterocycles enabling orthogonal site-selectivity and iterative C(sp3)–H cross-coupling sequences.
Notably, this method exhibits orthogonal site-selectivity to previously established HAT-mediated functionalizations, enabling iterative C(sp3)–H cross-coupling sequences. For instance, the chlorine-radical-mediated α-acylation product could undergo subsequent bromine-radical-mediated benzylic C–H acylation under our previously developed conditions, allowing rapid buildup of molecular complexity with precise stereocontrol. A key innovation lies in the synergistic interplay of thermodynamic and kinetic effects: the strong H–Cl bond (103 kcal/mol) provides a substantial driving force for HAT, while the high electrophilicity of the chlorine radical (3.8 eV) ensures kinetically selective abstraction from the most hydridic α-amino C–H bonds. Mechanistic studies revealed a Ni(I)/Ni(III) catalytic cycle analogous to our previously reported arylation platform (Figure 18).
Building on our halogen-radical-enabled metallaphotoredox platform for enantioselective C(sp3)–H cross-coupling, our group recently addressed the long-standing challenge of enantioselective C(sp3)–H alkylation for C(sp³)–C(sp³) bond formation by developing the first general protocol for the direct asymmetric (trideutero)methylation and alkylation of α-amino C(sp3)–H bonds (Figure 20)[78]. This work represents the first example of harnessing highly reactive methyl radicals in catalytic enantioselective transformations. The study marks a significant conceptual advance, moving from monoradical cross-coupling to a dual radical-radical cross-coupling paradigm wherein two transiently generated alkyl radicals, an α-amino radical derived from polarity-matched HAT by a bromine radical and an alkyl radical generated from a redox-active ester (RAE) via reductive decarboxylation, are asymmetrically coupled by a chiral nickel catalyst. The key innovation lies in the modular control of radical generation and cross-coupling through two discrete and cooperative catalytic cycles, enabling the use of highly reactive methyl radicals and other alkyl fragments that have proven elusive in asymmetric catalysis. The protocol exhibits exceptional scope, accommodating diverse acyclic and cyclic N-heterocycles (pyrrolidines, piperidines, azepanes, azocanes), a wide array of primary and secondary alkyl RAEs, and various functional groups, delivering α-alkylated products in good yields with excellent enantioselectivities (up to 98% ee). Notably, this method enables the asymmetric synthesis of challenging dialkyl carbinamines with minimally differentiated substituents (e.g., methyl versus ethyl) and facilitates late-stage diversification of complex bioactive molecules.
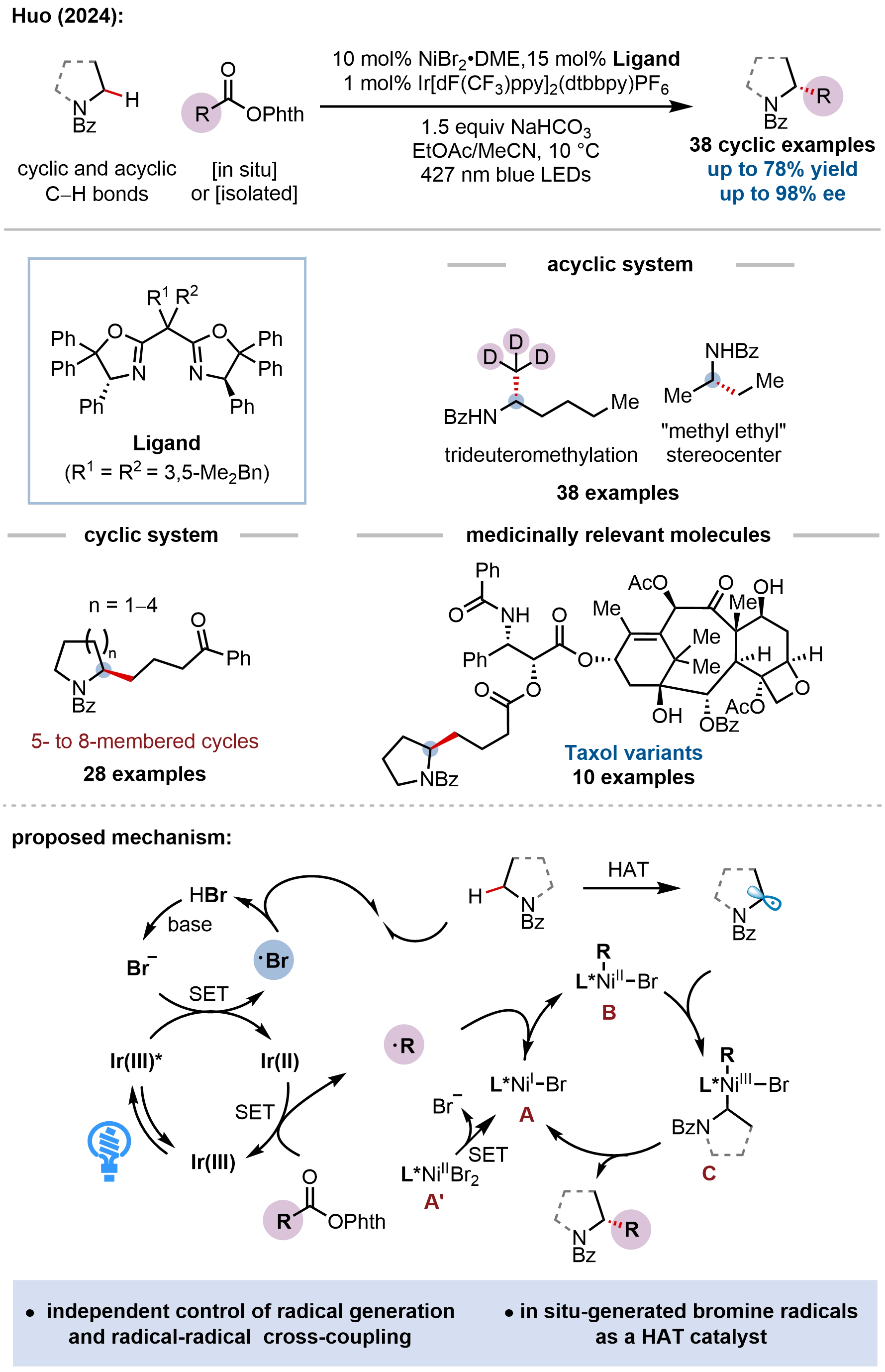
Figure 20. Enantioselective (trideutero)methylation and alkylation of α-amino C(sp3)–H bonds.
The mechanistic paradigm uniquely separates the activation of both coupling partners from the stereodetermining cross-coupling, thereby circumventing the traditional challenges associated with enantioselective radical-radical recombination. In this dual catalytic system, the excited photocatalyst simultaneously oxidizes bromide (dissociated from NiBr2·glyme) to generate an electrophilic bromine radical for HAT, and reduces the RAE to produce an alkyl radical via decarboxylation, maintaining redox neutrality. Simultaneously, the chiral nickel catalyst operates in concert with the photoredox cycle: Ni(I) selectively captures the
In a significant advance beyond amide-based substrates, our group recently addressed the long-standing challenge of direct enantioselective C(sp3)–H functionalization of N-alkyl anilines, Lewis-basic amines bearing free N–H bonds that are notoriously difficult to engage in transition-metal catalysis due to their propensity for catalyst deactivation (Figure 21)[79]. This work introduces a conceptually distinct metallaphotoredox platform that enables the first direct enantioselective α-C(sp3)–H (hetero)arylation of diverse N-alkyl anilines, providing modular access to enantioenriched α-aryl amines, a structural motif prevalent in pharmaceuticals yet previously inaccessible via asymmetric C–H RCC. The method exhibits exceptional substrate scope, accommodating diverse cyclic and acyclic N-alkyl anilines, a wide array of electronically varied (hetero)aryl bromides (including challenging electron-rich arenes), and complex bioactive molecules, delivering α-arylated products in good yields with excellent enantioselectivities (up to 97% ee) and site selectivity. The synthetic utility was compellingly demonstrated by streamlining the synthesis of several therapeutically relevant glucagon receptor antagonists, reducing multi-step sequences involving chiral resolution to concise two-step protocols with retained stereochemical integrity.
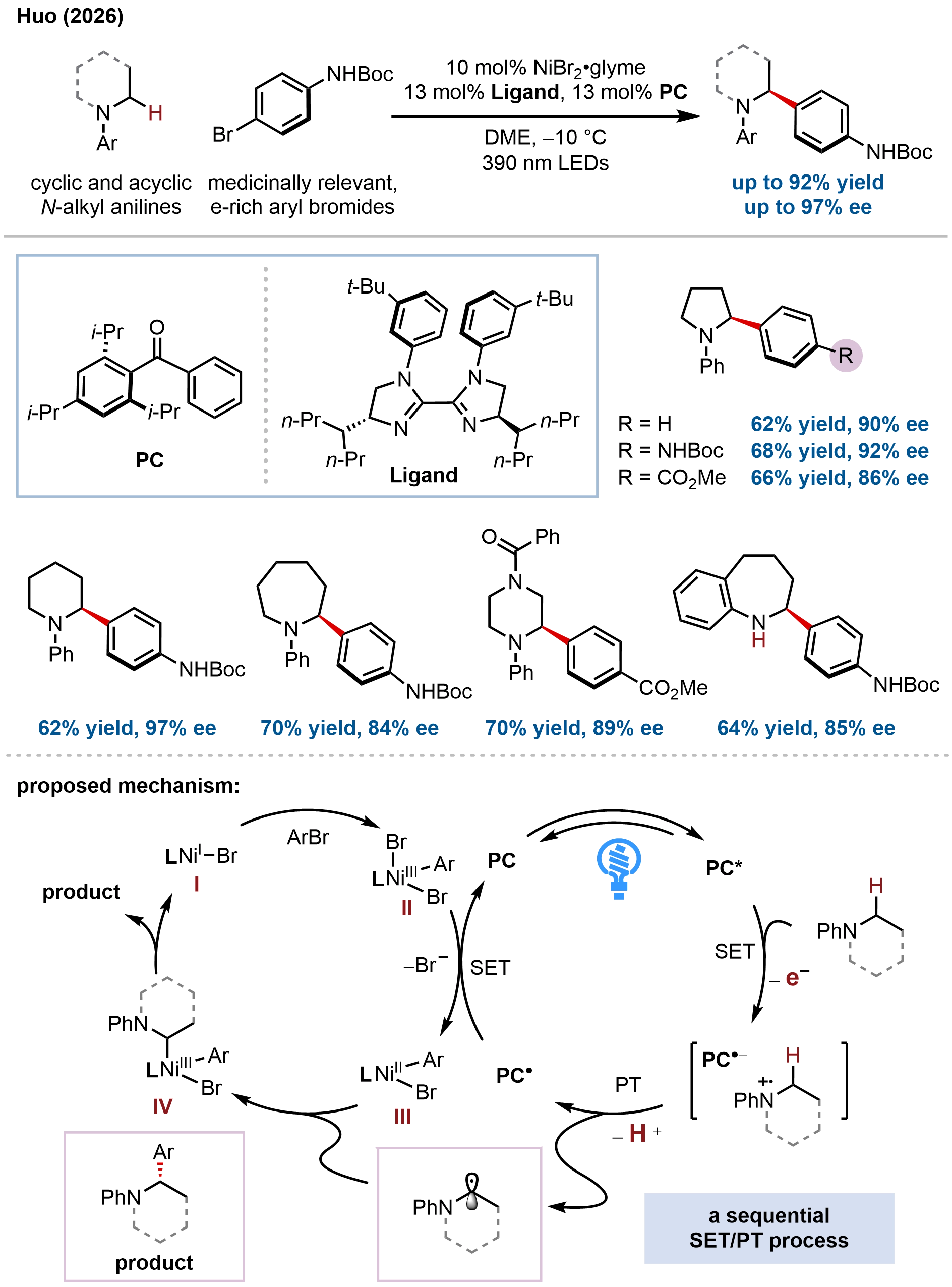
Figure 21. Direct enantioselective C(sp3)–H coupling of N-alkyl anilines via sequential SET/PT, overcoming challenges associated with Lewis-basic amine substrates.
The key innovation lies in a dual catalytic design that uniquely combines a sterically hindered aryl ketone photocatalyst with a chiral bis(imidazoline)/nickel catalyst, operating via a sequential SET and proton transfer (PT) mechanism, distinct from the HAT paradigms established in previous work. The sterically encumbered photocatalyst is critical to success: it slows undesired back-electron transfer from the reduced photocatalyst to the aniline radical cation, a well-recognized challenge in oxidative amine functionalization, by stabilizing the ketyl radical anion intermediate. This enables efficient generation of α-anilinoalkyl radicals via SET/PT, while the electron-rich chiral bis(imidazoline) ligand facilitates oxidative addition to electron-rich aryl bromides, a persistent challenge in metallaphotoredox catalysis. Mechanistic studies, including radical trapping, stoichiometric organometallic experiments with an independently synthesized Ni(II) aryl bromide complex, and cyclic voltammetry, support a Ni(I)/Ni(III) catalytic cycle wherein oxidative addition precedes radical capture, followed by reductive elimination to forge the C(sp3)–C(sp2) bond with high enantiocontrol. This work establishes a distinct platform for enantioselective functionalization of Lewis-basic amines and expands the repertoire of metallaphotoredox catalysis to underexplored substrate classes.
In terms of alkynylation, Zhang, Qi, Guo, and coworkers recently developed an elegant, photoinduced copper-catalyzed enantioselective α-C(sp3)–H alkynylation of cyclic amines via an intramolecular 1,5-HAT strategy, providing modular access to chiral propargyl amines (Figure 22)[80]. The key innovation lies in the cooperative action of a newly developed 2-chloro-substituted bisoxazoline diphenylamine (BOPA) ligand and BINOL as a crucial additive, which together enhance the reduction potential of the photoexcited copper acetylide complex to enable SET to the aryl iodide substrate. The reaction exhibits broad substrate scope, accommodating a diverse range of terminal alkynes (aryl, heteroaryl, alkyl, silyl, enyne) and various cyclic amines, including pyrrolidines, piperidines, azepanes, and notably challenging benzo-fused systems such as tetrahydroquinolines, benzomorpholines, and indolines, delivering the desired propargyl amines in moderate to good yields with excellent enantioselectivities (up to 94% ee) and, in the case of chiral substrates, exceptional diastereoselectivity (> 20:1 dr).
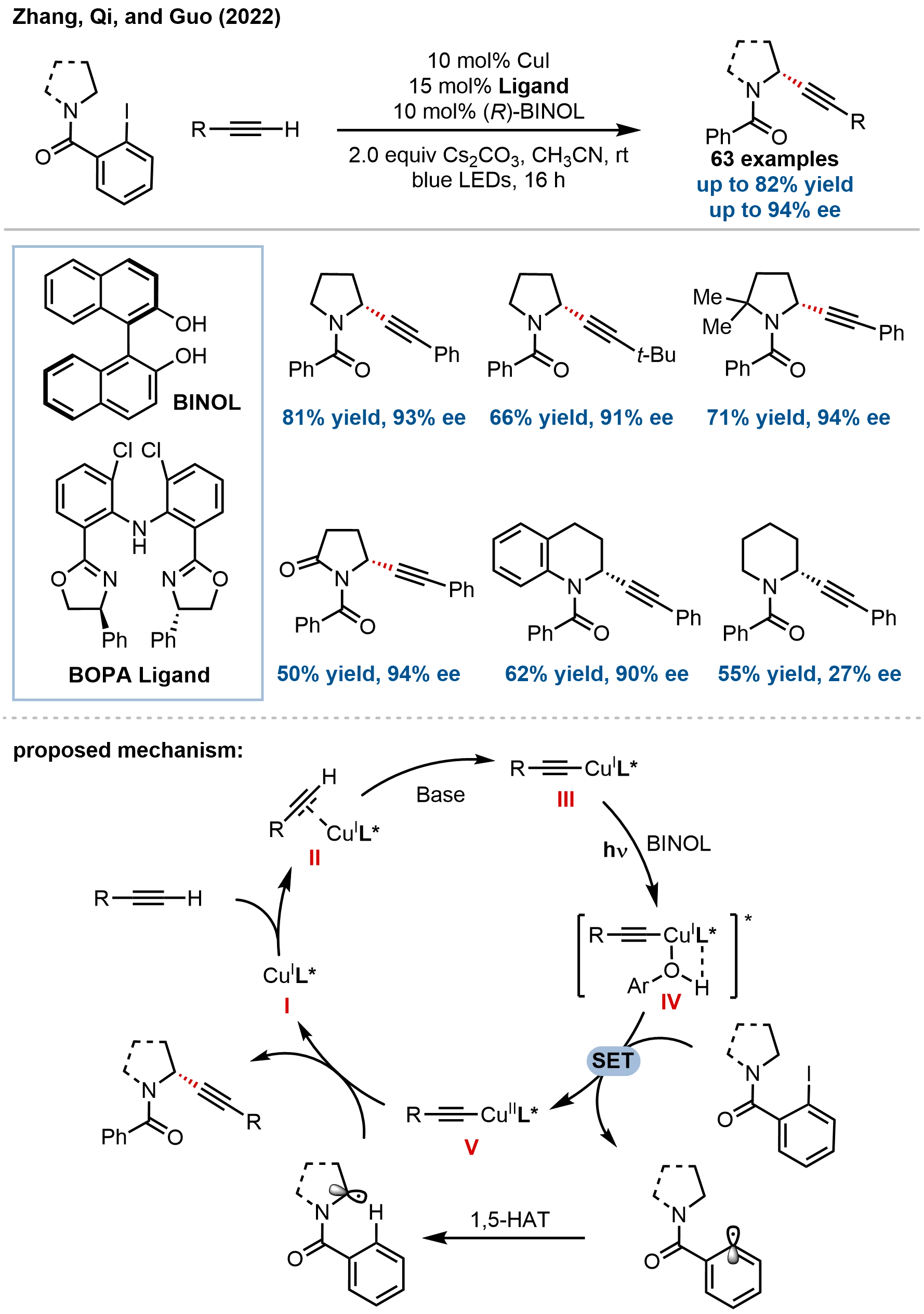
Figure 22. Photoinduced copper-catalyzed enantioselective α-C(sp3)–H alkynylation of cyclic amines via intramolecular 1,5-HAT. HAT: hydrogen atom transfer.
Mechanistic studies elucidated a dual-function copper catalytic cycle wherein the BOPA-coordinated copper(I) acetylide complex serves both as the photosensitizer and the coupling catalyst. The addition of BINOL was found to be critical, forming a
In addition to nitrogen-containing heterocycles, enantioselective C(sp3)–H RCC has been extended to oxygen cycles, with Kong and coworkers developing a general platform for the direct asymmetric functionalization of oxacycles via photo-HAT/nickel dual catalysis (Figure 23)[82]. This method addresses the challenge of achieving enantiocontrol in C(sp3)–H functionalization of substrates that lack both directing groups and significant steric differentiation. The key innovation lies in the synergistic combination of a commercially available diaryl ketone photocatalyst for HAT and a chiral PHOX/nickel catalyst for enantioselective cross-coupling, enabling the enantioselective C(sp3)–H arylation and alkenylation of diverse oxacycles. The reaction exhibits broad substrate scope, accommodating tetrahydrofuran, oxetane, oxane, γ-butyrolactone, and fused systems such as ambroxide, as well as a wide array of (hetero)aryl and alkenyl bromides bearing diverse functional groups, delivering α-functionalized products in good yields with high enantioselectivities (up to 94% ee). The synthetic utility was demonstrated through gram-scale reactions, cross-coupling of complex, biologically active molecules.
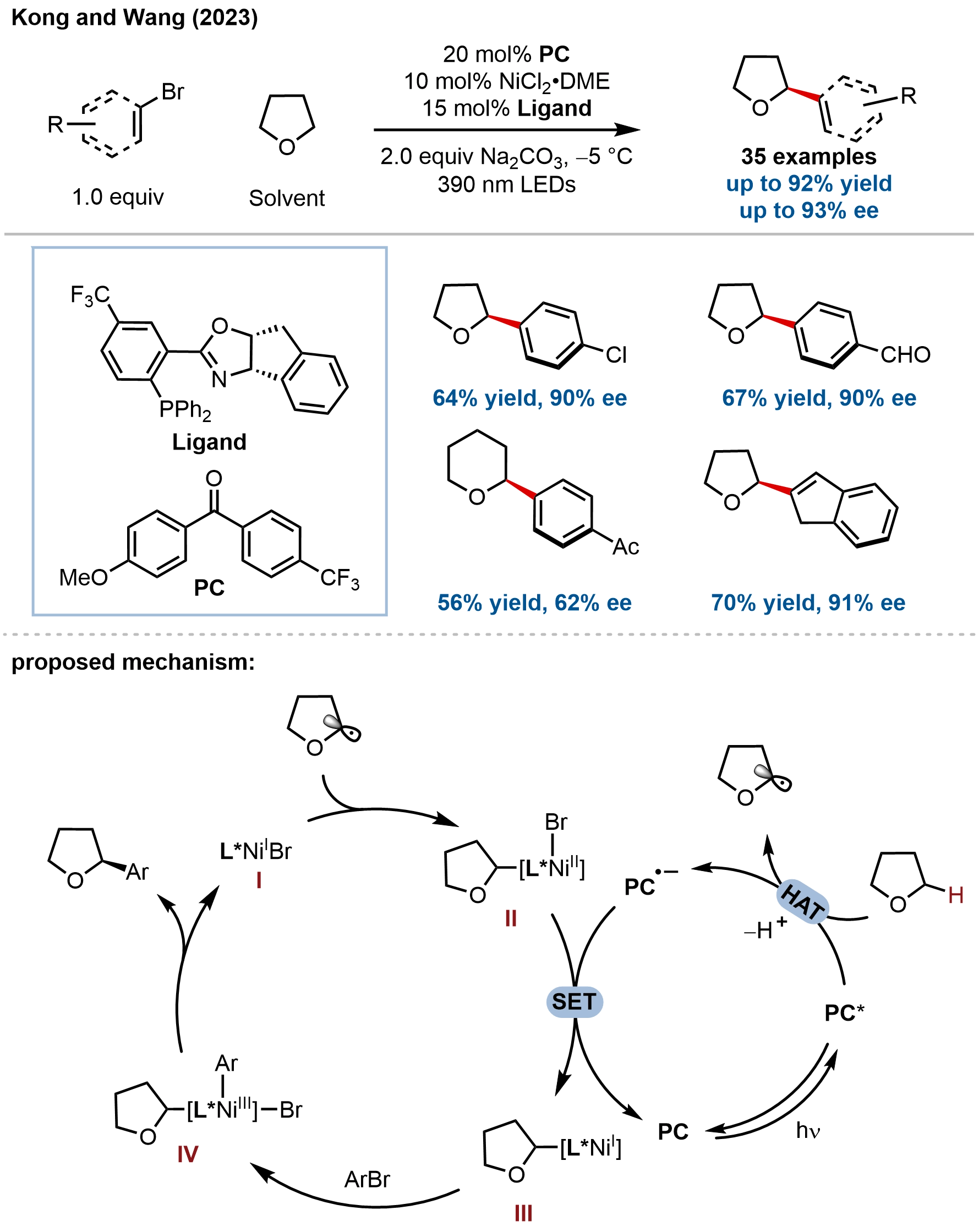
Mechanistic investigations, combining experimental studies and DFT calculations, elucidated a dual catalytic cycle distinct from chlorine-radical-mediated processes (Figure 18). The excited diaryl ketone undergoes triplet-sensitized HAT with the oxacycle to generate an α-oxy radical, while simultaneously reducing the Ni(II) precatalyst to Ni(I) via SET. The Ni(I) species preferentially captures the α-oxy radical over oxidative addition of aryl bromide, forming a key alkyl-Ni(II) intermediate that was independently synthesized and shown to be catalytically competent. DFT calculations identified radical addition to Ni(I) as the enantiodetermining step, with the chiral PHOX ligand creating a well-defined chiral environment wherein weak hydrogen-bonding interactions stabilize the favored transition state, while steric repulsion disfavors the alternative approach. The calculated energy difference (1.0 kcal/mol) aligns with the observed enantioselectivity, and the rate-determining step was identified as oxidative addition of the aryl bromide to the
Building on their photo-HAT/nickel dual catalysis platform, Kong and coworkers recently extended the substrate scope from simple oxacycles to acetonide-protected diols (Figure 24)[83]. The key innovation lies in temporarily masking the diol as an acetonide, which not only suppresses competitive O-arylation but also imparts the necessary selectivity for enantioselective C(sp3)–H cross-coupling. A switch from diaryl ketone to tetrabutylammonium decatungstate (TBADT) as the HAT photocatalyst proved critical, enabling efficient generation of α-oxy radicals from the masked diols. The reaction exhibits broad substrate scope, accommodating diverse (hetero)aryl and alkenyl bromides bearing various functional groups, delivering the corresponding products in good yields with excellent enantioselectivities (up to 97% ee). Furthermore, the method was extended to the diastereoselective synthesis of acetonide-protected diols containing two stereogenic centers (up to > 20:1 dr) via sequential C(sp3)–H arylation of enantioenriched substrates, providing access to 1,2-syn- and 1,3-syn-diaryl diols.
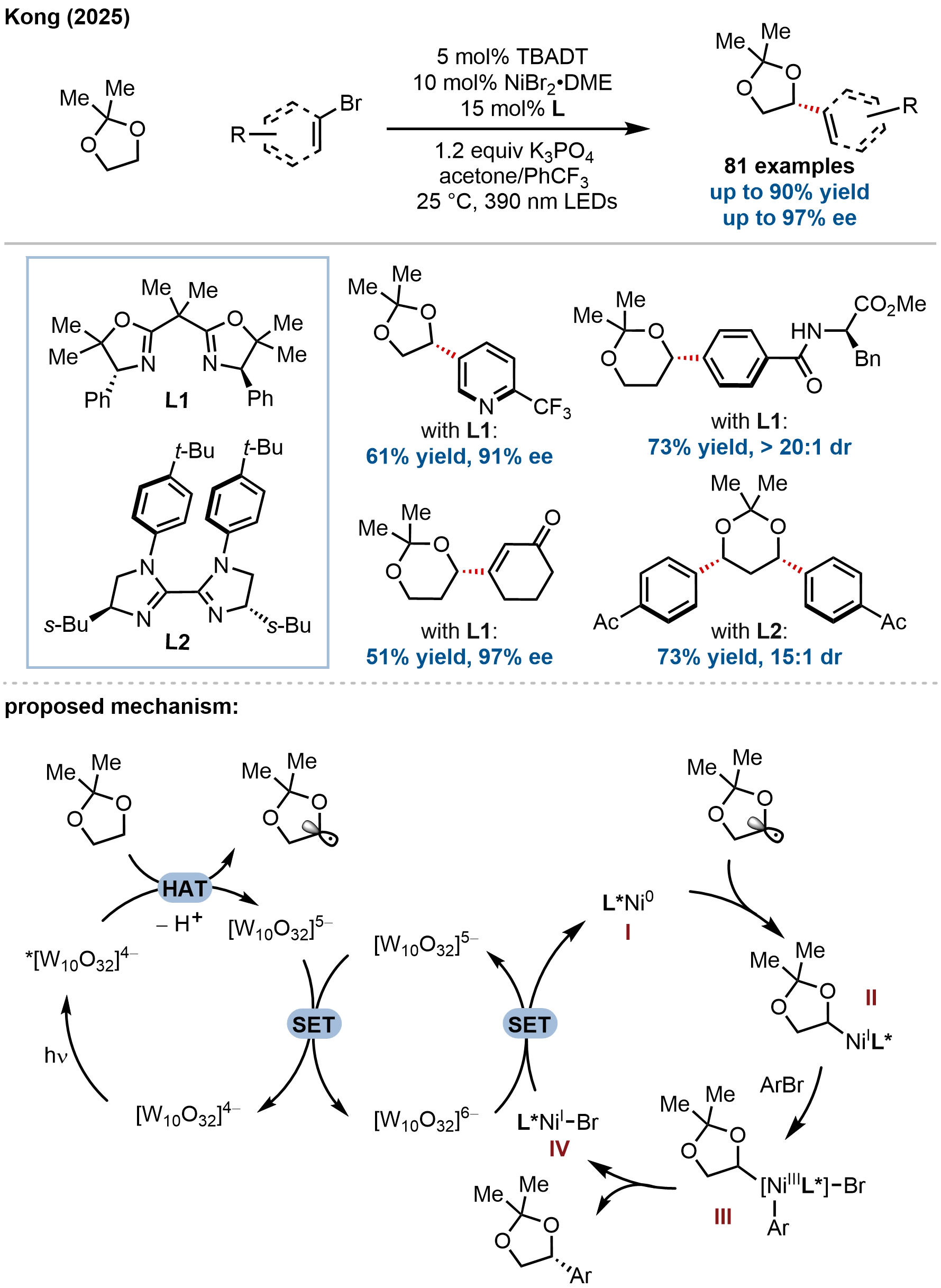
Figure 24. Nickel- and TBADT-catalyzed C(sp3)–H arylation of acetonide-protected diols. For clarity, only one catalytic cycle is depicted. An alternative Ni(0)-Ni(II)-Ni(III)-Ni(I) pathway involving an aryl-Ni(II) complex is omitted. TBADT: tetrabutylammonium decatungstate.
Mechanistic investigations suggested a dual catalytic cycle distinct from their previous ketone-based system. TBADT acts as the HAT photocatalyst, abstracting an α-hydrogen atom from the acetonide-protected diol to generate an α-oxy radical and the reduced decatungstate species. The resulting carbon-centered radical is captured by a Ni(0) or Ni(I) species. Two plausible pathways were considered: (i) radical addition to Ni(0) followed by oxidative addition of aryl bromide, or (ii) oxidative addition of aryl bromide to Ni(0) to form an aryl-Ni(II) intermediate, which then intercepts the radical. Stoichiometric experiments supported the catalytic competence of both alkyl-Ni(II) and aryl-Ni(II) complexes, suggesting that both pathways may operate concurrently. For clarity, only one catalytic cycle is depicted in Figure 24. CV studies confirmed that reduced TBADT possesses sufficient reducing potential to access both Ni(0) and Ni(I) oxidation states.
In 2025, Kong, Ping, and coworkers reported a nickel-catalyzed enantioselective C(sp3)–H alkylation of saturated heterocycles using olefins, representing a significant advance beyond the C(sp3)–C(sp2) cross-couplings discussed above (Figure 25)[84]. This work introduces a distinct thermochemical approach employing an oxidative HAT/reductive cross-coupling strategy, marking a fundamental departure from previously established photoinduced HAT systems. The key innovation lies in the use of dicumyl peroxide as both the HAT reagent and oxidant, generating reactive alkoxyl radicals to cleave α-C(sp³)–H bonds of nitrogen and oxygen heterocycles, while a chiral bisoxazoline/nickel catalyst orchestrates enantioselective coupling with olefins via a Ni–H migratory insertion manifold. The reaction exhibits broad substrate scope, tolerating a diverse array of olefins and saturated N- and
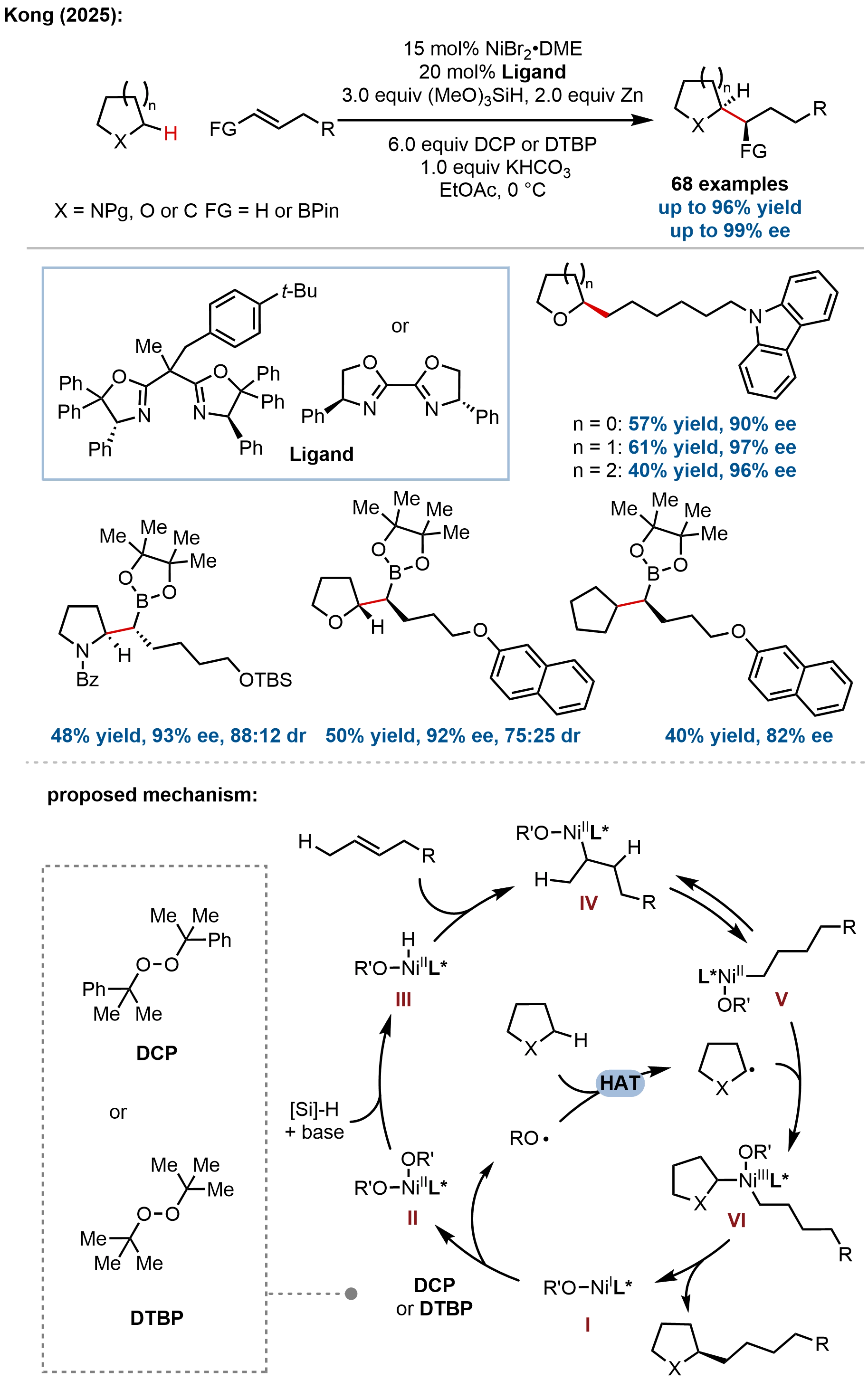
Figure 25. Nickel-catalyzed enantioselective C(sp3)–H alkylation of saturated heterocycles with olefins via oxidative HAT/reductive cross-coupling.
Mechanistic investigations elucidated a dual catalytic cycle distinct from photoredox-mediated processes. The reaction was proposed to proceed through: (i) Ni(I)-mediated peroxide reduction to generate alkoxyl radicals, (ii) HAT to form α-heterocycle radicals,
Very recently, the same group has demonstrated that the peroxide-based oxidative HAT/reductive cross-coupling strategy can also be applied to photochemical enantioselective acylation of diverse α-C(sp3)–H Bonds with aldehydes. In the context of saturated cyclic structures, it provides an alternative approach to enantioenriched cyclic α-amino ketones[85,86]. Concurrent with Kong’s
More recently, Maruoka, Xie, and coworkers developed an enantioselective C(sp3)–C(sp3) cross-coupling between oxacycles and alkylsilyl peroxides via dual photoredox/nickel catalysis, using chlorine radicals generated from the nickel precursor for the HAT process to activate tetrahydrofuran and related cyclic ethers[88]. The key innovation lies in the use of tunable alkylsilyl peroxides as radical precursors, whose reactivity can be modulated by varying the silicon substituents, enabling efficient enantioselective alkyl–alkyl radical coupling with good yields and enantioselectivities.
3. Conclusion and Perspective
Enantioselective RCC has emerged as a powerful and versatile strategy for the construction of enantioenriched saturated (hetero)cyclic skeletons that are difficult or impossible to access via traditional polar disconnection approaches. These methodologies exhibit excellent complementarity in both substrate substitution patterns and product architectures, leveraging diverse starting material types and employing either thermochemical or photocatalytic activation modes. This diversity has systematically expanded the methodological toolkit for asymmetric synthesis of functionalized saturated ring systems.
In terms of structural diversity, current methods have successfully encompassed a broad range of saturated ring systems, including small rings (three-membered), common rings (five- and six-membered), medium rings (seven- and eight-membered), and rigid bridged polycyclic architectures. Regarding heteroatom incorporation, these strategies extend beyond common nitrogen and oxygen heterocycles to include heterocyclic systems containing third-period and heavier main-group elements (sulfur, phosphorus, silicon, germanium), demonstrating excellent functional group tolerance and broad substrate scope. These transformations generally proceed under mild reaction conditions with high levels of enantiocontrol, providing reliable and practical tools for the efficient construction of complex chiral cyclic structures.
Despite this significant progress, the field of enantioselective RCC for saturated (hetero)cycle construction remains in its early stages, and several fundamental challenges persist. First, extending the methodology to macrocycles (≥ 9-membered rings) remains largely unexplored and represents a significant synthetic challenge due to entropic and conformational factors. Second, current methods predominantly focus on C(sp3)–C(sp2) bond formation, and expanding the scope to encompass the more challenging enantioselective C(sp3)–C(sp3) and C(sp3)–heteroatom cross-couplings would substantially broaden the synthetic utility of these approaches. Third, developing enantioselective three-component RCC involving diverse olefin partners would enable rapid assembly of molecular complexity and provide enhanced modularity. Fourth, the field is currently dominated by nickel and cobalt catalysis, and exploration of alternative transition metals (e.g., copper, iron, titanium) may unlock new reactivity patterns and mechanistic pathways. Fifth, discovering new mechanistic paradigms for direct enantioselective C(sp3)–H RCC beyond substrates containing α-heteroatoms would dramatically expand the substrate scope and practical applicability. Finally, conducting comprehensive mechanistic studies, including detailed kinetic analyses, spectroscopic investigations, and computational studies, is essential for rational catalyst design and reaction optimization. Addressing these challenges will likely catalyze transformative advances in the field, potentially revolutionizing access to enantioenriched (hetero)cyclic architectures with far-reaching applications in pharmaceutical discovery, agrochemical development, materials science, and chemical biology.
Acknowledgements
Deepseek was used for language refinement during preparation of the initial manuscript. The authors take full responsibility for the accuracy, originality, and integrity of all contents in the manuscript.
Authors contribution
Luo X, He G, Lin X: Conceptualization, methodology, writing-original draft.
Huang K, Xu Z: Writing-review & editing.
Huo H: Supervision, conceptualization, writing-review & editing.
Conflicts of interest
The authors declare no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
The work was supported by the National Key R&D Program of China (Grant Nos. 2023YFA1507202 and 2021YFA1502500), National Natural Science Foundation of China (Grant No. 22471228), Fundamental Research Funds for the Central Universities (Grant No. 20720240125), and the Innovative Research Fund for the Top Students Program in Chemistry.
Copyright
© The Author(s) 2026.
References
-
2. Lovering F. Escape from flatland 2: Complexity and promiscuity. MedChemComm. 2013;4(3):515.[DOI]
-
4. Jiang JK, Shen M, Thomas CJ, Boxer MB. Chiral kinase inhibitors. Curr Top Med Chem. 2011;11(7):800-809.[DOI]
-
5. Coleman PJ, Schreier JD, Cox CD, Breslin MJ, Whitman DB, Bogusky MJ, et al. Discovery of [(2R,5R)-5-{[(5-fluoropyridin-2-yl)oxy]methyl}-2-methylpiperidin-1-yl][5-methyl-2-(pyrimidin-2-yl)phenyl]methanone (MK-6096): A dual orexin receptor antagonist with potent sleep-promoting properties. ChemMedChem. 2012;7(3):415-424.
-
6. Yamamoto T, Matsuda H, Utsumi Y, Hagiwara T, Kanisawa T. Synthesis and odor of optically active rose oxide. Tetrahedron Lett. 2002;43(50):9077-9080.[DOI]
-
7. Wiesenfeldt MP, Nairoukh Z, Dalton T, Glorius F. Selective arene hydrogenation for direct access to saturated carbo- and heterocycles. Angew Chem Int Ed. 2019;58(31):10460-10476.[DOI]
-
8. Liu DH, Pflüger PM, Outlaw A, Lückemeier L, Zhang F, Regan C, et al. Late-stage saturation of drug molecules. J Am Chem Soc. 2024;146(17):11866-11875.[DOI]
-
10. Cabré A, Verdaguer X, Riera A. Recent advances in the enantioselective synthesis of chiral amines via transition metal-catalyzed asymmetric hydrogenation. Chem Rev. 2022;122(1):269-339.[DOI]
-
12. Deng Y, Cheng QQ, Doyle M. Asymmetric [3+3] cycloaddition for heterocycle synthesis. Synlett. 2017;28(14):1695-1706.[DOI]
-
14. Liu YZ, Song H, Zheng C, You SL. Cascade asymmetric dearomative cyclization reactions via transition-metal-catalysis. Nat Synth. 2022;1(3):203-216.[DOI]
-
17. Twilton J, Le C, Zhang P, Shaw MH, Evans RW, MacMillan DWC. The merger of transition metal and photocatalysis. Nat Rev Chem. 2017;1(7):52.[DOI]
-
18. Chan AY, Perry IB, Bissonnette NB, Buksh BF, Edwards GA, Frye LI, et al. Metallaphotoredox: The merger of photoredox and transition metal catalysis. Chem Rev. 2022;122(2):1485-1542.[DOI]
-
23. Li Y, Lu X, Fu Y. Recent advances in cobalt-catalyzed regio- or stereoselective hydrofunctionalization of alkenes and alkynes. CCS Chem. 2024;6(5):1130-1156.[DOI]
-
27. Yang PF, Shu W. Asymmetric alkyl-alkyl cross-coupling enabled by earth-abundant metal-catalyzed hydroalkylations of olefins. Chem Catal. 2023;3(4):100508.[DOI]
-
29. Golden DL, Suh SE, Stahl SS. Radical C(sp3)–H functionalization and cross-coupling reactions. Nat Rev Chem. 2022;6(6):405-427.[DOI]
-
30. Zhang C, Li ZL, Gu QS, Liu XY. Catalytic enantioselective C(sp3)–H functionalization involving radical intermediates. Nat Commun. 2021;12:475.[DOI]
-
31. Mukherjee K, Ben David A, Nikoghosyan H, Hakobyan R, Gevorgyan V. Light-induced transition-metal-catalysed hydrogen atom transfer in organic transformations. Nat Catal. 2025;8(11):1146-1158.[DOI]
-
32. Zhang J, Rueping M. Metallaphotoredox catalysis for sp3 C–H functionalizations through single-electron transfer. Nat Catal. 2024;7(9):963-976.[DOI]
-
34. Bellotti P, Huang HM, Faber T, Glorius F. Photocatalytic late-stage C–H functionalization. Chem Rev. 2023;123(8):4237-4352.[DOI]
-
37. Chen X, Kramer S. Photoinduced transition-metal-catalyzed enantioselective functionalization of non-acidic C(sp3)–H bonds. Chem Catal. 2024;4(1):100854.[DOI]
-
38. Zhang Z, Chen P, Liu G. Copper-catalyzed radical relay in C(sp3)–H functionalization. Chem Soc Rev. 2022;51(5):1640-1658.[DOI]
-
39. Reimann CE, Kim KE, Rand AW, Moghadam FA, Stoltz BM. What is a cross-coupling? An argument for a universal definition. Tetrahedron. 2023;130:133176.[DOI]
-
40. Gentry EC, Knowles RR. Synthetic applications of proton-coupled electron transfer. Acc Chem Res. 2016;49(8):1546-1556.[DOI]
-
41. Proctor RSJ, Colgan AC, Phipps RJ. Exploiting attractive non-covalent interactions for the enantioselective catalysis of reactions involving radical intermediates. Nat Chem. 2020;12(11):990-1004.[DOI]
-
42. Mondal S, Dumur F, Gigmes D, Sibi MP, Bertrand MP, Nechab M. Enantioselective radical reactions using chiral catalysts. Chem Rev. 2022;122(6):5842-5976.[DOI]
-
46. Capacci AG, Malinowski JT, McAlpine NJ, Kuhne J, MacMillan DWC. Direct, enantioselective α-alkylation of aldehydes using simple olefins. Nat Chem. 2017;9(11):1073-1077.[DOI]
-
52. Gao Z, Liu L, Liu JR, Wang W, Yang NY, Tao L, et al. Copper-catalysed synthesis of chiral alkynyl cyclopropanes using enantioconvergent radical cross-coupling of cyclopropyl halides with terminal alkynes. Nat Synth. 2025;4(1):84-96.[DOI]
-
53. Wong THM, Tong X, Fu GC. Nickel-catalyzed enantioconvergent arylation of unactivated cyclic electrophiles: Asymmetric synthesis of
3-substituted pyrrolidines. Angew Chem Int Ed. 2026.[DOI] -
56. Chen C, Guo W, Qiao D, Zhou J, Wang Y, Zhu S. Rapid access to chiral α-alkylated azacycles via NiH-catalyzed asymmetric migratory hydroalkylation of N-heterocyclic alkenes. CCS Chem. 2024;6(4):898-904.[DOI]
-
61. Lavrencic L, Dhawa U, Hu X. Nickel-hydride-catalyzed hydroalkylation of cyclic phosphinates: Generation of enantioenriched phosphorus and carbon stereocenters. ACS Catal. 2025;15(18):16018-16025.[DOI]
-
62. Hu X, Wang C, Yu L, Tong YZ, Li Z, Li Y, et al. Modular construction of α- or β-stereogenic organosilanes and organogermanes via enantioselective alkene hydroalkylation. Nat Synth. 2025;4:1442-1452.[DOI]
-
64. Liu XY, Wang JW, Liu D, Liu J, Zhao C, Chen Y, et al. Cobalt-catalyzed enantioselective hydroalkylation of oxa- or azabicyclic alkenes. J Am Chem Soc. 2025;147(40):36851-36861.[DOI]
-
67. Liu B, Liu D, Niu C, Xia Y, Lu X, Fu Y, et al. Cobalt-catalyzed stereoselective synthesis of chiral gem-difluorocyclopropanes with vicinal stereocenters. Org Chem Front. 2024;11(23):6617-6626.[DOI]
-
68. Ding C, Ren Y, Sun C, Long J, Yin G. Regio- and stereoselective alkylboration of endocyclic olefins enabled by nickel catalysis. J Am Chem Soc. 2021;143(48):20027-20034.[DOI]
-
72. Xu J, Li Z, Xu Y, Shu X, Huo H. Stereodivergent synthesis of both Z- and E-alkenes by photoinduced, Ni-catalyzed enantioselective
C(sp3)–H alkenylation. ACS Catal. 2021;11(21):13567-13574.[DOI] -
76. Huo H, Cheng B, Xu Z. Nickel-catalyzed, bromine-radical-promoted enantioselective C(sp3)–H cross-couplings. Synlett. 2025;36(7):781-787.[DOI]
-
78. Li J, Cheng B, Shu X, Xu Z, Li C, Huo H. Enantioselective alkylation of α-amino C(sp3)−H bonds via photoredox and nickel catalysis. Nat Catal. 2024;7(8):889-899.[DOI]
-
87. Chen S, Xiao J, Mu Y, Tan Y, Li X. Enantioselective radical–radical cross-dehydrogenative coupling of α-amino C(sp3)–H bonds and aldehydes enabled by photoredox and nickel catalysis. ACS Catal. 2026;16(7):7101-7111.[DOI]
-
88. Xie JH, Kato T, Maruoka K. Enantioselective C(sp3)–C(sp3) cross-coupling between oxacycles and alkylsilyl peroxides via a dual photoredox/nickel catalytic system. ACS Catal. 2026;16(6):5466-5473.[DOI]
Copyright
© The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Publisher’s Note
Science Exploration remains a neutral stance on jurisdictional claims in published
maps
and institutional affiliations. The views expressed in this article are solely those
of
the author(s) and do not reflect the opinions of the Editors or the publisher.
Share And Cite



Science Exploration Style
Luo X, He G, Lin X, Huang K, Xu Z, Huo H. Catalytic enantioselective construction of saturated (hetero)cycles via radical cross-coupling. Chiral Chem. 2026;2:202614. https://doi.org/10.70401/cc.2026.0025
Tips
Copy completed.
Submit a Manuscript
Author Instructions
Cite this Article
Article Metrics
0
View
0
Download
Cited
Article Updates

- Abstract
- Keywords
- 1. Introduction
- 2. Catalytic Enantioselective Construction of Saturated (Hetero)cycles via RCC
- 3. Conclusion and Perspective
- Acknowledgements
- Authors contribution
- Conflicts of interest
- Ethical approval
- Consent to participate
- Consent for publication
- Availability of data and materials
- Funding
- References
- Copyright
Science Exploration Style
Luo X, He G, Lin X, Huang K, Xu Z, Huo H. Catalytic enantioselective construction of saturated (hetero)cycles via radical cross-coupling. Chiral Chem. 2026;2:202614. https://doi.org/10.70401/cc.2026.0025
copy
Share Link
copy