Highly active nickel catalysts for enantioselective reductive alkenylation of N-sulfonyl aldimines
Mengxin Zhao
1

,
Lantian Sun
1,#
,
Bo Xiao
2,3,#
,
Jianrong Steve Zhou
1,*
*Correspondence to:
Jianrong Steve Zhou, State Key Laboratory of Chemical Oncogenomics, Shenzhen Key Laboratory of Chemical Genomics, School of Chemical Biology and Biotechnology, Peking University Shenzhen Graduate School, Shenzhen 518055, Guangdong, China.
E-mail: jrzhou@pku.edu.cn
Chiral Chem. 2026;2:202618. 10.70401/cc.2026.0029
Received: April 01, 2026Accepted: June 01, 2026Published: June 01, 2026
Abstract
Chiral nickel complexes of bis(oxazoline)s serve as highly active catalysts for an enantioselective reductive alkenylation of N-sulfonyl and
Graphical Abstract
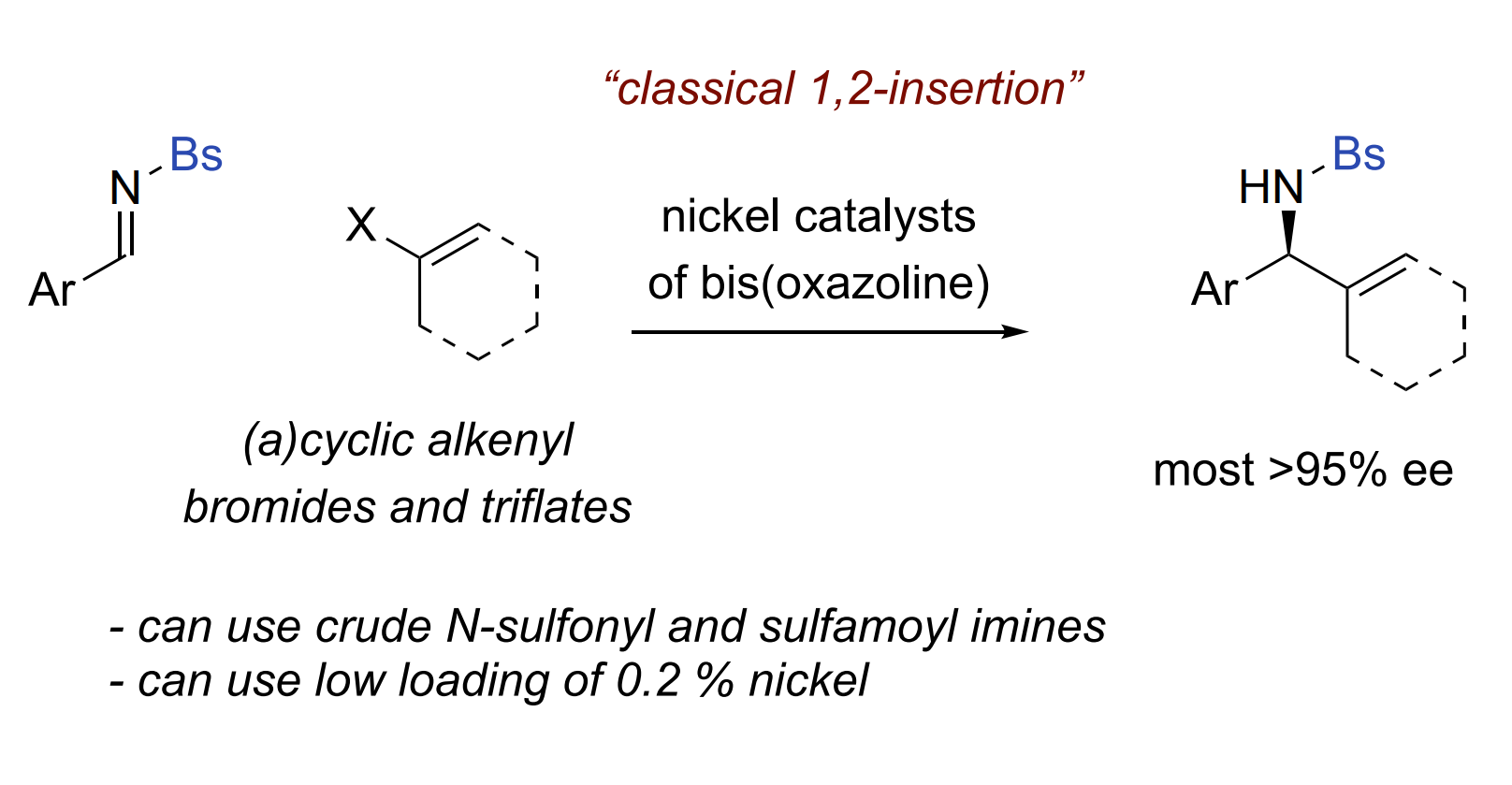
Keywords
Asymmetric catalysis, aldimine, alkenylation, nickel catalysis, reductive addition, chiral alkylamines
1. Introduction
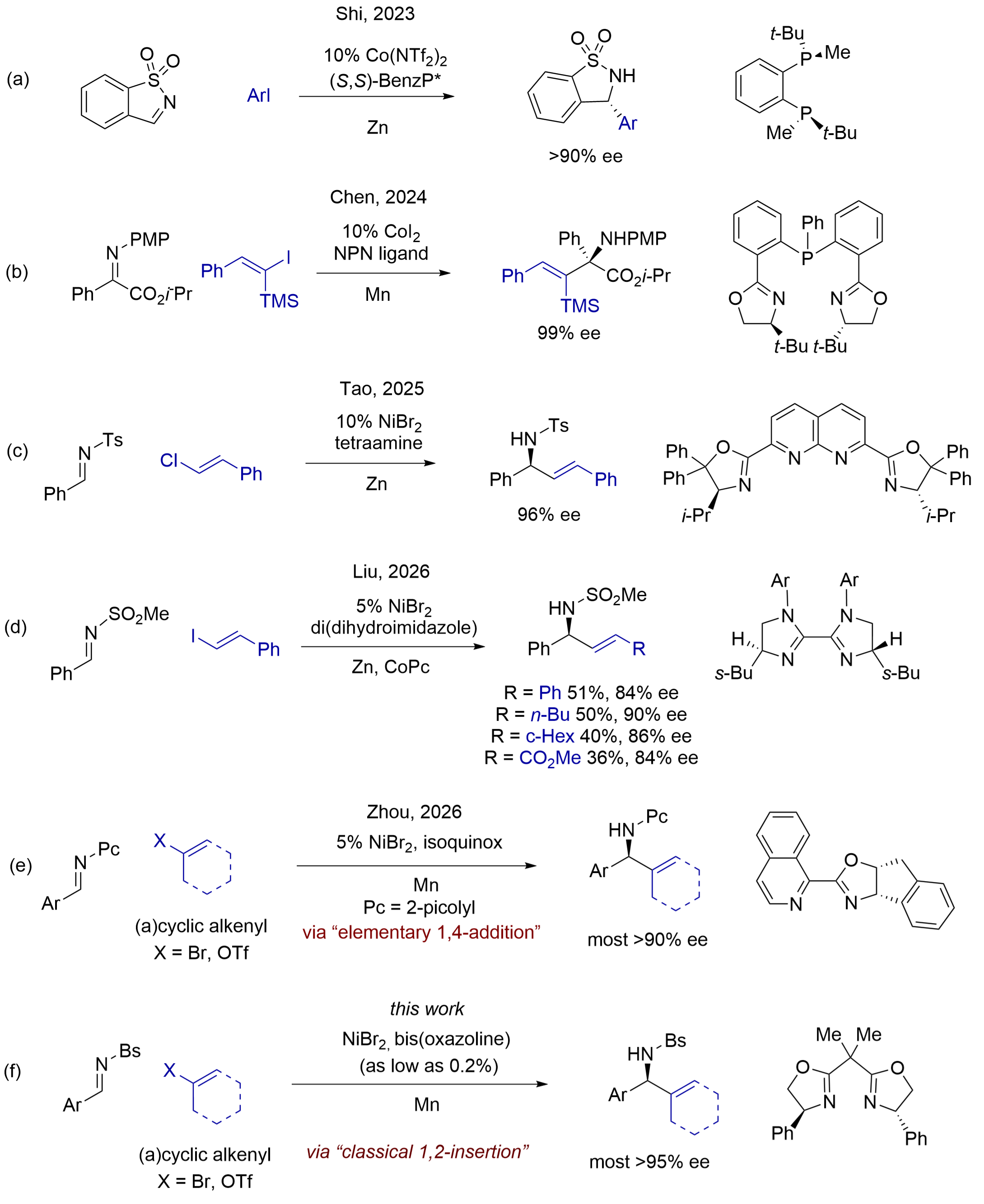
Aliphatic amines are important motifs in small-molecule medicines[1]. For example, chiral benzylic amines are present in cinacalcet, rivastigmine, and ontazolast. Thus, efficient stereoselective methods to prepare these chiral amines have been a longstanding quest among synthetic chemists[2]. Copper-catalyzed asymmetric imine addition of alkenyl metal reagents of Mg, Zn, Al, and Zr is often practiced, but the method is often limited by poor compatibility with sensitive functional groups[3]. Another family of benchtop-stable nucleophiles, alkenyl boronic acids and boronates, can add to various imines when promoted by chiral rhodium complexes[4]. Recently, cobalt- and nickel-catalysts were also reported for enantioselective addition of alkenyl boronic acids to cyclic N-sulfonyl imines[5].
In recent years, researchers have reported cobalt-[6] and nickel-catalyzed[7] enantioselective reductive addition of aryl and alkenyl halides to imines. These reductive methods directly use readily accessible alkenyl halides, sulphonates, and acetates, and avoid the preparation and use of organometallic reagents and organoboron reagents. For example, Shi et al. reported a cobalt-catalyzed asymmetric addition of aryl halides to a special type of activated cyclic aldimines, benzo[d]isothiazole 1,1-dioxides (Figure 1a), which are conformationally locked in (Z)-geometry[8]. Chen and coworkers have reported a cobalt-catalyzed asymmetric addition of alkenyl iodides to α-iminoesters for the formation of unnatural amino acids, and the iminoesters chelated on chiral cobalt complexes for asymmetric alkenyl transfer from the metal center (Figure 1b)[9]. Recently, Tao and coworkers disclosed that a chiral dinickel complex can selectively activate β-arylvinyl chlorides in asymmetric addition to N-tosyl aldimines (Figure 1c), but alkenyl halides having other substituents gave no products[10]. Very recently, Liu and coworkers reported that nickel complexes of bis(dihydroimidazole) promoted efficient asymmetric addition of aryl iodides to N-sulfonyl imines, but the addition of alkenyl iodides led to low-to-moderate yields and < 90% ees (Figure 1d)[11].
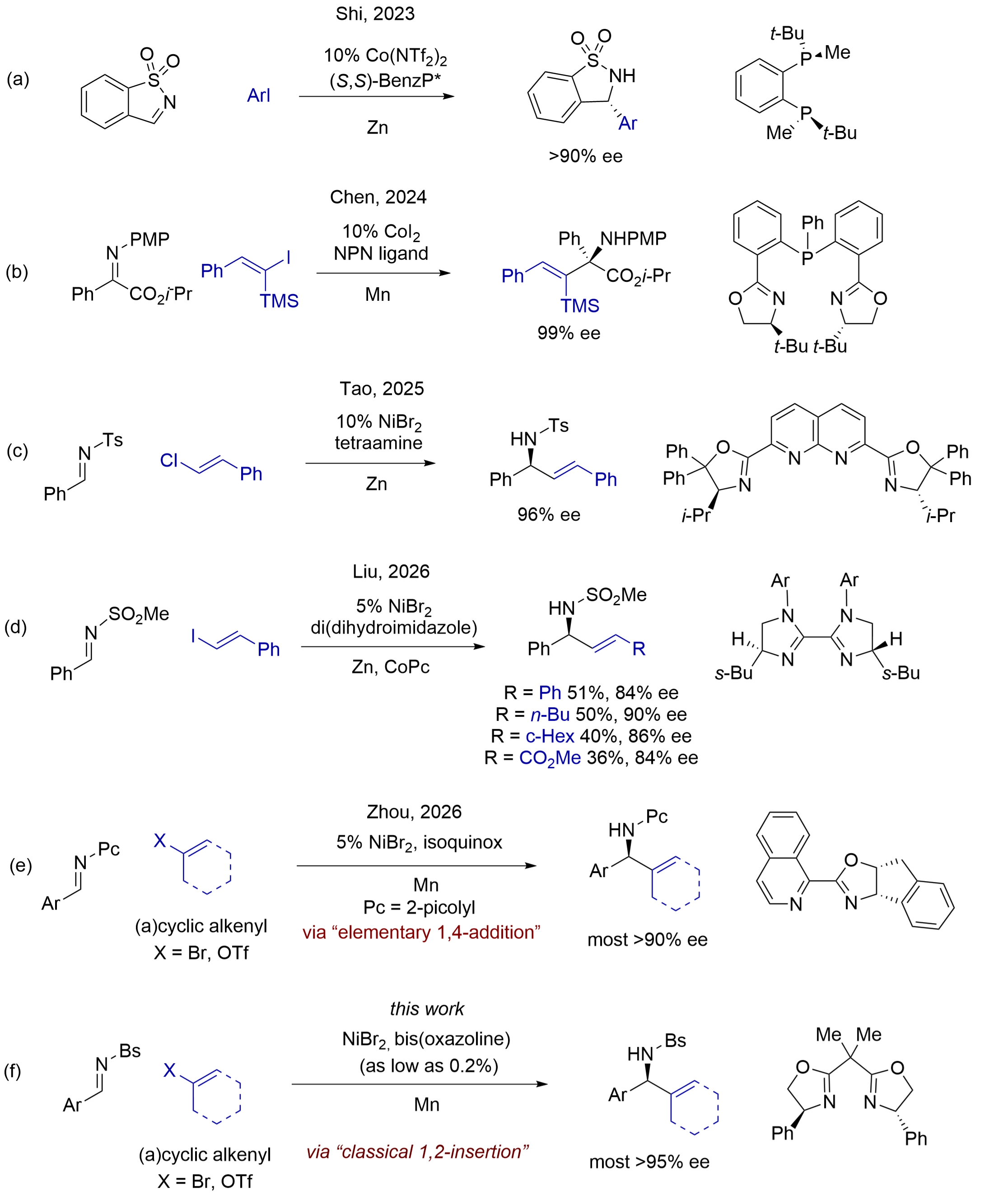
Figure 1. Nickel-catalyzed reductive addition of alkenyl halides and sulfonates to N-sulfonyl and sulfamoyl aldimines.
In 2023, we disclosed a nickel-catalyzed asymmetric addition of aryl halides to N-picolyl aldimines in excellent ees[12]. Recently, we successfully extended the asymmetric addition of alkenyl halides and triflates, having various substitution patterns, to N-picolyl imines (Figure 1e)[13]. These reactions proceed via a mechanism of elementary 1,4-addition on nickel catalysts. We also reported catalytic reductive addition of aryl and alkenyl halides to N-Boc aldimines lately[14].
We report here new catalytic methods for enantioselective alkenylation of aldimines having N-sulfonyl and sulfamoyl groups
2. Experimental Section
A general procedure for catalytic alkenylation: in an argon-filled glove box, NiBr2(DME) (1.5 mg, 0.005 mmol, 5 mol%),
3. Results and Discussion
3.1 Condition optimization
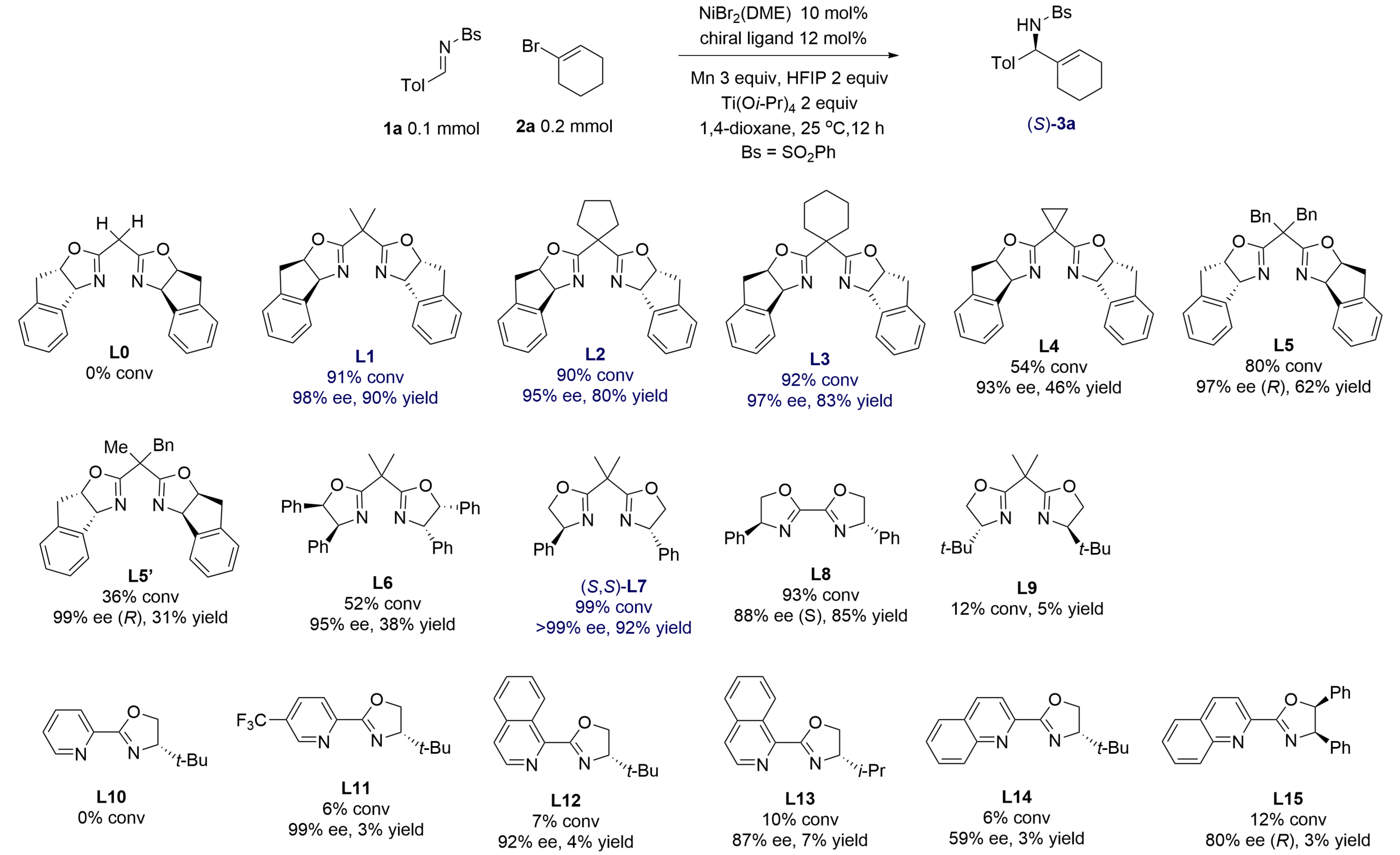
In a study of catalytic reductive alkenylation of aldimines having electron-withdrawing N-sulfonyl groups, we selected
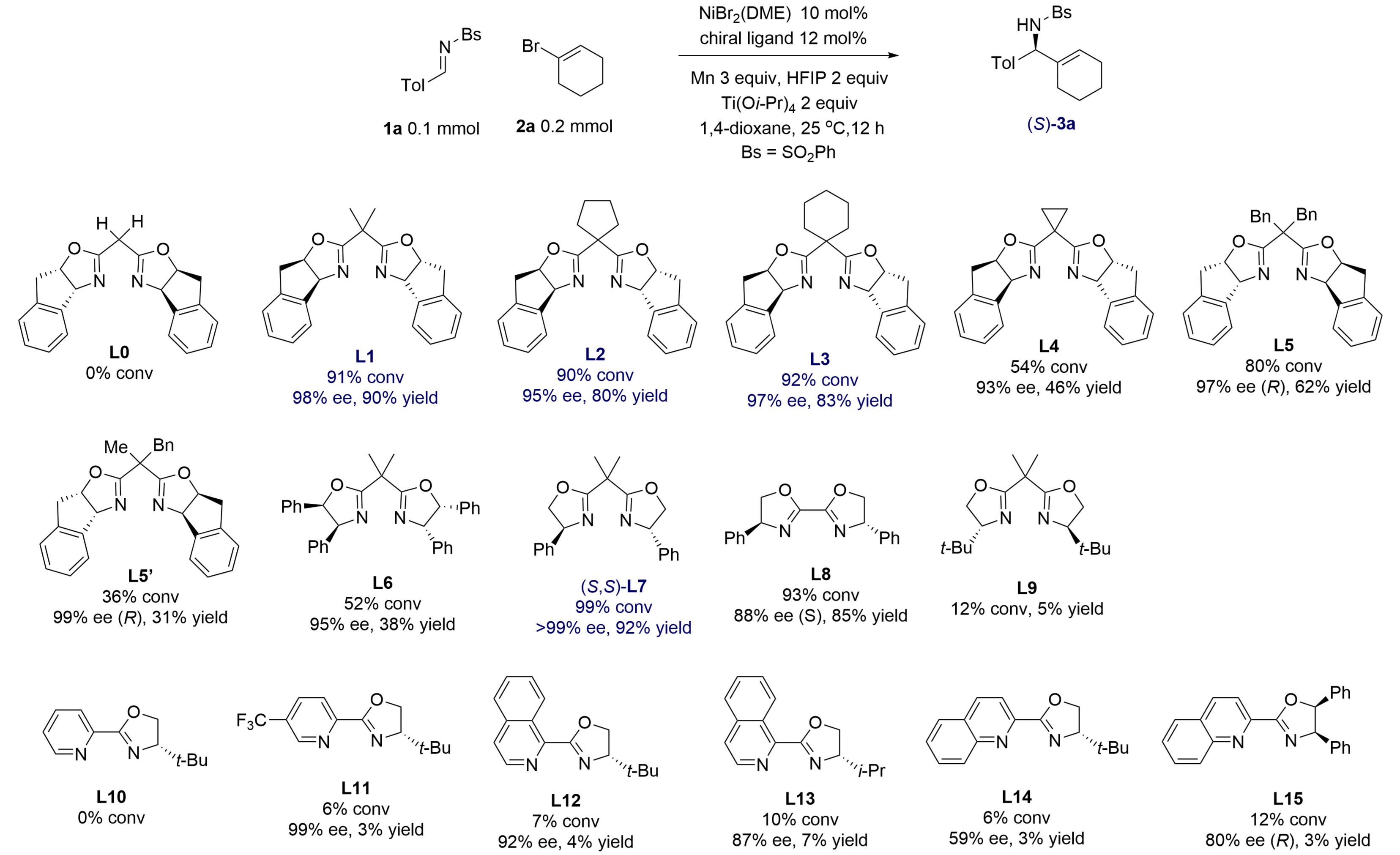
Figure 2. Optimization of ancillary ligands for a model alkenylation of aldimine 1a giving product 3a. Calibrated GC yields (Tol = 4-tolyl). GC: gas chromatography.
Among a series of chiral ligands, bis(oxazoline)s L1-L3, which featured two rigid indane-fused oxazoline rings and 1,1'-allkylene linkers, provided amine 3a in very high ees. For instance, the nickel complex of L1 yielded amine 3a in 98% ee and 90% yield. However, an analogous ligand L0 has a simple methylene linker showed no catalytic activity, which may be caused by a lower binding constant to nickel. Nickel complexes of di(oxazoline) L7 afforded the product in almost perfect enantiopurity (> 99% ee) and 92% yield, so it was chosen for the study of substrate scope. A di(oxazoline) ligand L8 with a conjugated p-system also showed high catalytic activity and gave 88% ee in the model reaction. However, a more hindered t-butyl-substituted ligand L9 did not form an active nickel catalyst. The absolute configuration of amine 3a was determined to be (S) by X-ray crystallography.
Previously, we reported that nickel complexes of chiral pyrox and (iso)quinox types having extensive p-conjugation gave excellent reactivity and > 90% ees in nickel-catalyzed reductive addition of aryl and alkenyl halides to conjugated enones[17], N-pyridyl aldimines[12,18], and N-Boc aldimines[14]. However, these ligands L10-L15 showed very low catalytic activity. Common chiral diphosphines BINAP, Segphos, Difluorphos, and t-butyl-substituted (diphenylphosphino)oxazoline (t-Bu-PHOX) failed to produce active nickel catalysts for the model reaction.
Titanium isopropoxide was an indispensable additive, and its absence led to complete failure of the reaction (Table 1, entry 1). The reaction proceeded well in several ethereal solvents, among which the best results of 92% yield and > 99% ee were obtained in
Table 1. Optimization of conditions for model alkenylation of aldimine 1a (calibrated GC yields).

| Entry | Change of conditions | Conv of 1a (%) | Yield of 3a (%) | Ee (%) |
| 1 | no Ti(Oi-Pr)4 | 32 | 0 | - |
| 2 | 1,4-dioxane (standard) | 99 | 92 | >99 |
| 3 | THF | 90 | 88 | 95 |
| 4 | 2-MeTHF | 91 | 86 | 93 |
| 5 | DME | 84 | 60 | 93 |
| 6 | DCM | 80 | 71 | 90 |
| 7 | DMF | 38 | 0 | - |
| 8 | DMSO | 40 | 0 | - |
| 9 | HFIP 0 equiv | 0 | 0 | - |
| 10 | CF3CH2OH 2 equiv | 3 | 0 | - |
| 11 | t-BuOH 1.5 equiv | 2 | 0 | - |
| 12 | H2O 1.5 equiv | 2 | 0 | - |
GC: gas chromatography; THF: tetrahydrofuran; DME: 1,2-dimethoxyethane; DCM: dichloromethane; DMF: N,N-dimethylformamide; DMSO: dimethyl sulfoxide;
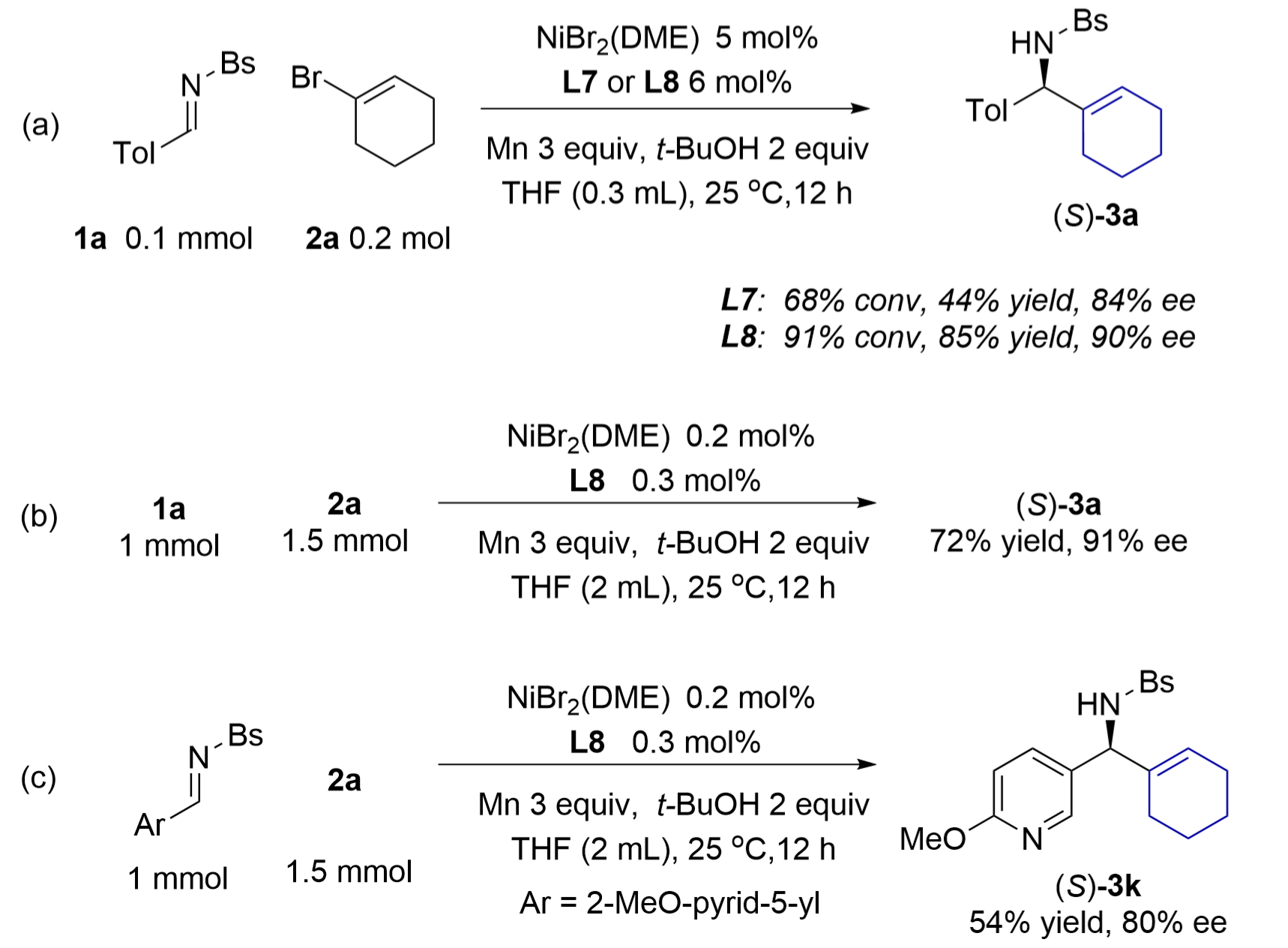
In the model reaction under the condition using 5 mol% nickel catalyst of bis(oxazoline) L7 and Ti(OR)4, a long induction period of hours was detected before initiating the catalytic process, and it was difficult to reduce the loading of the nickel catalyst. We found that another set of conditions without titanium alkoxide allowed the reactions to scale up with low loadings of nickel (Figure 3a). In the model reaction of 1a in THF, a 5 mol% nickel catalyst of L7 gave 68% conversion of 1a and amine 3a in 44% yield and 84% ee. Switching the chiral ligand to di(oxazoline) L8 improved the result to 85% yield and 90% ee. This new procedure was easily scaled up to 1 mmol using 0.2 mol% nickel catalyst of L8. Thus, 1 mmol of imine 1a provided amine 3a in 72% yield and 91% ee, while a methoxypyridyl imine gave amine 3k in 54% yield and 80% ee (Figure 3b,c). However, the ees of these two products were slightly lower than those obtained under the conditions using titanium alkoxide.
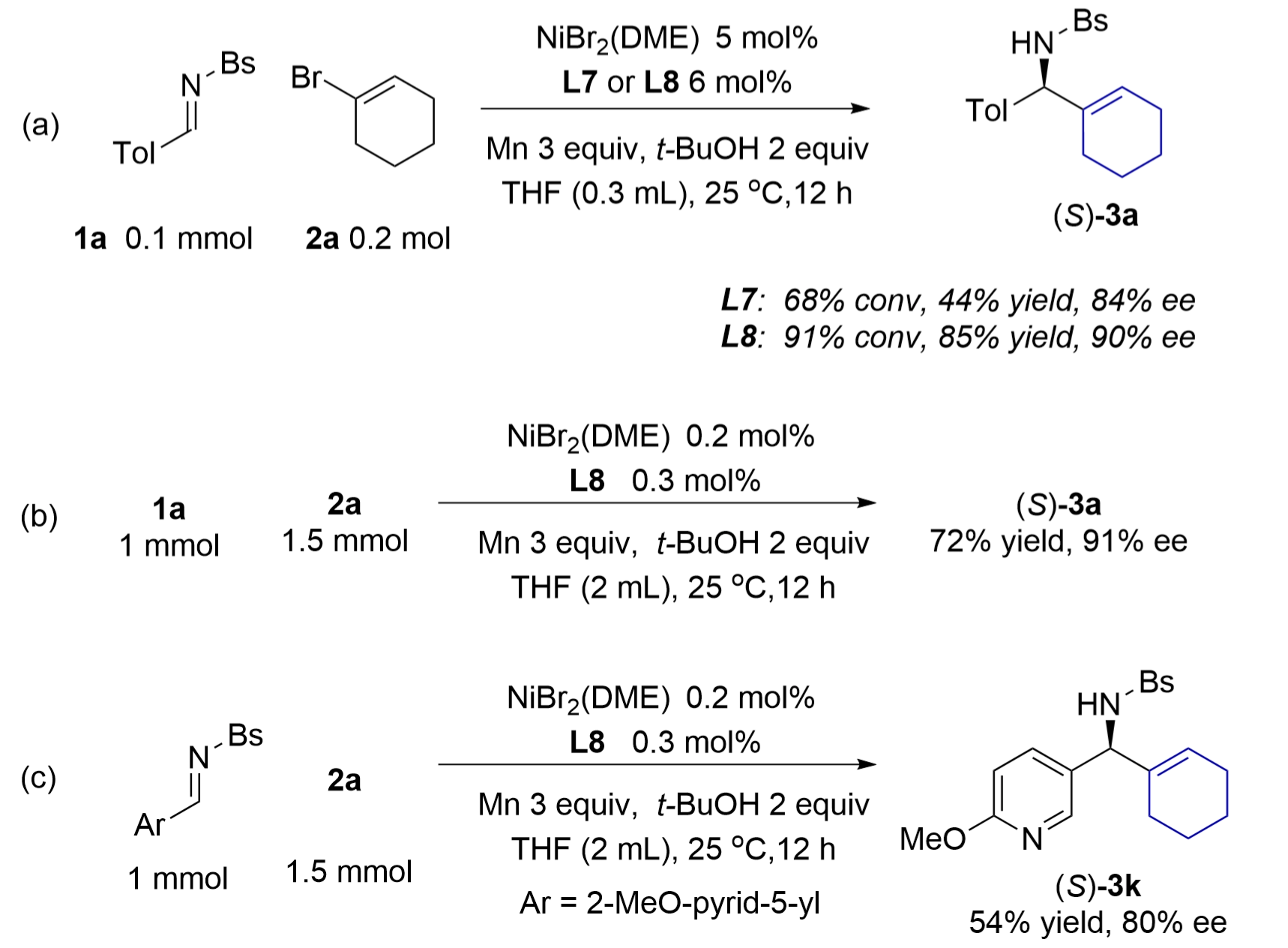
Figure 3. Catalytic alkenylation of aldimine 1a under conditions without titanium isopropoxide: (a) A reaction on a 0.1 mmol scale using 5% nickel catalyst; (b-c) Reactions on a 1 mmol scale using 0.2% nickel catalyst. DME: 1,2-dimethoxyethane; THF: tetrahydrofuran.
3.2 The reaction scope
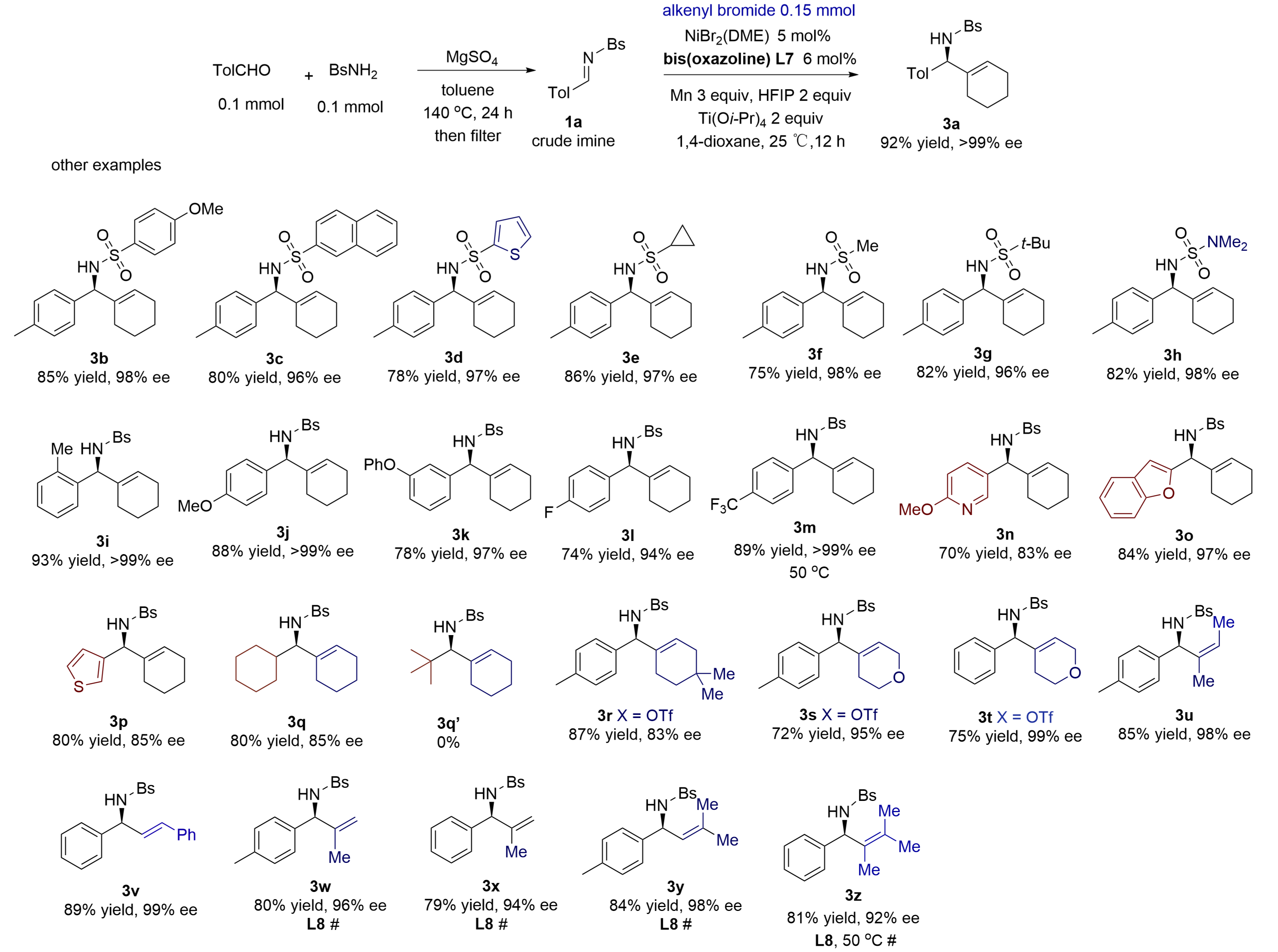
The crude N-sulfonyl aldimines were easily prepared via heating aldehydes and sulfonamides with MgSO4 in toluene in a sealed tube at 140 °C (external temperature in an oil bath). Crude aldimines were obtained after filtration and concentration, and they were used directly in reductive alkenylation without further purification. The results are summarized in Figure 4. Various substituents can be present on the N-sulfonyl groups of aldimines (3a-3f), including anisyl, naphthyl, thienyl, cyclopropyl, methyl, and t-butyl. Among them, the t-butylsulfonyl group in benzylic amine 3g is easily removable by the treatment of HCl, while the N-2-thiophenesulfonyl group is susceptible to reductive cleavage by magnesium dust in methanol. An N-carbamoyl aldimine reacted to provide amine 3h in 82% yield and 95% ee. The N-carbamate group is removable by acids[21].
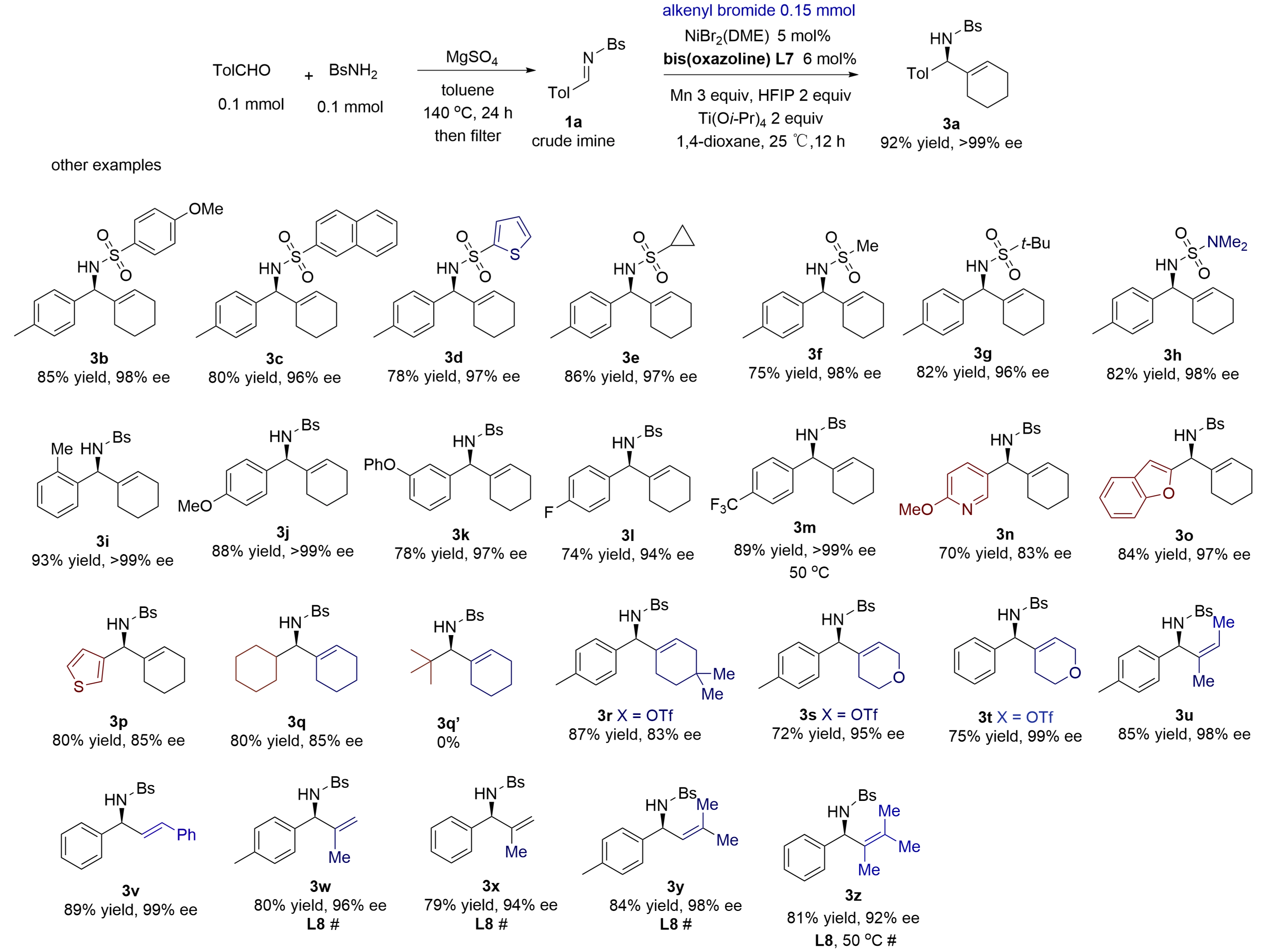
Figure 4. One-pot synthesis and alkenylation of N-sulfonyl aldimines using alkenyl bromides and triflates. Isolated yields from reactions of 0.2 mmol of aldehydes and
MgSO4 efficiently promoted the condensation of BsNH2 with various aldehydes. The resulting crude imines were directly used in catalytic alkenylation, producing chiral benzylic amines in high yields and excellent ees. A range of aromatic imines having
N-Bs-cyclohexylimine was converted to amine 3q in 80% yield and 85% ee. However, N-Bs-t-butyl aldimine completely failed to give amine 3q' due to severe steric effects. The condensation of primary aliphatic aldehydes with Bs-amine failed to give clean samples of N-Bs-aldimines in the presence of MgSO4, Ti(Oi-Pr)4, or 4Å molecular sieve, owing to side reactions from the aldol reaction. The catalytic alkenylation failed completely when applied to the N-sulfonyl ketimine of acetophenone.
A diverse set of alkenyl bromides and triflates possessing different steric and electronic properties participated in reductive alkenylation in excellent ees under the standard conditions. The examples of triflates included 1-cyclohexenyl bromide having
Ligand re-optimization was often required to achieve high levels of ees for asymmetric transfer of other alkenyls having other α- or cis-β-substituents. Thus, under the conditions using ligand L8 without titanium isopropoxide, several acyclic alkenyl bromides gave good yields and excellent 92-98% ees, including 2-propenyl bromide (3w and 3x), 1-isobutenyl bromide (3y) and trimethylvinyl bromide (3z). For example, hindered trimethylvinyl bromide provided amine 3z in 81% yield and 92% ee under the condition without titanium isopropoxide at 50 oC. The standard conditions using the titanium alkoxide gave low yields, in comparison. Cyclohexenyl chloride did not react, while trans-β-styrenyl iodide gave a low yield of the amine due to fast homocoupling.
3.3 Synthetic applications
The alkenyl groups in the chiral amines are easily converted to aliphatic groups. These alkylated compounds may not be easily prepared from catalytic asymmetric alkylation reactions otherwise (Figure 5). For example, Pd/C-catalyzed hydrogenation with additive Et3N[22] efficiently hydrogenated the cyclohexenyl ring in 3a, affording chiral benzylic amine 4a in almost qualitative yield. Notably, the N-benzenesulfonyl group of 3a was reductively cleaved by SmI2 to provide unprotected benzylic amine 4b in 80%

{kind=link}
{kind=link}
{kind=link}
{kind=link}
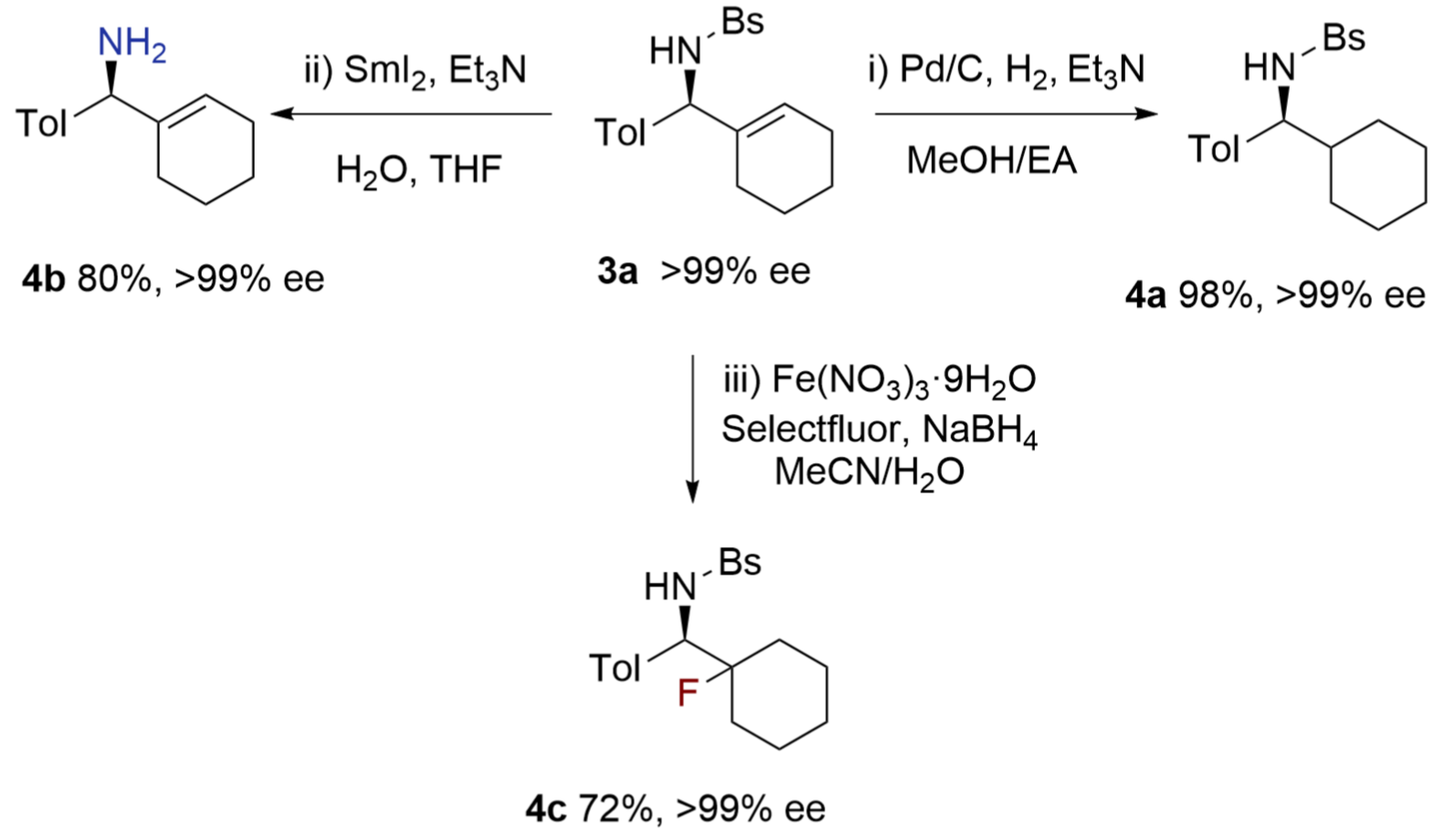{kind=link}
We envisioned that catalytic hydrogenation of olefins and arenes in these products would allow access to chiral methylamines having two aliphatic substituents, which are difficult to prepare otherwise[27]. Thus, N-Bs-amine 3u was converted to N-Cbz-amine 4d after SmI2 deprotection, full hydrogenation of the olefin and arene by Adom’s catalyst[28] and N-Cbz protection (Figure 6a). Similarly, amines 3a and 3q were saturated by Adom’s catalyst, affording N-Cbz-amines 4e (dr 1.4:1) and 4f in good yields (Figure 6b,c). The
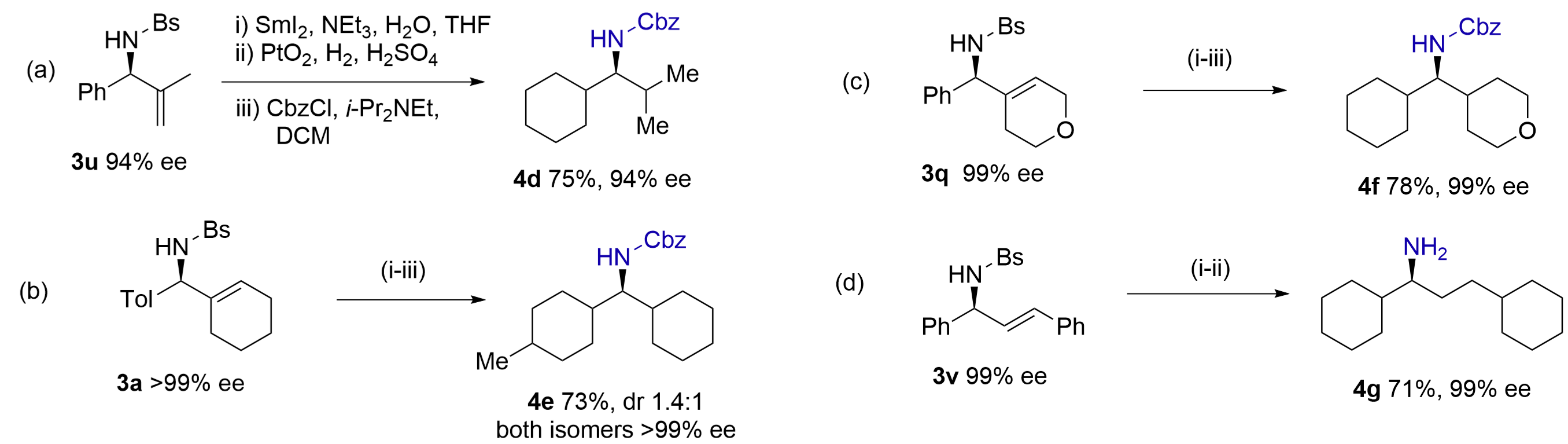{kind=link}
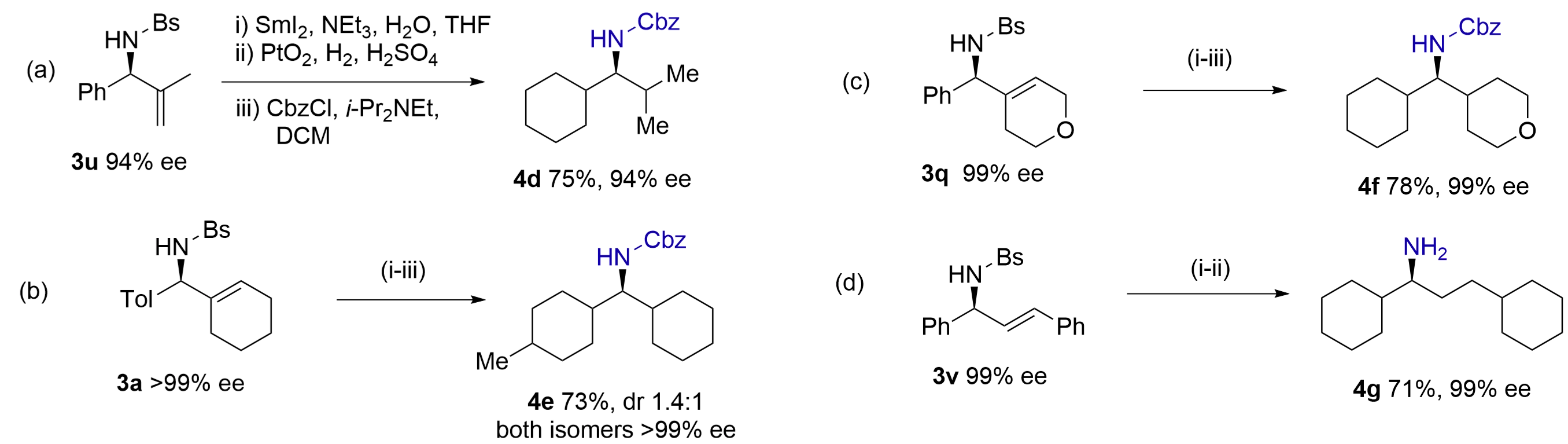
Figure 6. Catalytic hydrogenation of olefins and arenes to access methylamines having α,α-dialkyl substituents in high ees.
3.4 Computational mechanistic studies
For the alkenyl insertion of the neutral complex (1-cyclohexenyl)nickel of ligand L7 into (E)-N-Bs-imine 1a, we have identified 12 ground-state conformers and 12 transition states (see the Supplementary materials). Importantly, the C2-symmetric ligand bis(oxazoline) L7, upon chelation to nickel, forms an unsymmetrical puckered ring. It is evident that one central methyl group of L7 points up vertically while the other points back in the coordination plane, as shown in Figure 7. Consequently, the two phenyl substituents of L7 are dispositioned in different orientations in space, the right phenyl ring is pointing downward vertically while the left phenyl is tilted close off the coordination plane.
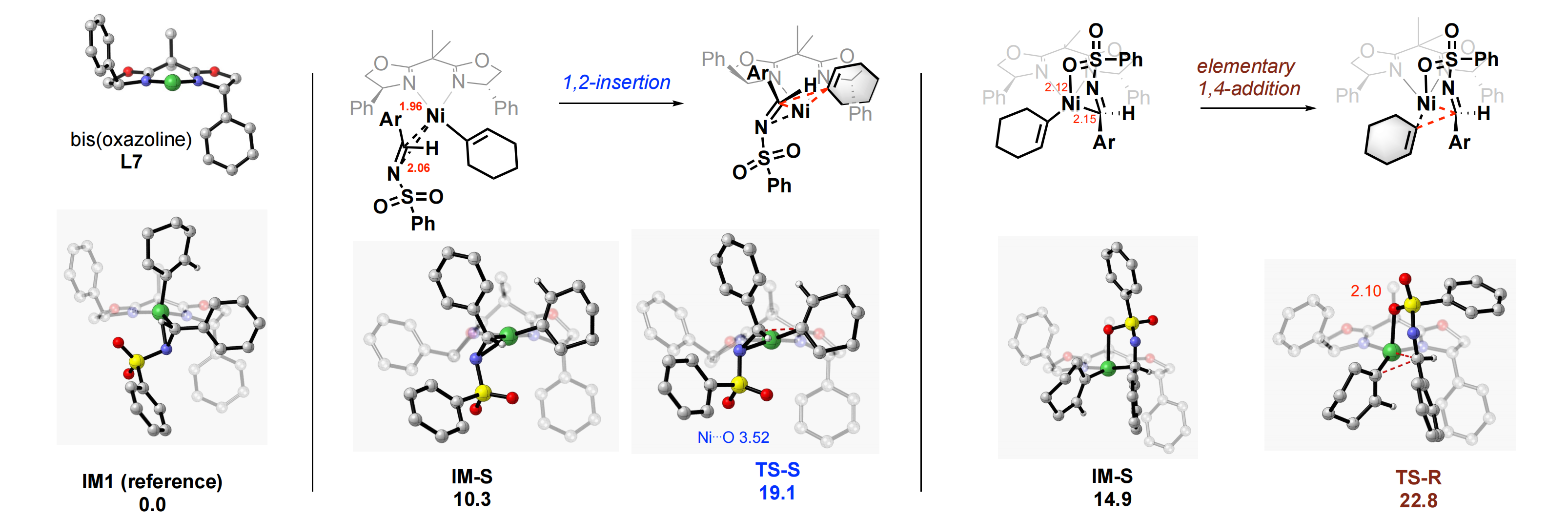{kind=link}
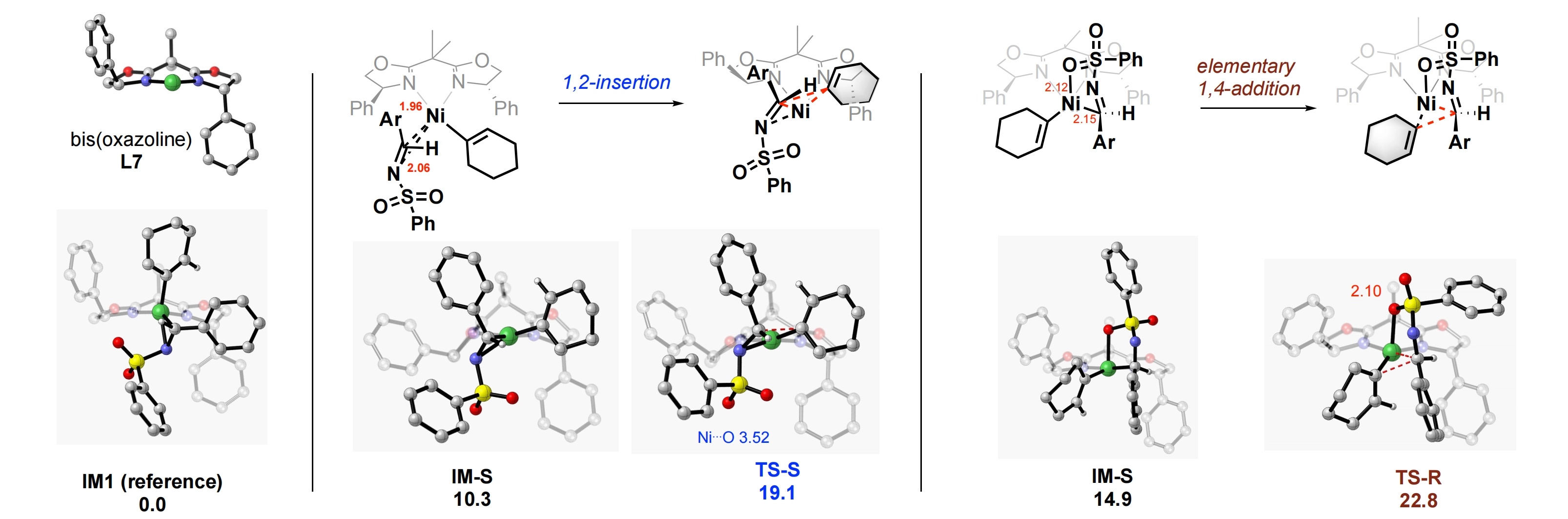
Figure 7. Calculated ground states and corresponding transition structures of the lowest-energies for asymmetric alkenyl addition of neutral complex of L7 (cyclohexenyl)nickel to (E)-N-benzenesulfonyl imine 1a.
We chose to use the lowest-energy ground state IM1 having an h2-imine coordination as the reference structure, for comparison of energies of different transition states. The two lowest-energy TSs leading to the major (S)-isomer and minor (R)-isomer are shown in Figure 7. The Gibbs free energy of TS-S is 19.1 kcalmol-1, which is 3.7 kcalmol-1 lower than that of TS-R. It is consistent with the almost perfect enantioinduction observed experimentally using the nickel catalyst. Notably, the insertion barrier of TS-S from its own ground-state IM-S is only 8.8 kcalmol-1. The 4-membered core structure of TS-S is close to a planar classical 1,2-insertion. The nickel(I) ion is 3.52 Å away from the closest sulfonyl oxygen, way beyond the sum of the van der Waas radii of the two atom/ion. A closer examination of structure TS-R leading to the minor isomer, however, reveals a nonplanar 6-membered cyclic TS featuring a short Ni-O bond distance of 2.10 Å, which is reminiscent of a mechanism of "elementary 1,4-addition". Other insertion structures can be found in the SI.
4. Conclusion
We report here nickel-catalyzed asymmetric reductive alkenylations of aromatic aldimines featuring various N-sulfonyl and sulfamoyl groups. The reactions produce chiral benzylic amines in excellent stereoselectivity, with most examples having > 95% ees. These reactions offer several practical advantages over existing catalytic alkenylations using organometallic reagents and alkenyl boronic acids: nearly neutral conditions, excellent compatibility with polar groups and heterocycles such as pyridine, thiophene, and benzofuran. A broad scope of both cyclic and acyclic alkenyl bromides and sulfonates are suitable electrophiles with different substituents. One procedure was scalable with low loadings of nickel catalysts (as low as 0.2 mol%). Moreover, olefinic groups in products can be readily converted to other aliphatic groups via Pd/C hydrogenation and iron-catalyzed radical hydrofluorination, for example, offering access to other types of chiral benzylic amines. Additionally, complete hydrogenation of arenes and olefins using Adom’s catalyst provided chiral methylamines carrying two different aliphatic groups with exceptionally high ees. They are challenging to prepare by other efficient methods. Importantly, density functional theory (DFT) calculations revealed that in the reactions of N-sulfonyl imines, the major pathway proceeds through classical 1,2-migratory insertion of alkenyl nickel species via planar four-membered-ring transition states.
Supplementary materials
The supplementary material for this article is available at: Supplementary materials.
Authors contribution
Zhou JS: Conceptualization, supervision, writing-original draft, writing-review & editing.
Zhao M: Methodology, investigation, data curation.
Sun L: Investigation, data curation.
Xiao B: Investigation, data curation.
Conflicts of interest
Jianrong Steve Zhou is an Advisory Editor of Chiral Chemistry. The other authors declare no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
This work was supported by the National Natural Science Foundation of China (Nos. W2431014 and 22271007), Peking University Shenzhen Graduate School, Shenzhen Bay Laboratory Supercomputing Center and Shenzhen Polytechnic University Startup fund (6025310040K).
Copyright
© The Author(s) 2026.
References
-
1. (a) Roughley SD, Jordan AM. The medicinal chemist’s toolbox: An analysis of reactions used in the pursuit of drug candidates. J Med Chem. 2011;54(10):3451-3479.
-
2. (a) Li W, Zhang X. Stereoselective formation of amines. Berlin: Springer; 2014.[DOI](b) Trowbridge A, Walton SM, Gaunt MJ. New strategies for the transition-metal catalyzed synthesis of aliphatic amines. Chem Rev. 2020;120(5):2613-2692.[DOI](c) Mondal S, Roy D, Panda G. Critical view on the recent enantioselective synthesis of alcohols, amines and related molecules having tertiary benzylic stereocenter. Tetrahedron. 2018;74(36):4619-4703.[DOI](d) Cabré A, Verdaguer X, Riera A. Recent advances in the enantioselective synthesis of chiral amines via transition metal-catalyzed asymmetric hydrogenation. Chem Rev. 2022;122(1):269-339.[DOI](e) Kobayashi S, Mori Y, Fossey JS, Salter MM. Catalytic enantioselective formation of C–C bonds by addition to imines and hydrazones: A ten-year update. Chem Rev. 2011;111(4):2626-2704.[DOI]
-
3. (a) Ge L, Harutyunyan SR. Asymmetric nucleophilic addition to ketones and ketimines and conjugate addition reactions. In: Akiyama T, Ojima I, editor. Catalytic Asymmetric Synthesis. Hoboken: Wiley; 2022.[DOI](b) Rong J, Collados JF, Ortiz P, Jumde RP, Otten E, Harutyunyan SR. Catalytic enantioselective addition of Grignard reagents to aromatic silyl ketimines. Nat Commun. 2016;7:13780.[DOI](c) Alexakis A, Krause N, Woodward S. Copper-catalyzed asymmetric synthesis. New York: John Wiley & Sons; 2014.[DOI]
-
4. (a) Shintani R, Takeda M, Soh YT, Ito T, Hayashi T. Rhodium-catalyzed asymmetric addition of potassium organotrifluoroborates to N-sulfonyl ketimines. Org Lett. 2011;13(12):2977-2979.(b) Gopula B, Chiang CW, Lee WZ, Kuo TS, Wu PY, Henschke JP, et al. Highly enantioselective Rh-catalyzed alkenylation of imines: Synthesis of chiral allylic amines via asymmetric addition of potassium alkenyltrifluoroborates to N-tosyl imines. Org Lett. 2014;16(2):632-635.(c) Qian XW, Xue ZJ, Zhao Q, Cui Z, Chen YJ, Feng CG, et al. Enantioselective rhodium-catalyzed alkenylation of aliphatic imines. Org Lett. 2017;19(20):5601-5604.[DOI](d) Li Y, Liu B, Xu MH. Rhodium-catalyzed enantioselective alkenylation of cyclic ketimines: Synthesis of multifunctional chiral α, α-disubstituted allylic amine derivatives. Org Lett. 2018;20(8):2306-2310.[DOI](e) Luo Y, Carnell AJ, Lam HW. Enantioselective rhodium-catalyzed addition of potassium alkenyltrifluoroborates to cyclic imines. Angew Chem Int Ed. 2012;51(27):6762-6766.(f) Brak K, Ellman JA. Asymmetric synthesis of alpha-branched allylic amines by the Rh(I)-catalyzed addition of alkenyltrifluoroborates to N-tert-butanesulfinyl aldimines. J Am Chem Soc. 2009;131(11):3850-3851.
-
5. (a) Huang Y, Huang RZ, Zhao Y. Cobalt-catalyzed enantioselective vinylation of activated ketones and imines. J Am Chem Soc. 2016;138(20):6571-6576.(b) Quan M, Wang X, Wu L, Gridnev ID, Yang G, Zhang W. Ni(II)-catalyzed asymmetric alkenylations of ketimines. Nat Commun. 2018;9:2258.[DOI]
-
6. (a) Le Gall E, Haurena C, Sengmany S, Martens T, Troupel M. Three-component synthesis of alpha-branched amines under Barbier-like conditions. J Org Chem. 2009;74(20):7970-7973.(b) Pignon A, Le Gall E, Martens T. A new manganese-mediated, cobalt-catalyzed three-component synthesis of (diarylmethyl)sulfonamides. Beilstein J Org Chem. 2014;10:425-431.[DOI]
-
7. (a) Turro RF, Brandstätter M, Reisman SE. Nickel-catalyzed reductive alkylation of heteroaryl imines. Angew Chem Int Ed. 2022;61(38):e202207597.(b) Heinz C, Lutz JP, Simmons EM, Miller MM, Ewing WR, Doyle AG. Ni-catalyzed carbon–carbon bond-forming reductive amination. J Am Chem Soc. 2018;140(6):2292-2300.[DOI]
-
9. Xia T, Wu Y, Hu J, Wu X, Qu J, Chen Y. Cobalt-catalyzed asymmetric aza-nozaki–hiyama–kishi (NHK) reaction of α-imino esters with alkenyl halides. Angew Chem Int Ed. 2024;63(7):e202316012.[DOI]
-
11. Liu YC, Li JY, University W, Hu LW, University W, et al. Ni-catalyzed asymmetric reductive arylation and alkenylation ofN-sulfonyl imines. Org Lett. 2026;28(2):782-787.[DOI]
-
13. Zhao M, Xiao B, University SP, Sun L, et al. Nickel-catalyzed enantioselective reductive alkenylation ofN-pyridyl aldimines via a mechanism of elementary 1, 4-addition. Org Lett. 2026;28(5):1720-1726.[DOI]
-
14. Wang X, Xiao B, University SP, Jiang T, et al. Catalytic enantioselective addition of aromatic, heteroaryl, and alkenyl halides toN-boc andN-cbz aldimines: Entatic states in transition metal catalysis. ACS Catal. 2026;16(6):6041-6056.[DOI]
-
16. (a) Li L, Liu YC, Shi H. Nickel-catalyzed enantioselective α-alkenylation of N-sulfonyl amines: Modular access to chiral α-branched amines. J Am Chem Soc. 2021;143(11):4154-4161.[DOI](b) Bishop HD, Zhao Q, Uyeda C. Catalytic asymmetric synthesis of zinc metallacycles. J Am Chem Soc. 2023;145(37):20152-20157.(c) Maji K, Palai A, Mallick D, Maji B. Cobalt-catalyzed enantioselective reductive coupling of imines and internal alkynes. Angew Chem Int Ed. 2025;64(13):e202424394.[DOI](d) Ortiz E, Shezaf J, Chang YH, Krische MJ. Enantioselective metal-catalyzed reductive coupling of alkynes with carbonyl compounds and imines: Convergent construction of allylic alcohols and amines. ACS Catal. 2022;12(14):8164-8174.[DOI]
-
17. (a) Zhang L, Zhao M, Pu M, Ma Z, Zhou J, Chen C, et al. Nickel-catalyzed enantioselective reductive conjugate arylation and heteroarylation via an elementary mechanism of 1, 4-addition. J Am Chem Soc. 2022;144(44):20249-20257.(b) Zhao M, Zhang L, Zhou JS. Enantioselective reductive conjugate alkenylation of α, β-unsaturated ketones and amides via nickel catalysis. ACS Catal. 2024;14(8):6228-6235.[DOI]
-
18. Zhao M, Xiao B, Sun L, Zhou JS. Nickel-catalyzed enantioselective reductive alkenylation of N-pyridyl aldimines via a mechanism of elementary 1, 4-addition. Org Lett. 2026;28(5):1720-1726.[DOI]
-
21. (a) Paul J, Luong Van My L, Behar-Pirès M, Guillaume C, Léonel E, Presset M, et al. Co(I)-catalyzed [3 + 2] annulation of o-haloaryl imines with alkenes for the synthesis of indanamines. J Org Chem. 2018;83(7):4078-4086.[DOI]
-
23. Ankner T, Hilmersson G. Instantaneous deprotection of tosylamides and esters with SmI2/amine/water. Org Lett. 2009;11(3):503-506.[DOI]
-
24. (a) Barker TJ, Boger DL. Fe(III)/NaBH4-mediated free radical hydrofluorination of unactivated alkenes. J Am Chem Soc. 2012;134(33):13588-13591.
-
25. Zhao M, Zhang L, Zhou JS. Enantioselective reductive conjugate alkenylation of α, β-unsaturated ketones and amides via nickel catalysis. ACS Catal. 2024;14(8):6228-6235.[DOI]
-
26. (a) Giardina GAM, Raveglia LF, Grugni M, Sarau HM, Farina C, Medhurst AD, et al. Discovery of a novel class of selective non-peptide antagonists for the human neurokinin-3 receptor. 2. J Med Chem. 1999;42(6):1053-1065.[DOI](b) Scott LJ. Repaglinide: A review of its use in type 2 diabetes mellitus. Drugs. 2012;72(2):249-272.[DOI]
-
27. (a) Cogan DA, Liu G, Ellman J. Asymmetric synthesis of chiral amines by highly diastereoselective 1, 2-additions of organometallic reagents to N-tert-butanesulfinyl imines. Tetrahedron. 1999;55(29):8883-8904.[DOI](b) Liu G, Cogan DA, Ellman JA. Catalytic asymmetric synthesis of tert-butanesulfinamide. Application to the asymmetric synthesis of amines. J Am Chem Soc. 1997;119(41):9913-9914.[DOI](c) Wang M, Liu S, Liu H, Wang Y, Lan Y, Liu Q. Asymmetric hydrogenation of ketimines with minimally different alkyl groups. Nature. 2024;631(8021):556-562.[DOI](d) Qian D, Bera S, Hu X. Chiral alkyl amine synthesis via catalytic enantioselective hydroalkylation of enecarbamates. J Am Chem Soc. 2021;143(4):1959-1967.(e) Wang JW, Li Y, Nie W, Chang Z, Yu ZA, Zhao YF, et al. Catalytic asymmetric reductive hydroalkylation of enamides and enecarbamates to chiral aliphatic amines. Nat Commun. 2021;12(1):1313.
-
28. Hilgraf R, Pfaltz A. Chiral bis(N-sulfonylamino)phosphine- and TADDOL-phosphite-oxazoline ligands: Synthesis and application in asymmetric catalysis. Adv Synth Catal. 2005;347(1):61-77.[DOI]
Copyright
© The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Publisher’s Note
Science Exploration remains a neutral stance on jurisdictional claims in published
maps
and institutional affiliations. The views expressed in this article are solely those
of
the author(s) and do not reflect the opinions of the Editors or the publisher.
Share And Cite



Science Exploration Style
Zhao M, Sun L, Xiao B, Zhou JS. Highly active nickel catalysts for enantioselective reductive alkenylation of N-sulfonyl aldimines. Chiral Chem. 2026;2:202618. https://doi.org/10.70401/cc.2026.0029
Tips
Copy completed.
Submit a Manuscript
Author Instructions
Cite this Article
Article Metrics
0
View
0
Download
Cited
Article Updates

Science Exploration Style
Zhao M, Sun L, Xiao B, Zhou JS. Highly active nickel catalysts for enantioselective reductive alkenylation of N-sulfonyl aldimines. Chiral Chem. 2026;2:202618. https://doi.org/10.70401/cc.2026.0029
copy
Share Link
copy