Advances in the synthesis of chiral bridged-cyclic phosphorus compounds and the applications in asymmetric catalysis
Fu-She Han
*

*Correspondence to:
Fu-She Han, College of Chemistry and Material Science, Shandong Agricultural University, Taian 271018, Shandong, China.
E-mail: fshan@sdau.edu.cn
Chiral Chem. 2026;2:202623. 10.70401/cc.2026.0030
Received: April 27, 2026Accepted: June 09, 2026Published: June 09, 2026
Abstract
Due to the conformational rigidity, the bridged-cyclic chiral phosphorus compounds have been demonstrated to show enantioinducing ability generally superior to conformationally flexible chiral phosphorus compounds as ligands or organocatalysts in catalytic asymmetric reactions. However, investigations into the bridged-cyclic chiral phosphorus compounds are relatively much rarer than the non-bridged-cyclic phosphorus compounds resulting from the paucity of efficient synthetic methods. In order to draw the attention of researchers who are interested in asymmetric synthesis and catalysis to this issue, in this review, we intend to summarize the progress in the synthesis of bridged-cyclic chiral phosphorus compounds and their applications in catalytic asymmetric reactions. We wish this review article could serve as a useful guide and stimulate, to some extent, the interests of asymmetric catalysis scholars to devote to this challenging but greatly important field.
Graphical Abstract
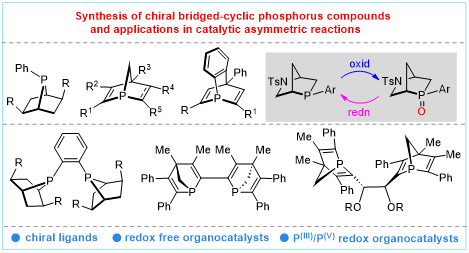
Keywords
Bridged-cyclic compounds, chiral phosphorus compounds, asymmetric catalysis, Diels-Alder reaction, transannulation, annulation, Morita-Baylis-Hillman reaction, Staudinger-aza-Wittig reaction
1. Introduction
In catalytic asymmetric synthesis and reaction, the chiral phosphorus compounds are indisputably the most frequently used ligands or organocatalysts either in scientific research or industrial applications[1-6]. Earlier investigations have been mainly concentrated on the phosphorus compounds with backbone chirality such as central, axial, planar, or helical chirality. Whereas the past decade has witnessed a surge in the development and catalytic applications of P-stereogenic phosphorus compounds in which the chiral environments are located on the phosphorus atoms. As comprehensively summarized in a very recent review[1,2], both the categories of chiral phosphorus compounds have demonstrated extensive applicability and excellent performance either as ligands of a rich range of transition metals or as organocatalysts. On the one hand, they have been extensively used as privileged chiral ligands of a broad array of transition metals such as Co, Rh, Ir, Ni, Pd, Pt, Cu, Au, and Ru in transition-metal-catalyzed asymmetric reactions such as hydrogenation, carbon−carbon and carbon-heteroatom (e.g., C–B, C–Si, C–N, C–P, and C–O etc.) bond-forming reactions. On the other hand, they also could serve as powerful chiral organocatalysts in a broad variety of organocatalyzed asymmetric reactions such as [m+n] annulation, reduction of ketones, kinetic resolution of secondary alcohols, Morita-Baylis-Hillman reaction, Staudinger-aza-Wittig reaction, and so on.
Owing to the paramount importance, the investigations into the chiral phosphorus compounds, including the development of new synthetic methodologies for the synthesis of chiral phosphorus compounds with structural diversity and exploration of new applications in catalytic asymmetric reactions, have attracted the widest and sustained attention from the global community of asymmetric catalysis and synthesis. This has led to the publication of many comprehensive reviews over the past decades[1-6]. However, it is noted that a special review article focused on the bridged-cyclic chiral phosphorus compounds is absent. Probably, this may result from the fact that, due to the paucity of efficient synthetic methods, the bridged-cyclic chiral phosphorus compounds are much rarer compared with the non-bridged-cyclic compounds. However, it has been demonstrated that, thanks to the rigid conformation, the bridged-cyclic chiral phosphorus compounds usually exhibit catalytic performance superior to that of non-bridged-cyclic compounds. As such, we think it is important to provide a review article concentrating on the synthesis and application of bridged-cyclic chiral phosphorus compounds. It is anticipated that this review can not only provide a deeper understanding of the current state of the art for this topic, but more to the point, can stimulate the development of conceptually new methodologies for efficient synthesis of bridged-cyclic chiral phosphorus, and consequently, advancing the chemistry of asymmetric catalysis.
2. Synthesis of Bridged-Cyclic Chiral Phosphorus Compounds
As mentioned above, the synthesis of bridged-cyclic chiral phosphorus compounds is considerably challenging. To date, the reported methods have mainly relied on chiral substrate-controlled[7] and chiral auxiliary-aided asymmetric synthesis, and optical resolution of racemic products. The progress will be summarized in this section.
2.1 Chiral substrate-controlled asymmetric synthesis
2.1.1 Synthesis through Diels-Alder (D-A) reaction
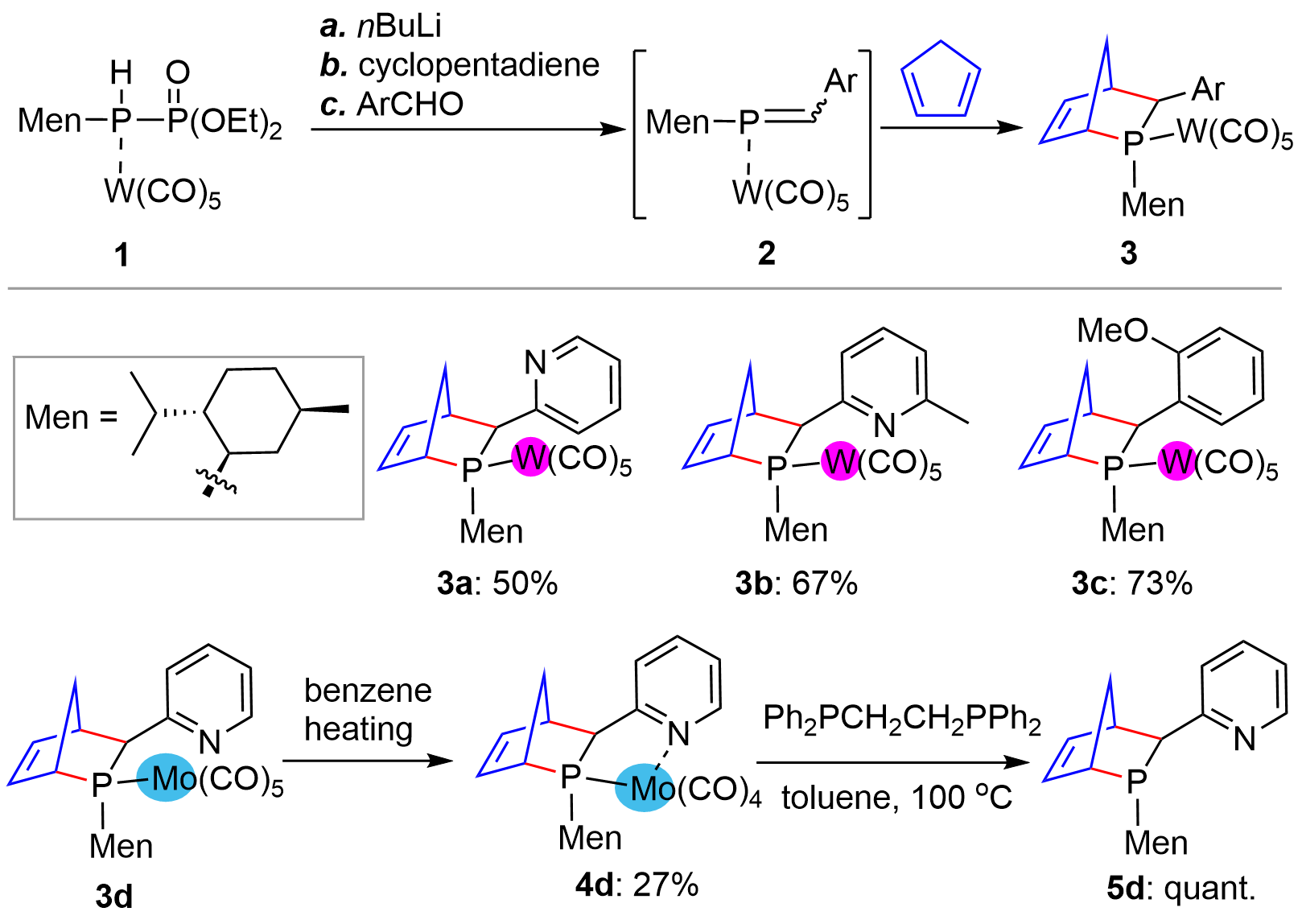
The chiral substrate-controlled synthesis of bridged-cyclic chiral phosphorus compounds could be traced back to more than three decades ago. In a 1992 report by Mathey’s group[8], the authors developed a D-A reaction of cyclopenta-1,3-diene with L-menthyl phosphaalkene P-W(CO)5 complexes 2 (Figure 1), generated in situ from complex 1 and aryl formaldehyde under the effect of nBuLi base, providing the P-stereogenic [2.2.1]-bridged bicyclic P-W complexes 3. Moderate to high yields with almost one pure diastereoisomer were obtained as seen from a few examples 3a-3c. The method could also be extended to a P-Mo phosphaalkene complex, giving bicyclic P-Mo complex 3d, which, in contrast to P-W complexes, could facilitate the decomplexation to provide metal free compound 5d via 4d.
{kind=link}
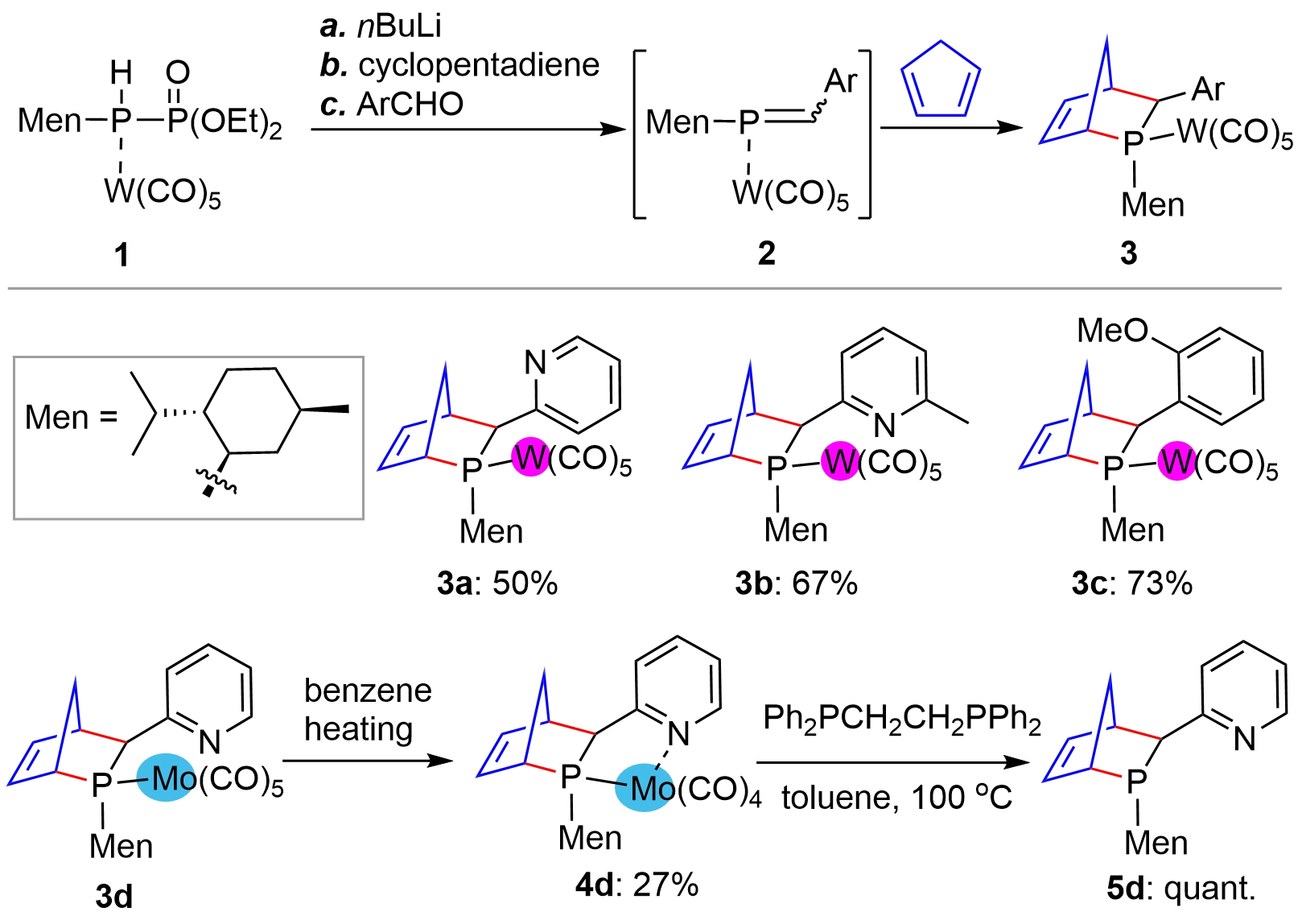
Figure 1. D-A reaction for the synthesis of P-stereogenic bridged bicycles from phosphaalkene. D-A: Diels-Alder.
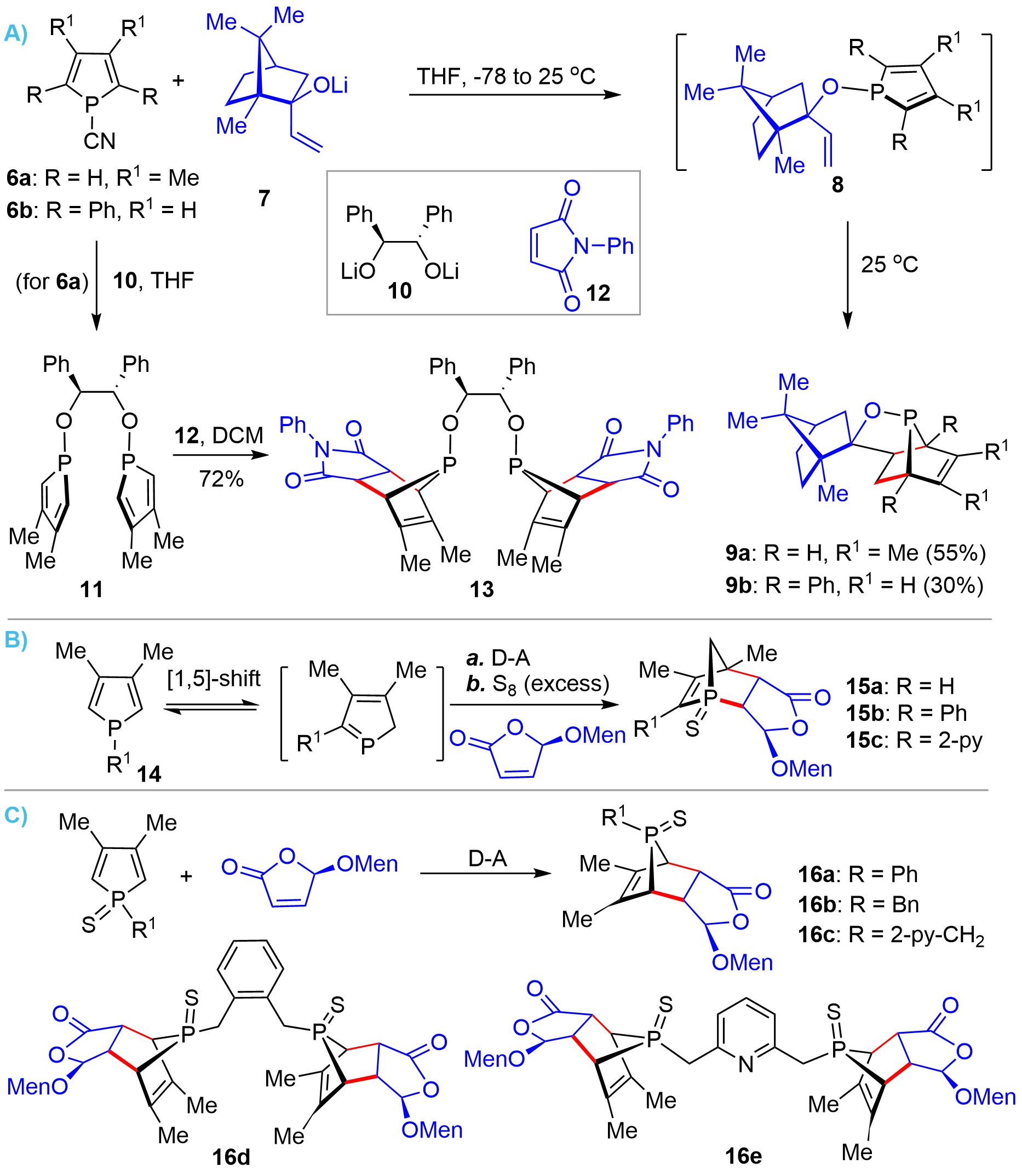
On the basis of their early work, in 2002, the Mathey’s group developed an intramolecular D-A reaction of 1-alkoxy substituted phospholes 8 (Figure 2A), which could be readily prepared from 1-cyano-phosphole 6 and a chiral allyloxy lithium 7, for the synthesis of bridged-pentacyclic chiral phosphinites 9[9]. It was found that incorporating 1-alkoxy substituents could dramatically enhance the Diels-Alder reactivity of phospholes as compared to an alkyl substituent. It was demonstrated that this protocol could also be reliably extended to the synthesis of bridged-cyclic bisphosphinite 13 from 6a via intermediate 11 in high yield[10]. Following these pioneering works, Hey-Hawkins achieved the synthesis of some structural analogues of bridged bicyclic P-stereogenic phosphorus compounds from (5R)-(L-menthyloxy)-2-(5H)-furanone (MOxF) via intermolecular D-A reactions[11,12]. Interestingly, the authors observed that the trivalent phospholes 14 underwent [1,5]-sigmatropic shift and produced bridged-head phospha-bicyclic compounds 15 (Figure 2B). Whereas the pentavalent sulfide derivatives proceeded via normal D-A reactions to afford the regioisomers 16 (Figure 2C).
{kind=link}
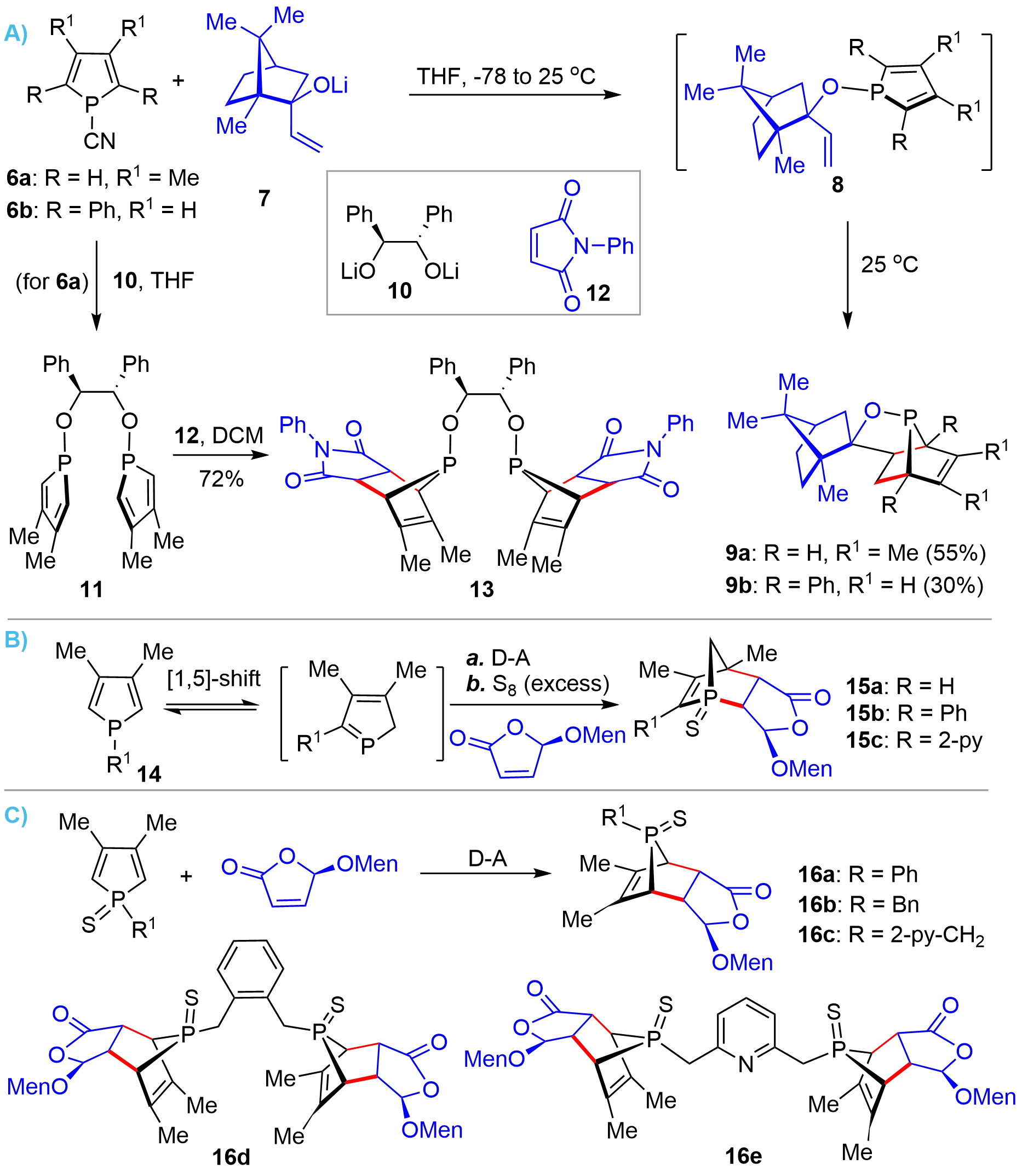
Figure 2. D-A reaction for the synthesis of bridged bicyclic chiral phosphorus compounds from phospholes. D-A: Diels-Alder.
2.1.2 Synthesis through transannular substitution reaction
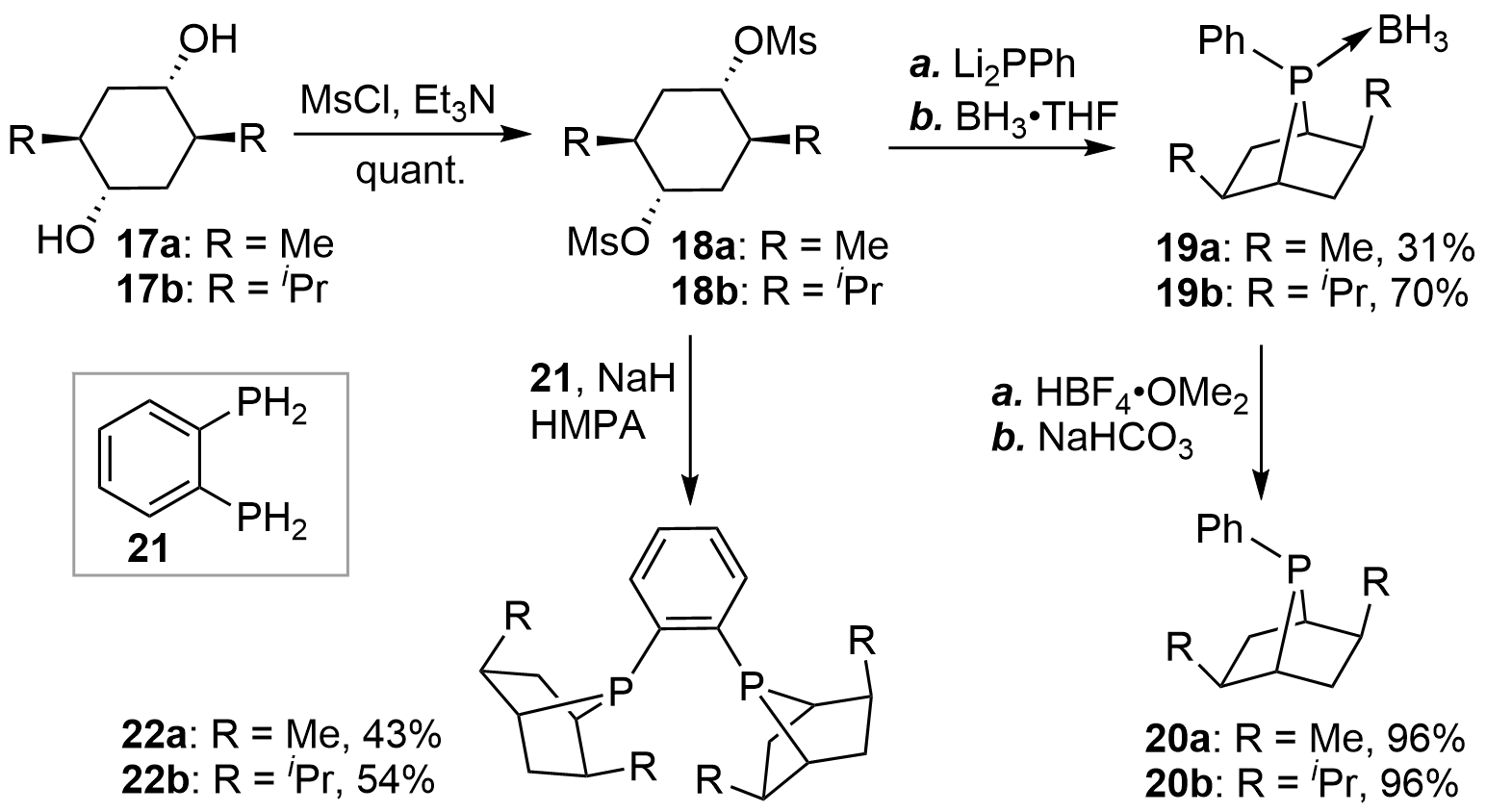
In 1997, the team of Zhang[13,14] synthesized a novel type of [2.2.1]-bridged bicyclic chiral monophosphines 20a and 20b via a transannular substitution. As shown in Figure 3, the synthesis commenced with the optically pure diols 17 obtained from commercial sources. Activation of the hydroxyl groups with Ms afforded 18 in almost quantitative yields. Double nucleophilic substitution of 18 by Li2PPh generated the corresponding bicyclic phosphines, which were then trapped with BH3•THF to afford the air-stable boron complexes of monophosphines 19. Finally, deprotection of BH3 with a strong acid produced the desired products 20 in high yields. According to similar procedures, the authors also synthesized the diphosphines 22 (PennPhos)[15].
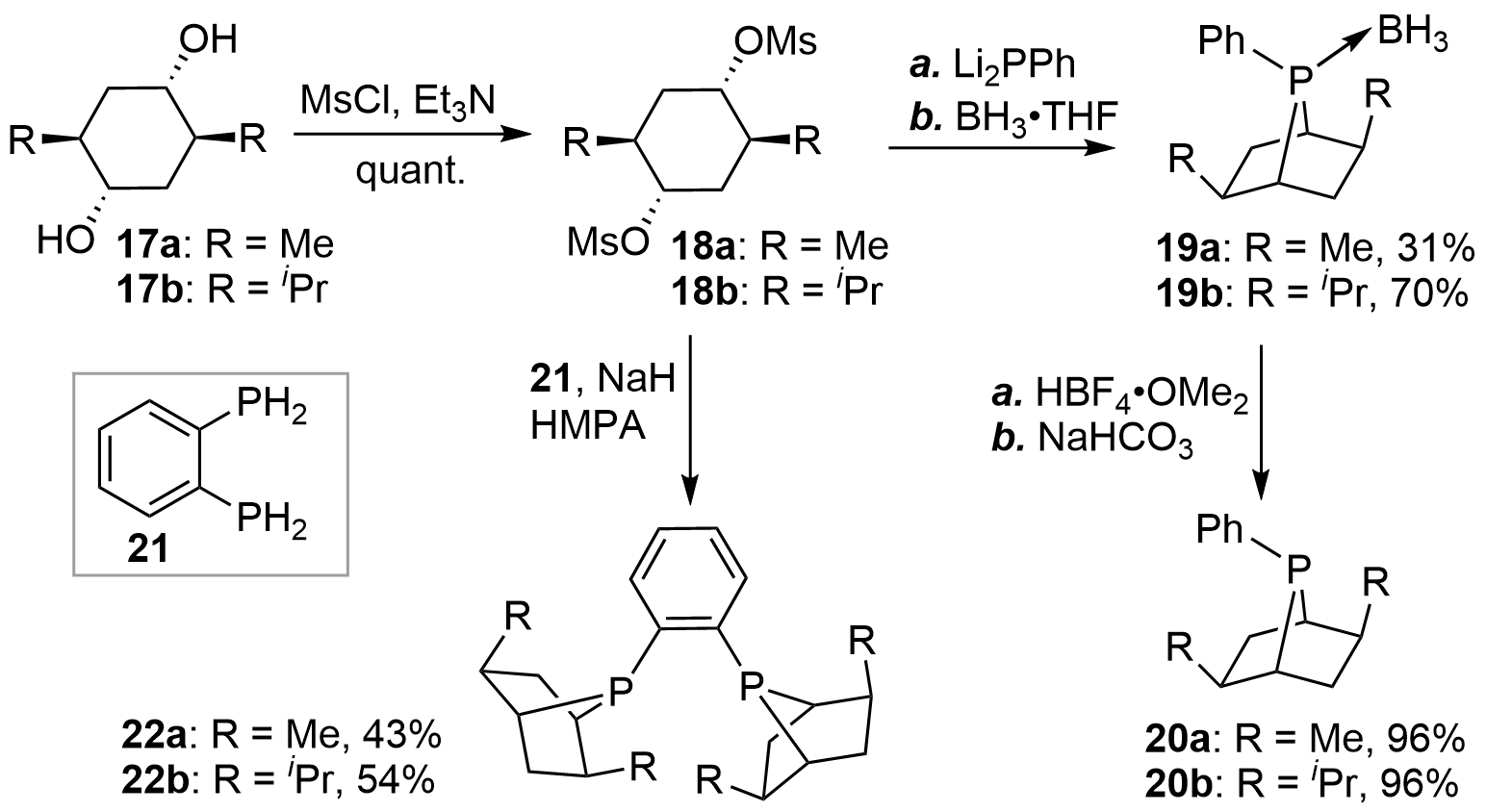{kind=link}
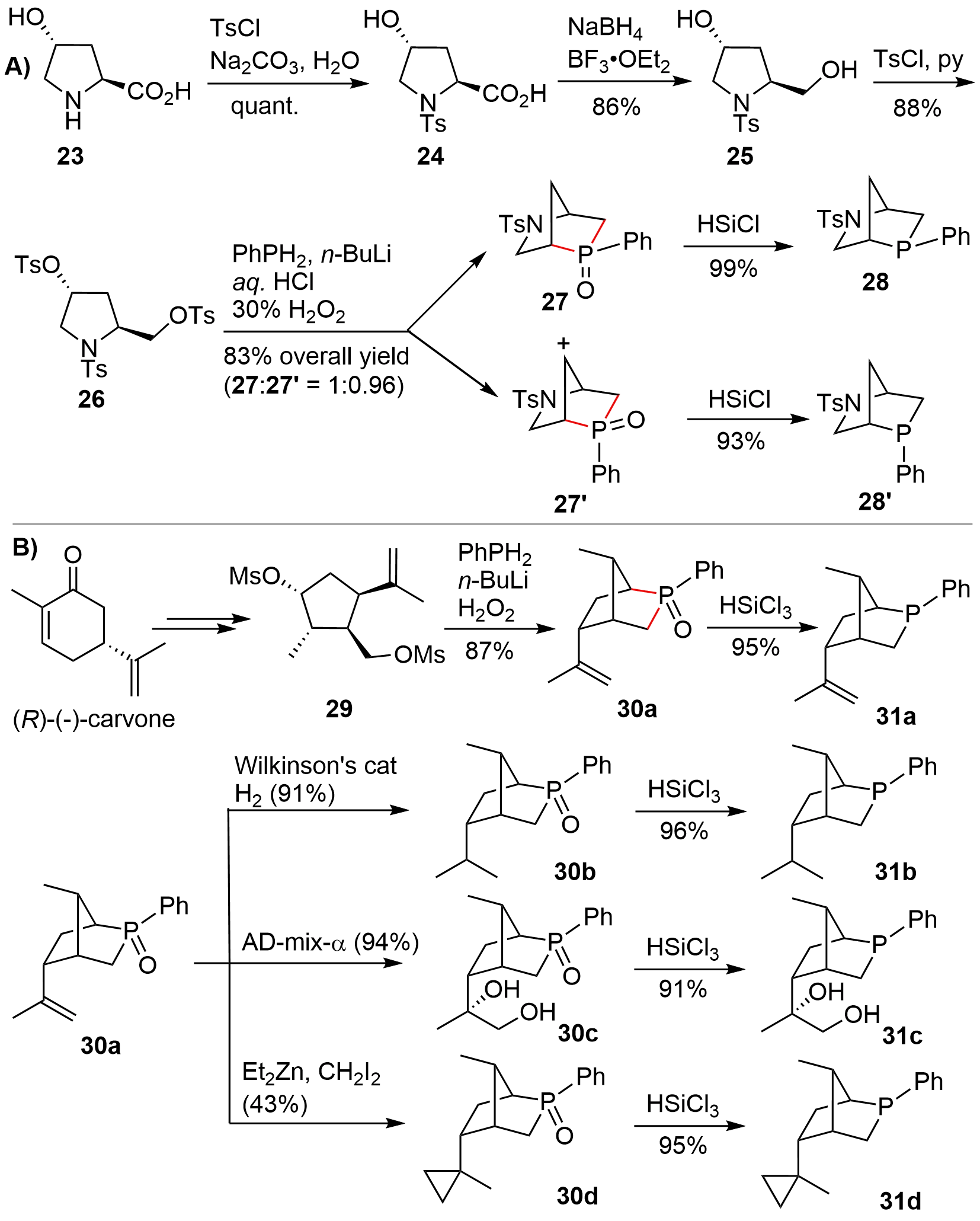
In 2014, Kwon and co-workers[16] disclosed a relatively more efficient route to access aza-[2.2.1] bridged bicyclic P-stereogenic compounds from trans-4-hydroxy-L-proline 23 (Figure 4A). The amino group in hydroxyproline 23 was chemoselectively protected using TsCl to give 24, whose carboxylic acid functionality was then reduced to deliver diol 25. Then activation of the two hydroxy groups with Ts afforded the tritosylated 26. Transannular disubstitution of 26 with the in situ generated PhPLi2 gave a ca. 1:1 mixture of the exo- and endo-P-phenylphosphines 28 and 28’, which was oxidized to the corresponding oxides 27 and 27’ to facilitate isolation. After isolating the oxide isomers, each isomer was reduced to phosphines 28 and 28’ in high yields. Based on this synthetic route, Kwon could also synthesize a P-stereogenic bicyclic variant from (R)-(-)-carvone[17] (Figure 4B). Of note is that, compared with the disubstitution of hydroxyproline 26, only one isomer 30a was obtained during the disubstitution of 29, presumably resulting from the presence of a 2-propenyl group, which might control the stereochemistry at the newly formed phosphorus center. By using 30a as a common intermediate, various bicyclic P-chiral phosphines 31a–31d were also smoothly synthesized. It should be mentioned that, following Kwon’s synthetic route, some other analogues were also prepared by other research teams in the later studies. These works will not be discussed in this section because of the similarity of the synthetic procedures. They will be suitably discussed in the part of asymmetric catalytic reactions (vide infra).
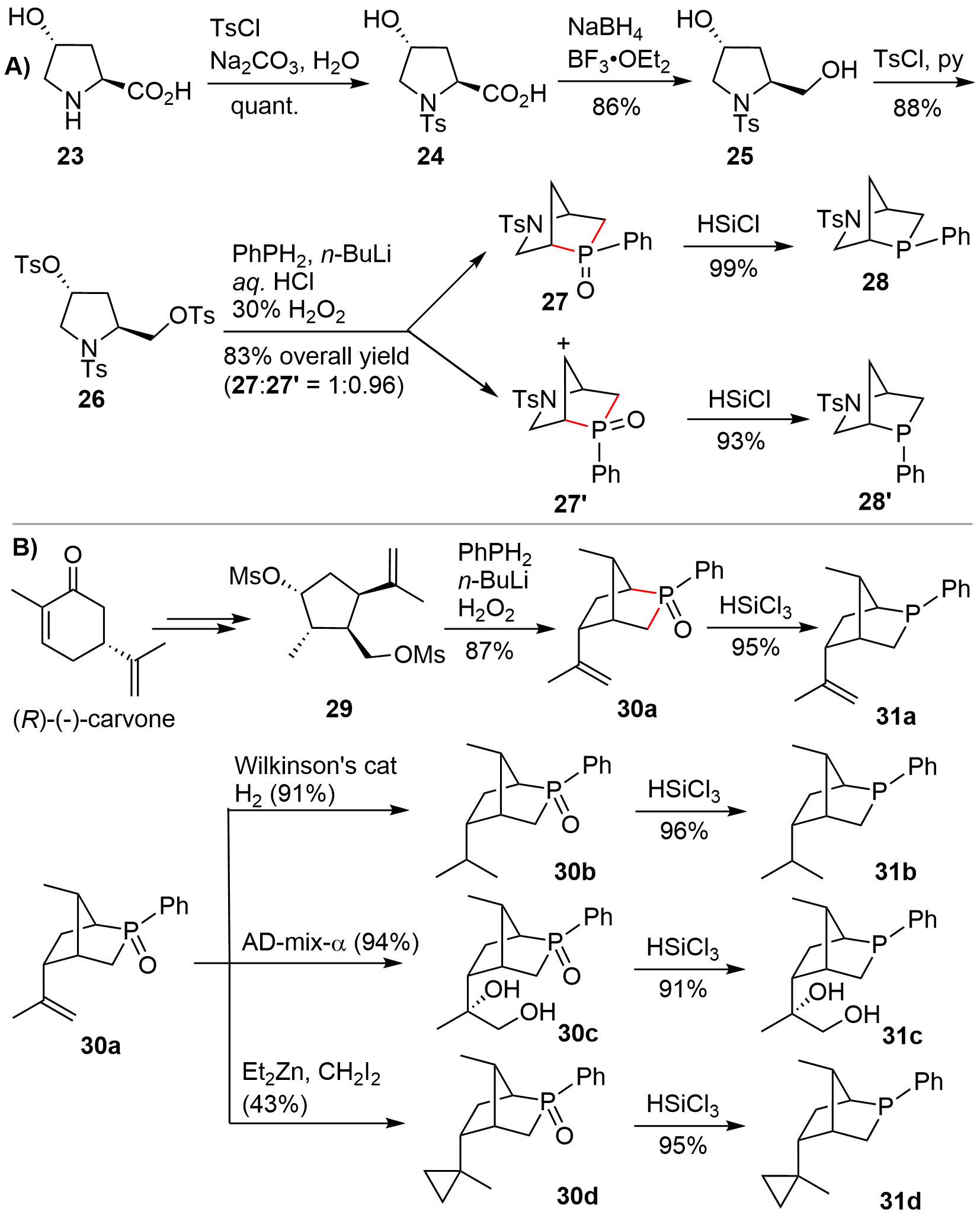{kind=link}
2.2 Chiral metal-template-aided asymmetric synthesis
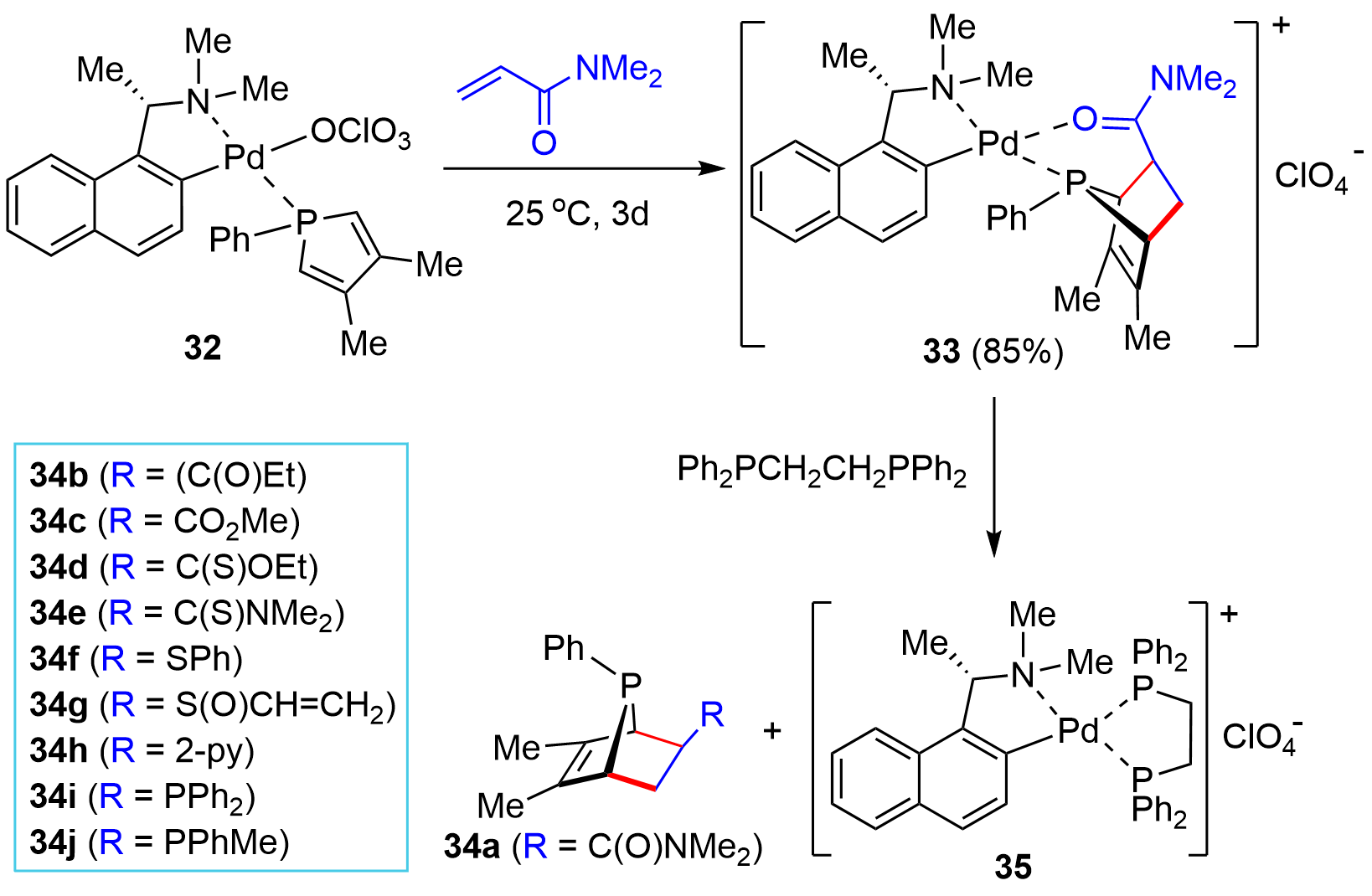
In an effort toward synthesizing P-stereogenic bridged-cyclic compounds, in 1996, Leung and co-workers reported an interesting protocol by means of a chiral palladium complex-template-controlled strategy[18]. Namely, the D-A reaction of a chiral metal complex 32 and N,N-dimethylacrylamide proceeded smoothly under mild conditions to afford the cycloaddition complex 33 (Figure 5), which upon treating with 1,2-bis(diphenylphosphino)ethane (DPPE) liberated the exo-bicyclic P-chiral product 34a as a single diastereoisomer and the DPPE complex 35. Further investigations indicated that the inert phosphole became reactive when it was coordinated onto a Pd(II) ion. Moreover, the presence of a functional group in the dienophile that could interact with a Pd(II) ion as well as a perchlorato counter anion, which might be readily replaced by the coordination groups in the dienophile, was critical for obtaining high exo-cycloaddition selectivity. The method allowed for a flexible synthesis of a variety of P-stereogenic bridged-cyclic compounds such as 34b–34j with exo-selectivity. The progress related to this method has been summarized in an elegant 2004 Account by Leung[19]. Readers interested in this method are referred to that Account since very few significance advancements have been made since that time.
{kind=link}
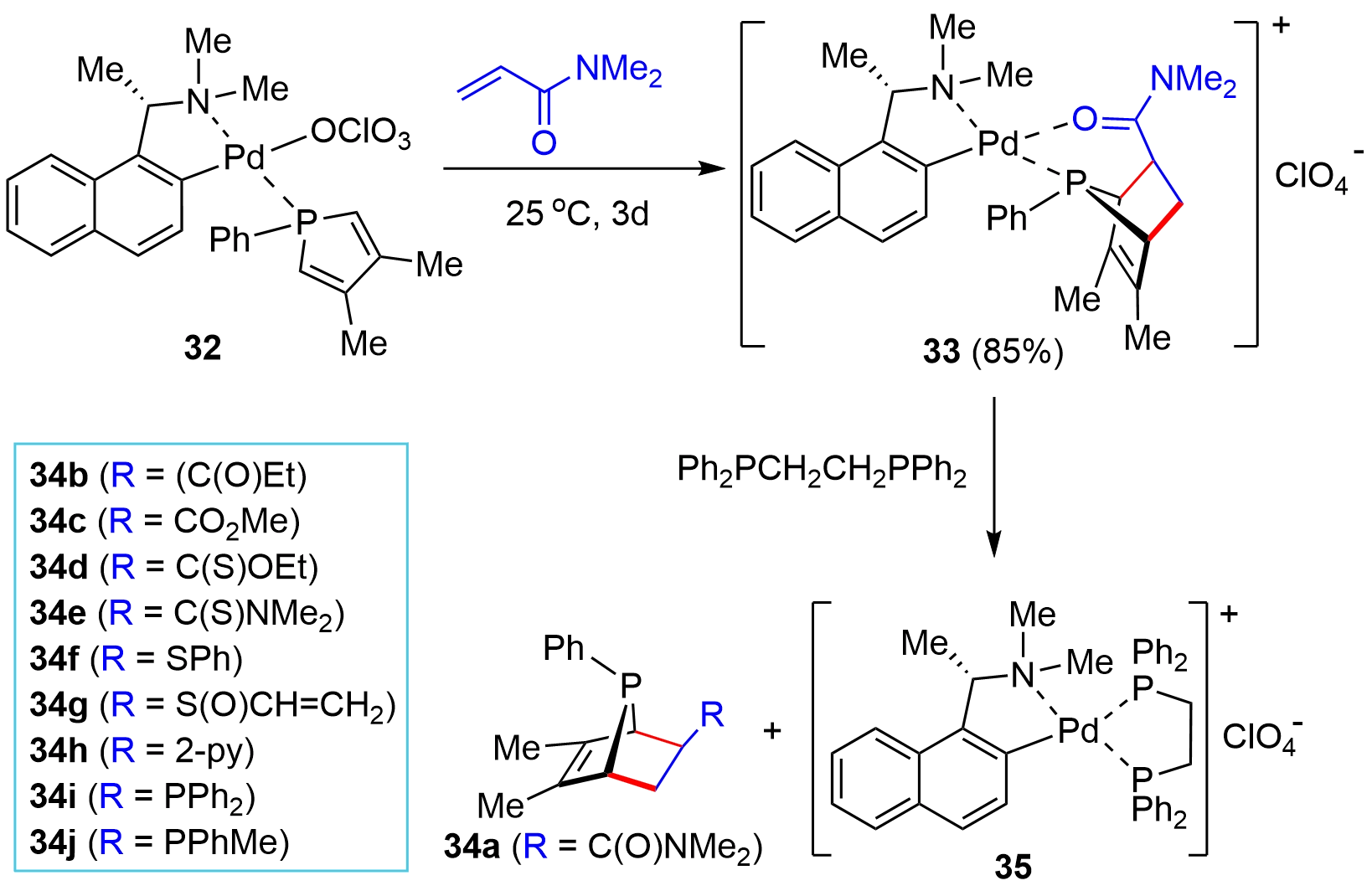
Figure 5. Chiral metal-template-controlled synthesis of bridged-cyclic P-chiral phosphines.
2.3 Preparation of chiral bridged-cyclic phosphorus compounds by optical resolution
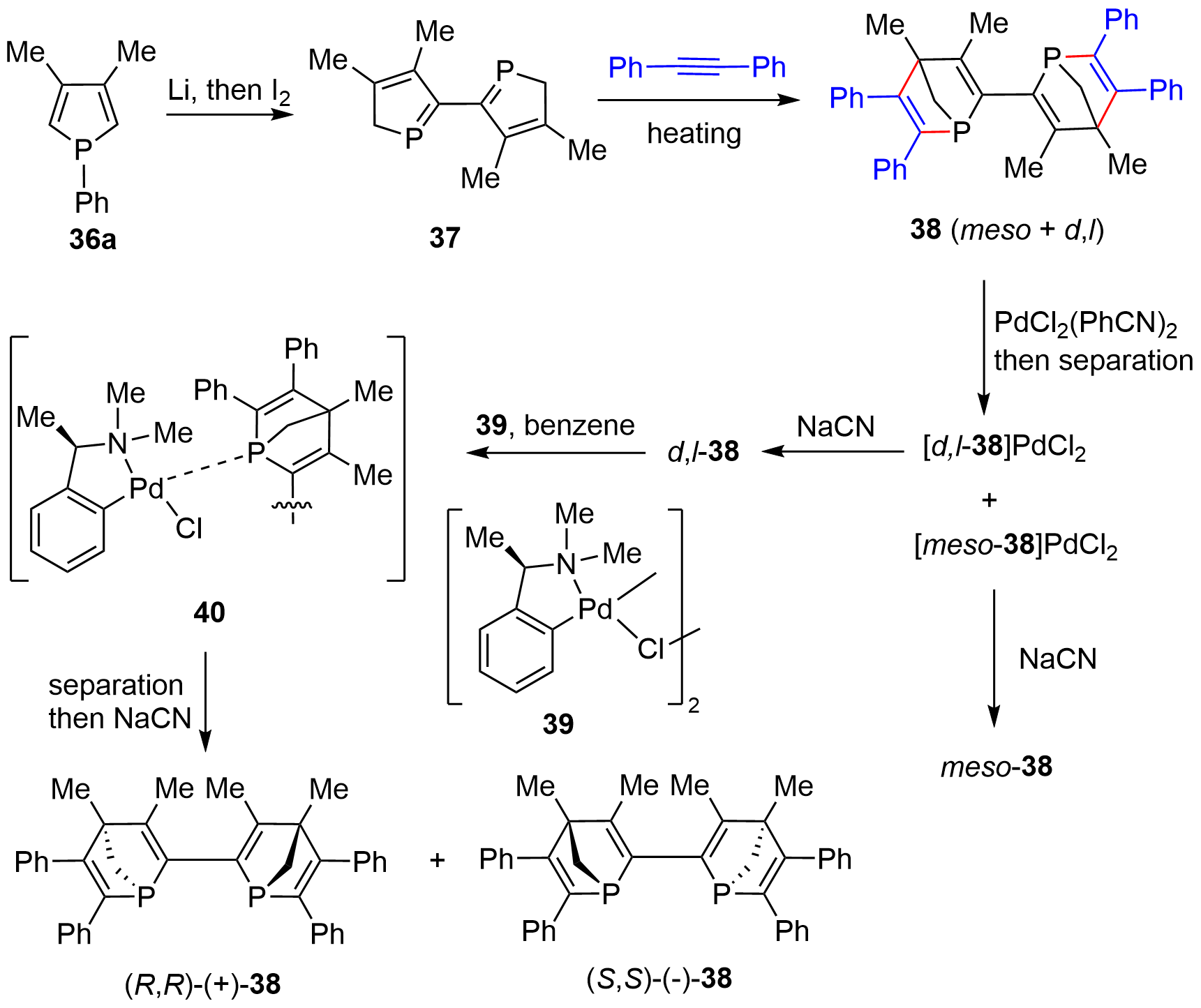
In early 1997, Mathey and coworkers prepared the optically active bis-[2.2.1]-bridged cycles with bridgehead phosphorus chiral centers[20]. As schematically shown in Figure 6, the racemic 38 was synthesized from phosphole 36a via the lithium metal-promoted P–Ph bond cleavage followed by an I2-mediated coupling to give dimerized product 37. Double D-A reaction of 37 with diphenylacetylene yielded a mixture of meso- and d,l-38, which was then separated through column chromatography on silica gel after converting the mixture into the palladium complexes. The [d,l-38]PdCl2 complex was then liberated by NaCN to give free d,l-38, which was then resolved with a stoichiometric amount of chiral palladium complex 39. Again, upon separation of the enantiomeric complex 40 followed by decomplexation with NaCN, a couple of optically active compounds, (R,R)-(+)-38 and (S,S)-(-)-38, were obtained.
{kind=link}
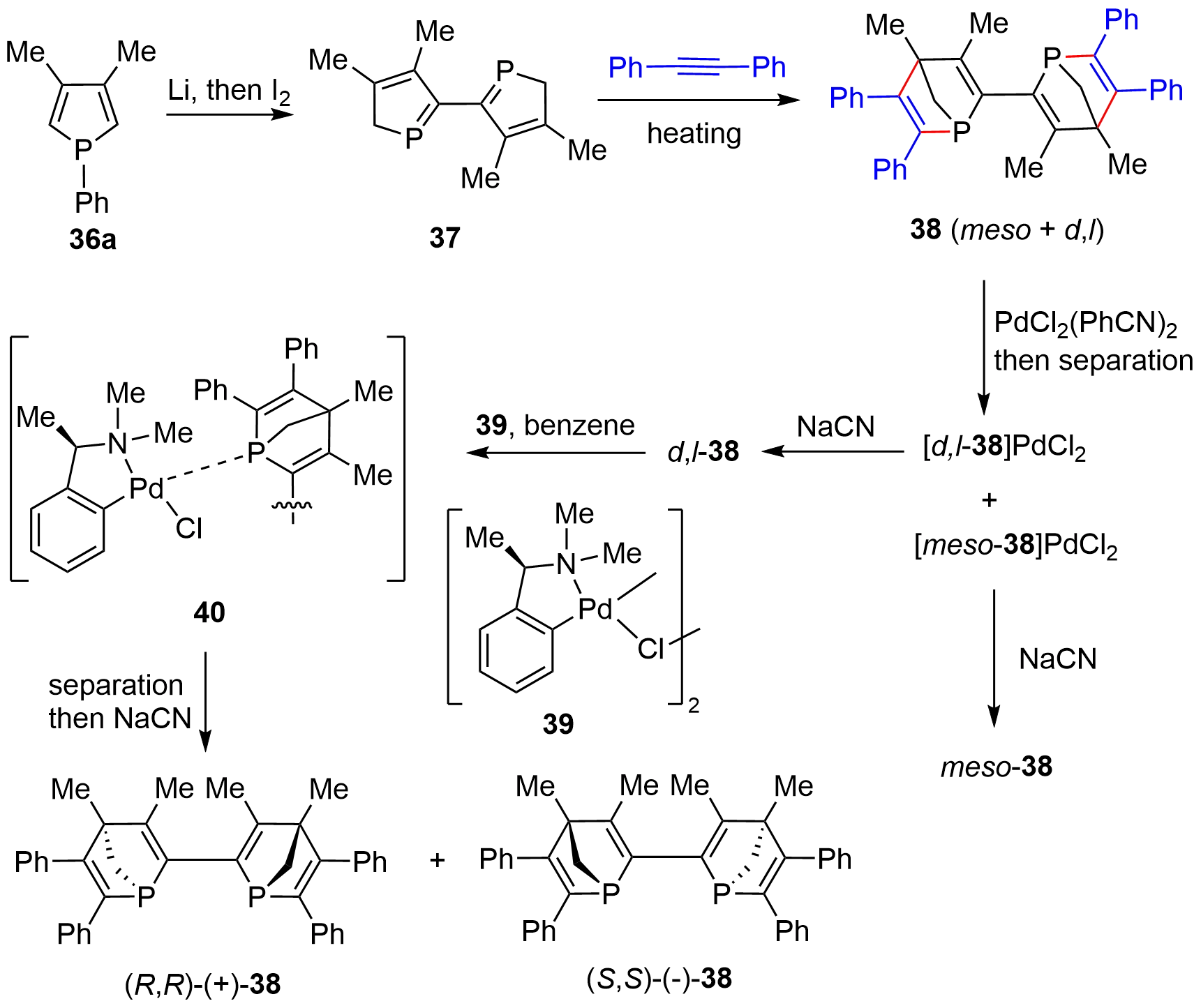
Figure 6. Synthesis of optically active bis-[2.2.1]-bridged cycles using chiral palladium-complex as resolving agent.
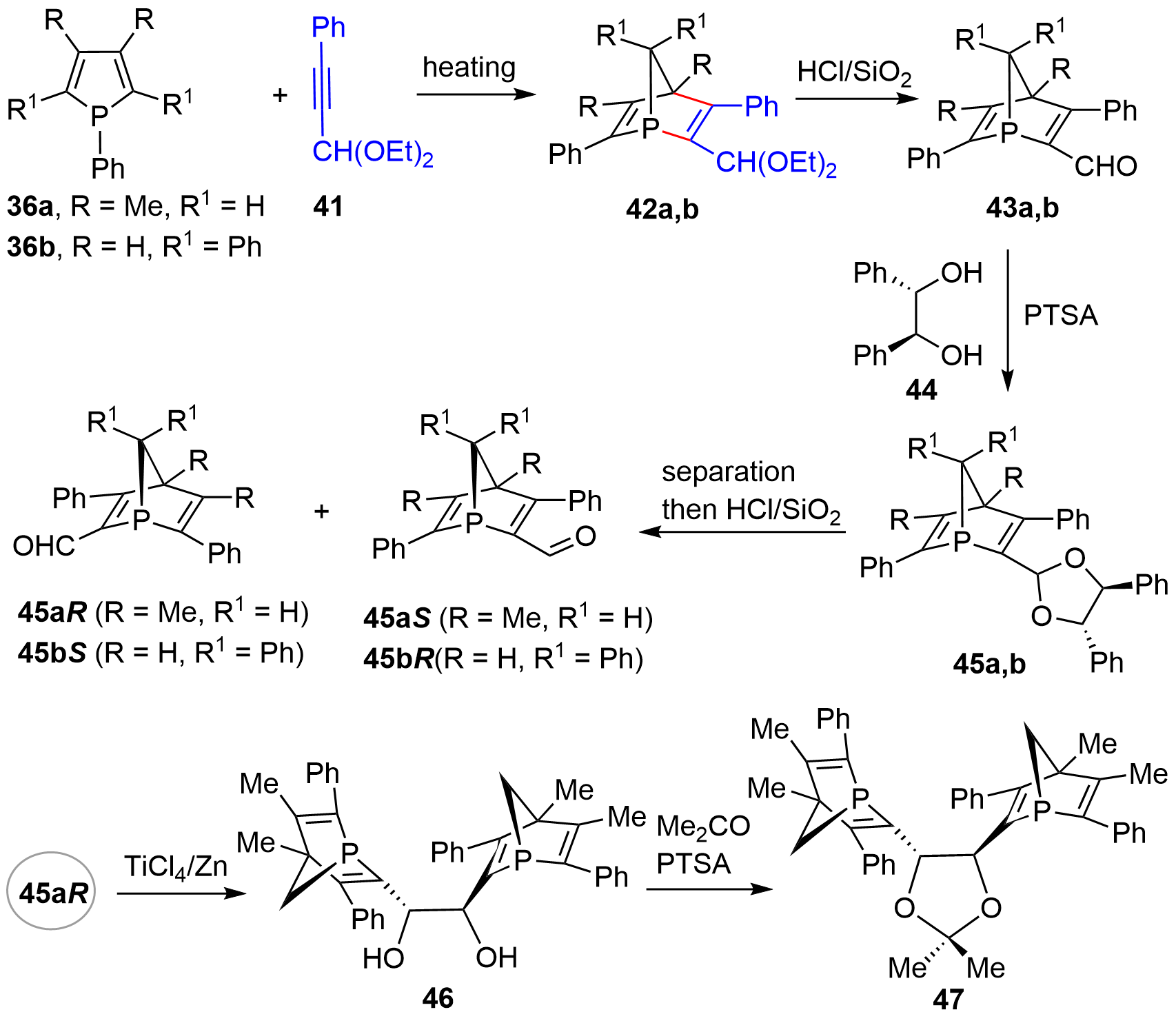
Three years later, the same group described an alternative resolution method for obtaining chiral P-stereogenic bicyclic compounds that avoided the use of a noble metal as a resolving agent[21]. As shown in Figure 7, the racemic bicycles 43 were synthesized via a two-step procedure involving D-A reaction of 36 and 41 to give bicyclic acetal 42, which upon acid-mediated hydrolysis afforded aldehyde 43. Using the readily available and cheap chiral diol 44 as a resolving agent, four optically active P-stereogenic bicyclic compounds 45aR, 45aS, 45bR, and 45bS were harvested. Moreover, bis-bicyclic derivatives 46 and 47 could be uneventfully synthesized from the mono-bicyclic compound[22].
{kind=link}
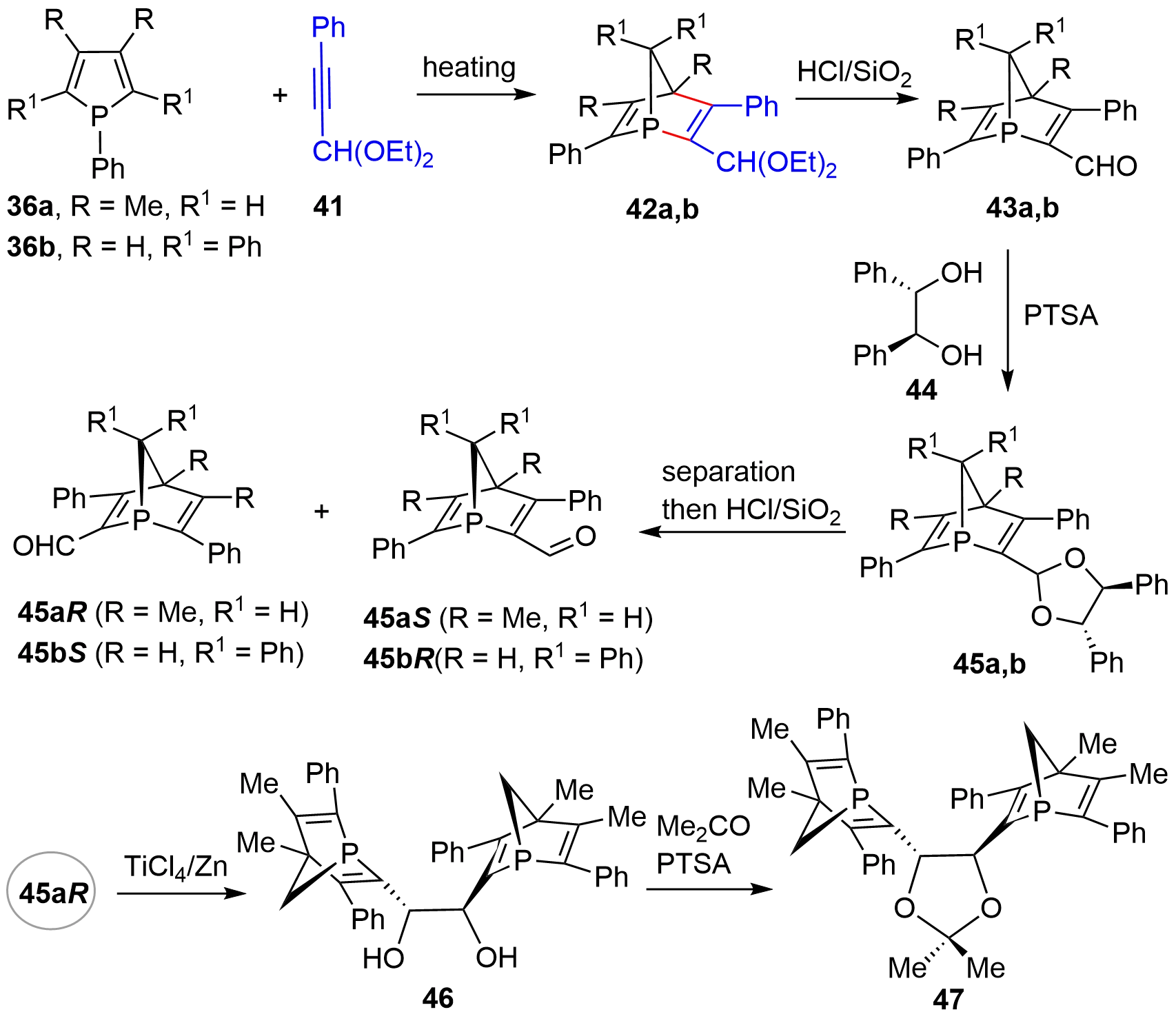
Figure 7. Synthesis of optically active [2.2.1]-bridged cycles using chiral diol as resolving agent.
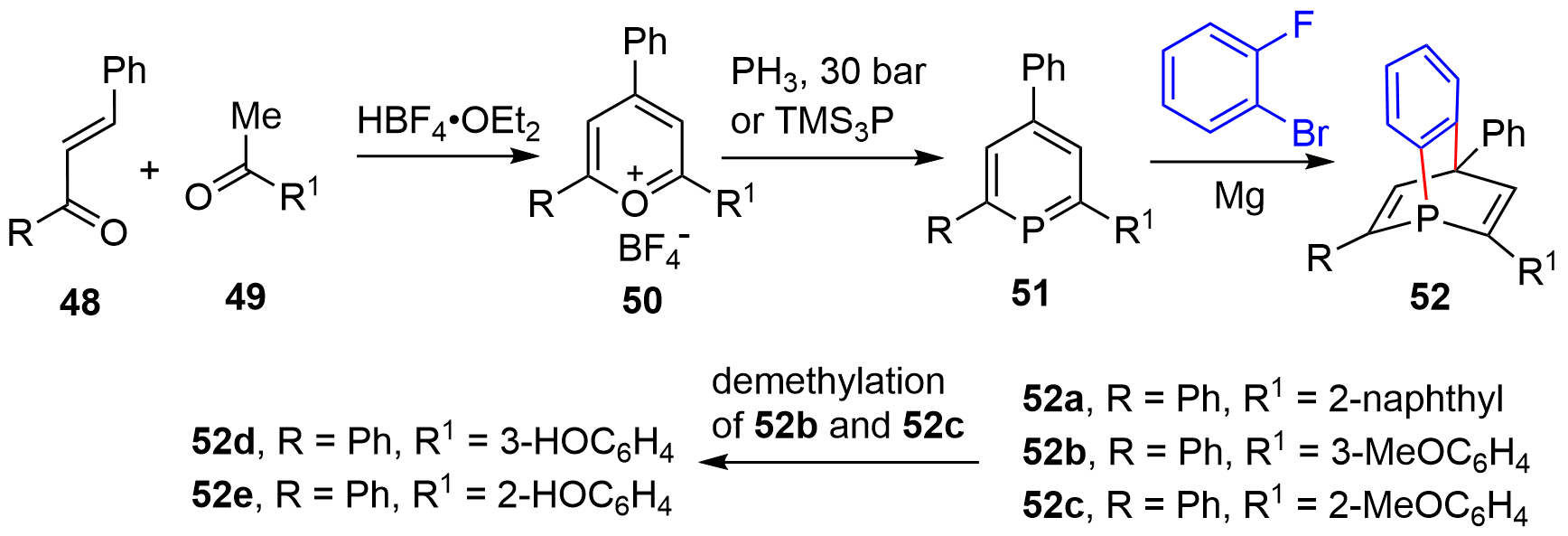
By employing the six-membered phosphabenzenes, Breit and coworkers achieved the synthesis of optically pure [2.2.2]-bicyclic phosphabarrelenes[23]. Namely, the acid-mediated condensation of chalcones 48 with acetophenones 49 generated pyrylium salts 50 (Figure 8). Transformation to phosphabenzenes 51 was executed by treating 50 with either phosphine at elevated pressure or tris(trimethylsilyl)phosphine. Finally, the D-A reaction of 51 with benzyne, generated from 1-bromo-2-fluorobenzene and magnesium, accomplished the synthesis of [2.2.2]-bicyclic phosphorus compounds 52a–52c. Further demethylation of 52b and 52c gave 52d and 52e, respectively. Each racemic 52 was resolved by high performance liquid chromatography (HPLC) to afford the corresponding enantiomers.
{kind=link}
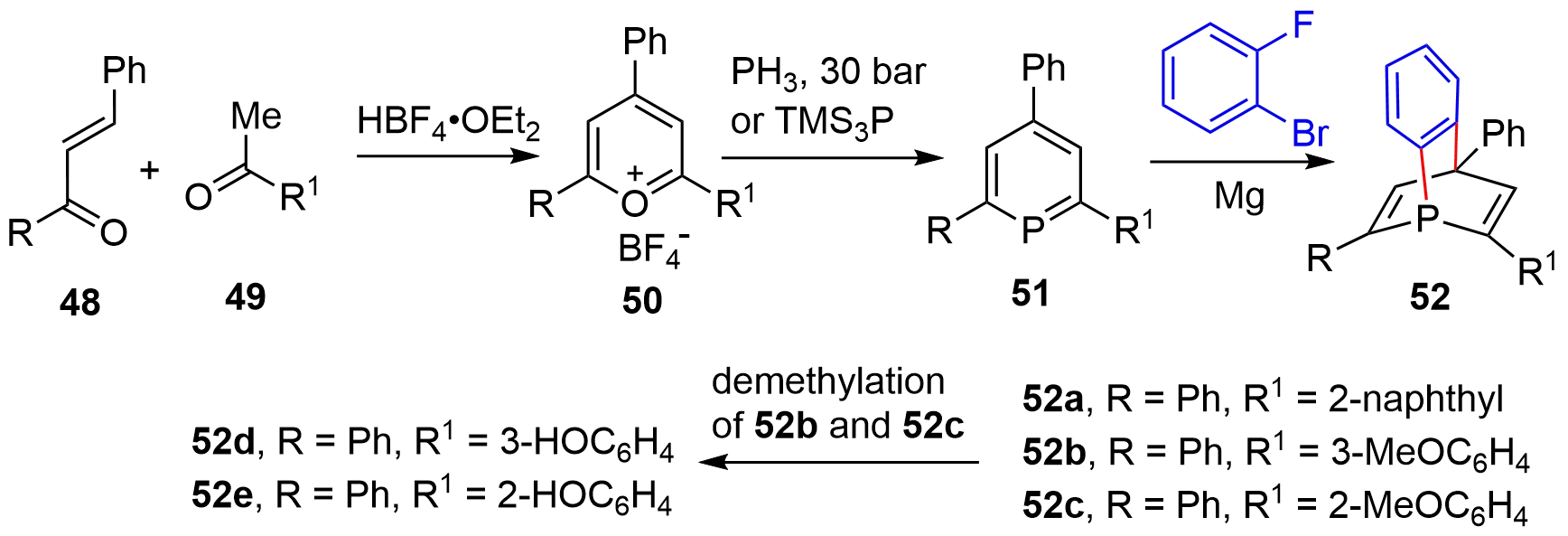
Figure 8. Synthesis of optically active [2.2.2]-bridged cycles through HPLC resolution. HPLC: high performance liquid chromatography.
3. Application of Bridged-Cyclic Chiral Phosphorus Compounds in Asymmetric Catalysis
3.1 Application as chiral ligands of transition-metals
3.1.1 Transition-metal-catalyzed asymmetric hydrogenation
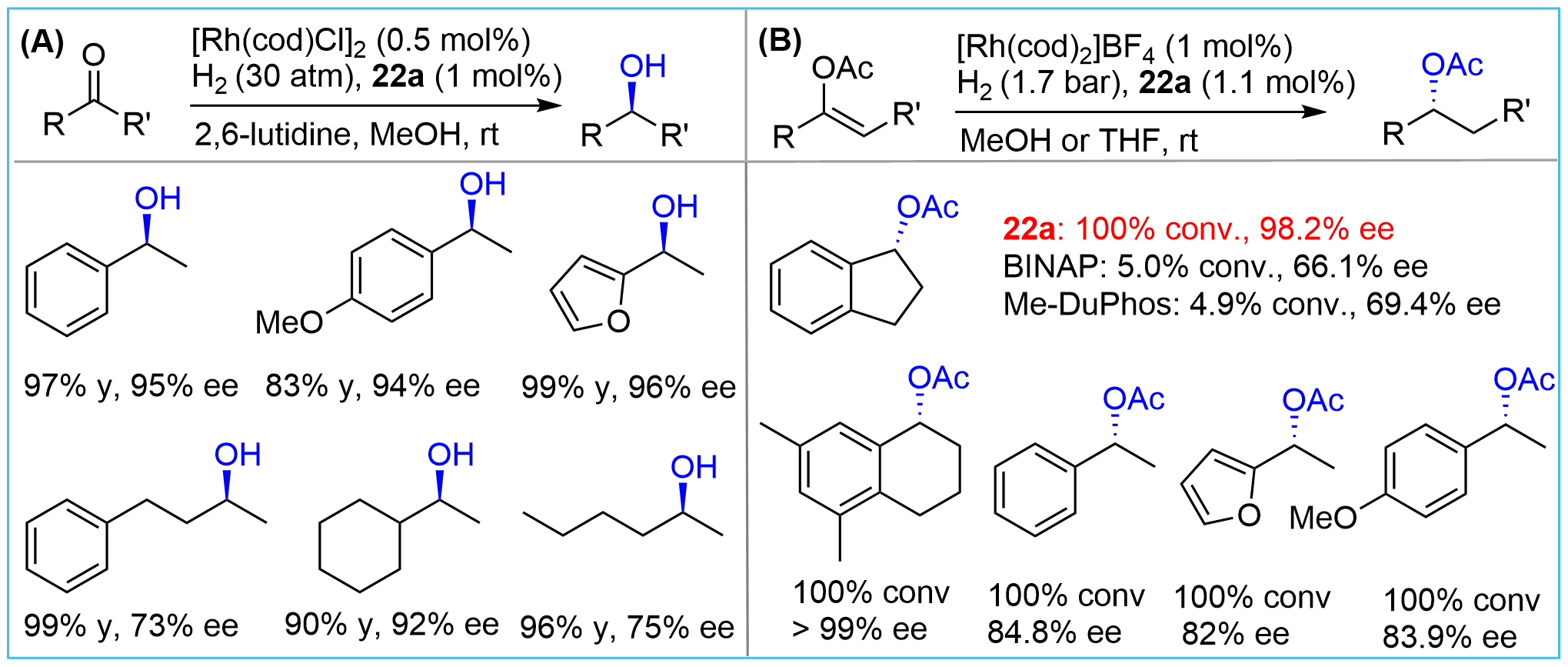
After synthesizing the bridged-cyclic chiral phosphines, Zhang and co-workers tested the efficiency of their new compounds as chiral ligands for transition-metal-catalyzed hydrogenation of various types of substrates[15,24]. Using their bis-bicyclic phosphine 22a (Figure 3), hydrogenation of simple ketones[15] and enolates[24], which are challenging substrates due to the lack of heteroatoms as anchoring sites, could be achieved with high efficiency (Figure 9). Good enantioselectivity for dialiphatic ketones was also observed although the ee values were somewhat lower than those for the arylaliphatic ketones (Figure 9A). Notably, a parallel comparison of the hydrogenation of enol acetate derived from 1-indanone showed that ligand 22a provided excellent conversion and enantioselectivity (Figure 9B). In stark contrast, the bidentate 2,2’-bis(diphenylphosphino)-1,1’-binaphthyl 2,2’-bis (diphenylphosphino)-1,1’-binaphthyl (BINAP) and 1,2-bis[(2R,5R)-2,5-dimethylphospholano]benzene (Me-DuPhos) showed a significantly decreased efficiency both for ee values and reactivity, exemplifying the crucial role of conformational rigidity. Further study demonstrated that 22a could also serve as an effective ligand for the hydrogenation of Rh-catalyzed enamides[25].
{kind=link}
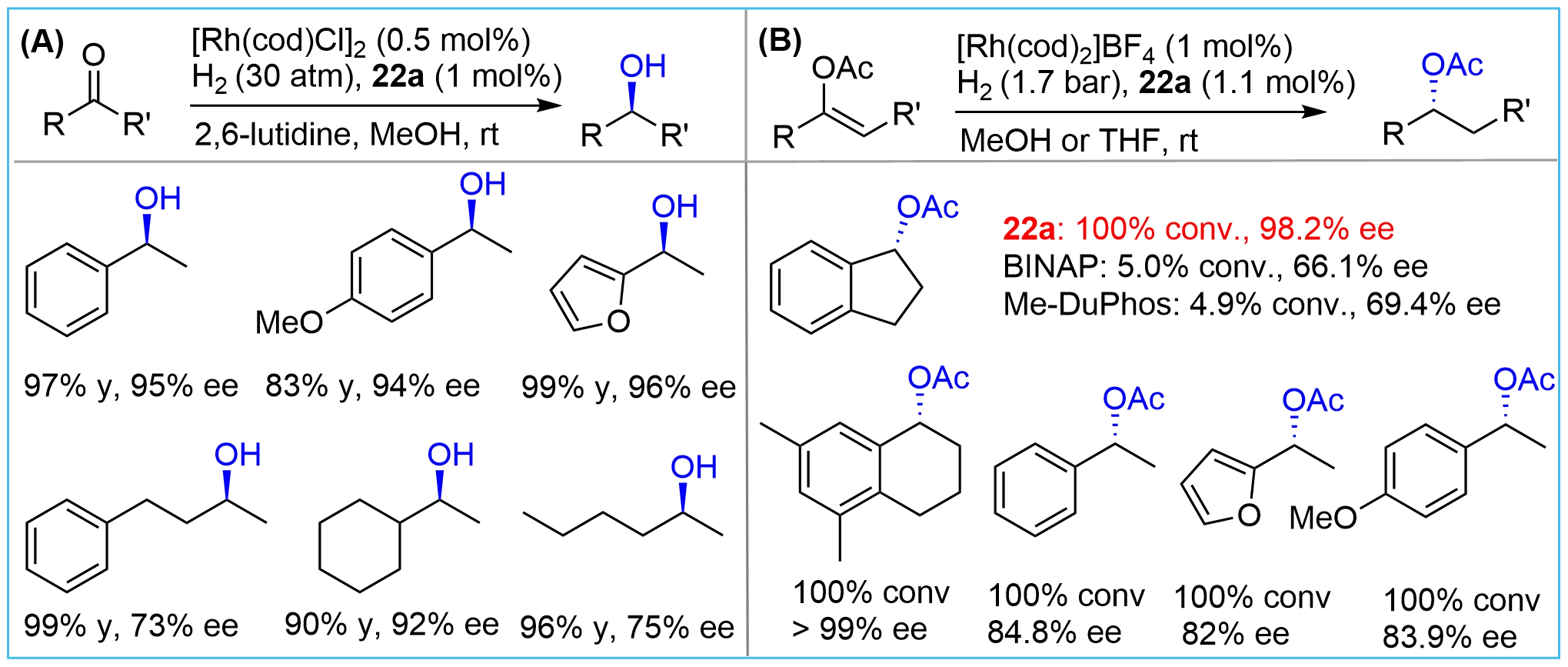
Figure 9. Rh-catalyzed asymmetric hydrogenation of ketones and enolates using phosphine 22a as ligand.
Mathey compared the efficiency of bis-bicyclic chiral phosphines 38 (Figure 6) with BINAP, {[2,2-dimethyl-1,3-dioxolane-4,5-diyl]dimethanediyl}bis(diphenylphosphane) (DIOP), and 2,2'-bis(dicyclohexylphosphino)-6,6'-dimethyl-1,1'-biphenyl) (BICHEP) using the Rh-catalyzed hydrogenation of several olefins and ketones[20]. Similar or slightly better results were obtained from these structurally different ligands. In a later study[10,22], the authors investigated the Rh-catalyzed hydrogenation of two functionalized olefins using bis-bicyclic phosphinite 13 (Figure 2) and phosphines 46 and 47 (Figure 7) developed by themselves as ligands. The results showed that 13 and 46 were highly efficient for both substrates in terms of conversion rate and enantioselectivity (Figure 10). In addition, investigations into the hydrogenation of several ketones and imines revealed low to moderate enantioselectivity although quantitative conversions were also observed. It should be mentioned that Breit and coworkers also evaluated the efficiency of bridged-bicylic chiral phosphines derived from their [2.2.2]-bicyclic phosphabarrelenes in Rh-catalyzed hydrogenation of enones[23]. However, only low stereoselectivity was obtained in most cases.
{kind=link}
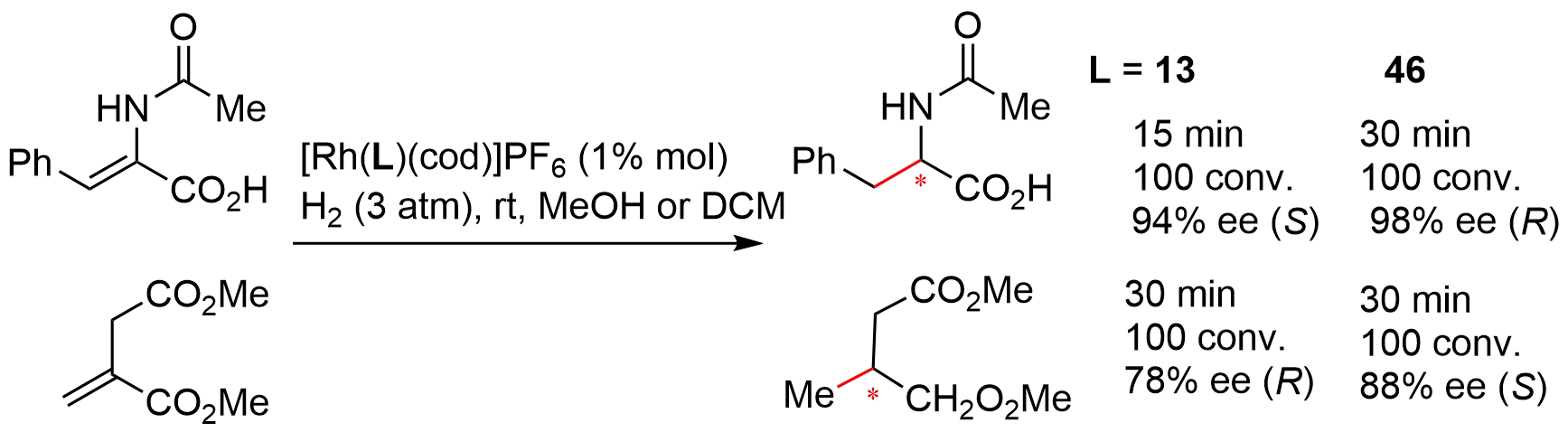
Figure 10. Rh-catalyzed asymmetric hydrogenation of functionalized olefins.
3.1.2 Transition-metal-catalyzed other asymmetric reactions
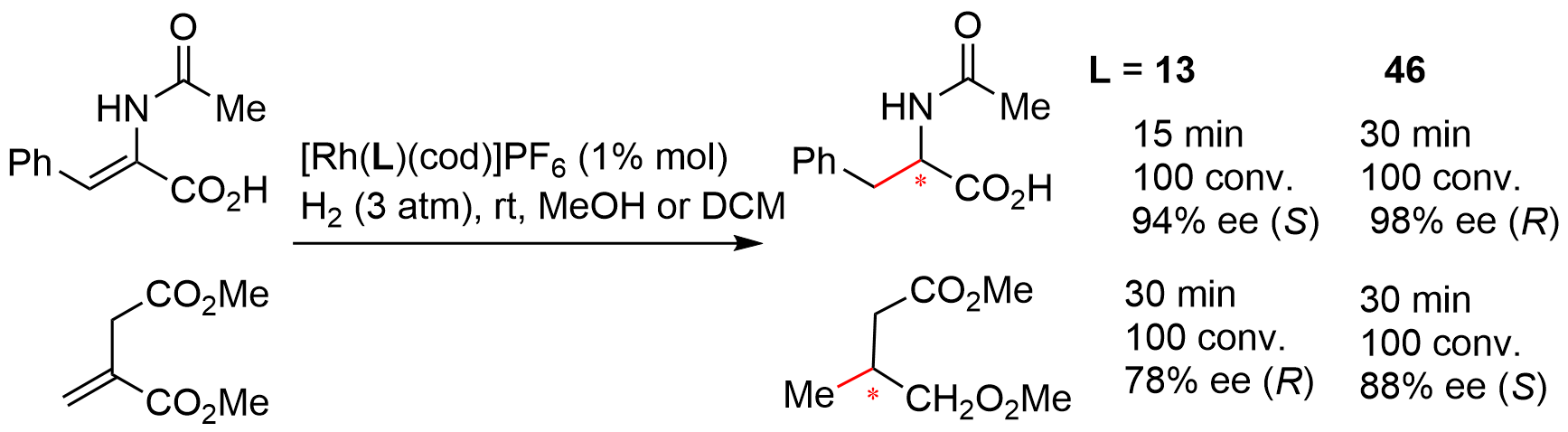
In 1997, Zhang and coworkers tested the efficiency of bridged-bicyclic chiral monophosphines synthesized in his group as chiral ligand for Pd-catalyzed allylic alkylation of 1,3-diphenyl-2-propenylacetate 53 as a template substrate[13]. Ligand 20a afforded the alkylated products 54 with excellent enantioselectivity as well as conversions under the optimized conditions (Figure 11). Of note is that, although many bidentate ligands displayed excellent performance for this reaction, simple chiral monophosphine ligands were usually less effective[26]. Therefore, these results further demonstrated the advantages of conformational rigidity in the ligand design. In addition, Mathey also briefly examined the performance of bis-bicycle 46 for Pd-catalyzed allylic alkylation with 53; the product ent-54a was obtained in 95% yield with 86% ee[22].
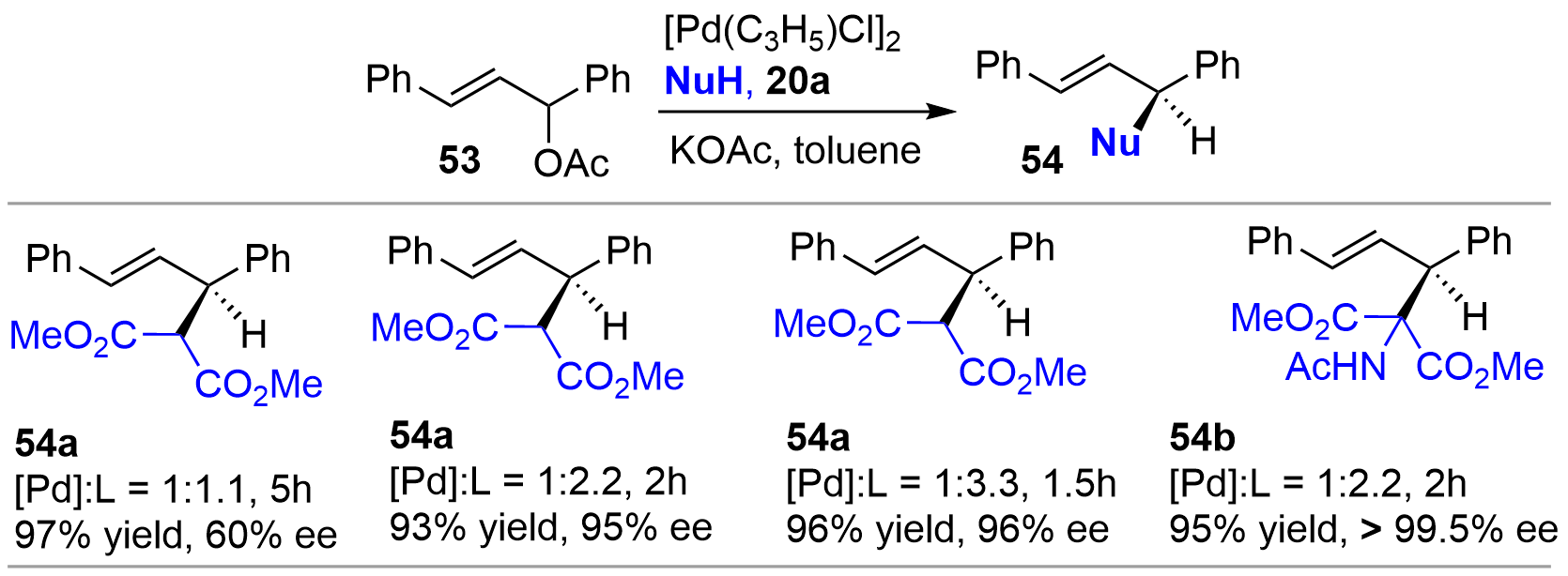{kind=link}
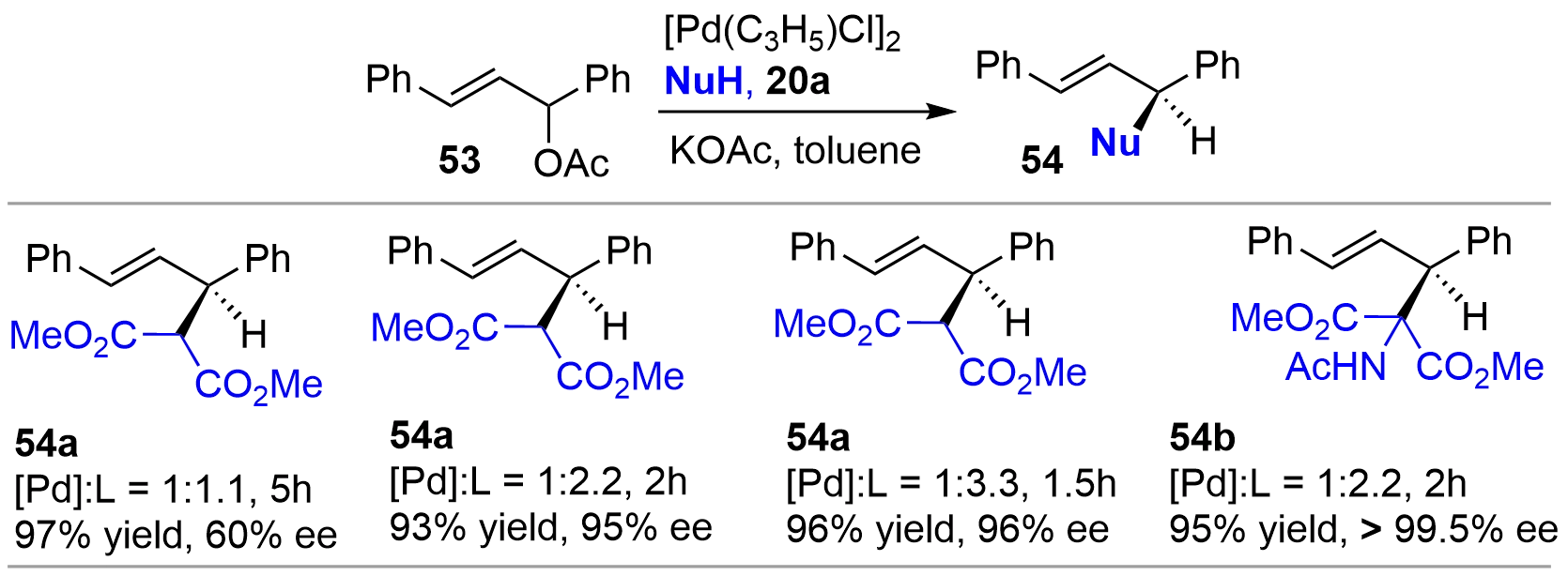
Figure 11. Pd-catalyzed asymmetric allylic alkylation using chiral monophosphine 20a as ligand.
In 2000, based on the similar method reported by Mathey as shown in Figure 7, Gilbertson synthesized a series of [2.2.1]-bicyclic P-chiral phosphine-oxazole derivatives 55a–d[27] (Figure 12). Using these phosphines as chiral ligands, the Pd-catalyzed asymmetric alkylation of 53 was implemented. Excellent conversion as well as ee values were harvested for ent-54a from 55a and 55b. However, 55c and 55d afforded much poorer stereoselectivities.
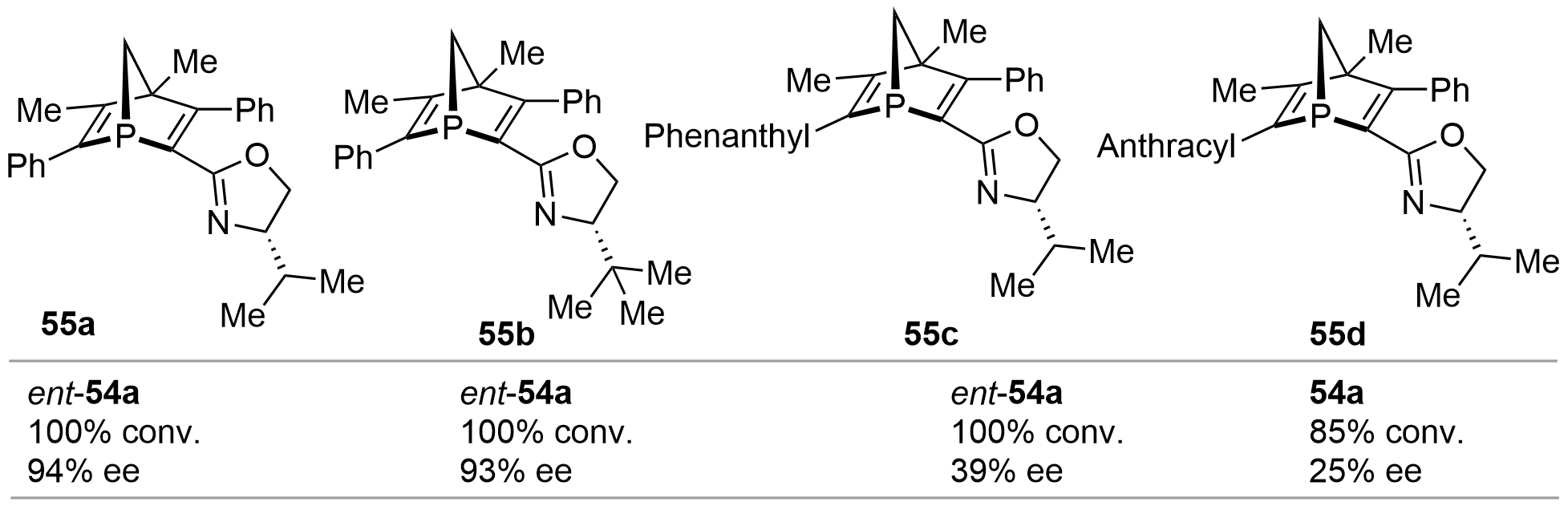{kind=link}
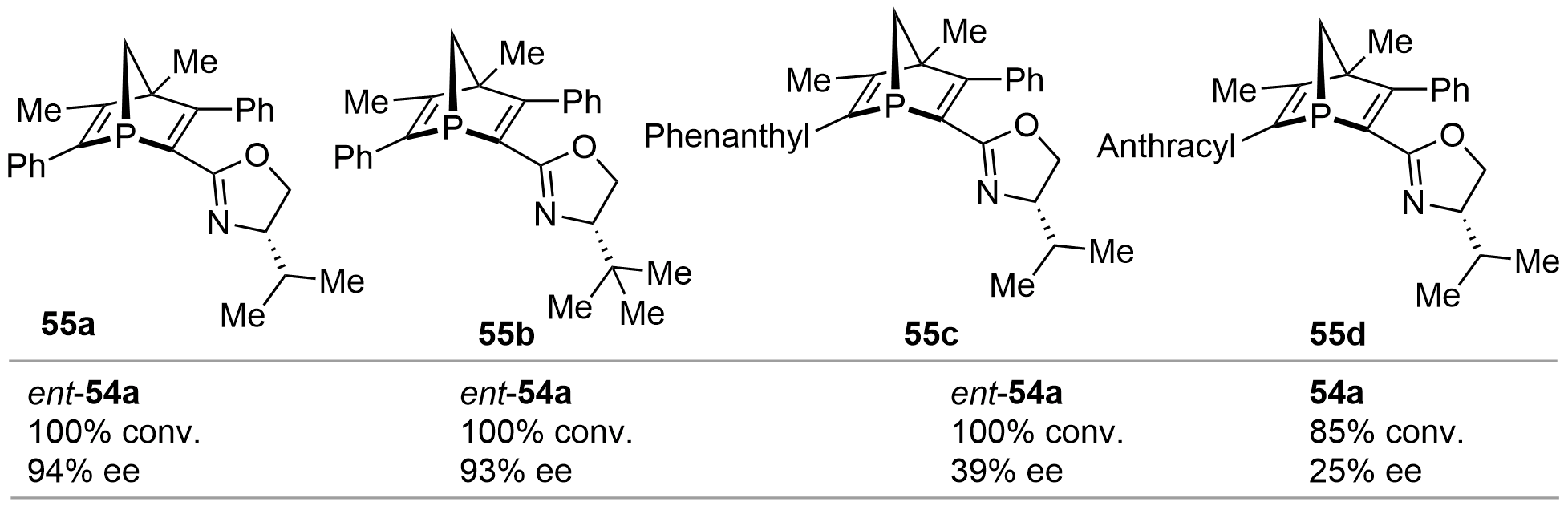
Figure 12. Pd-catalyzed asymmetric allylic alkylation using chiral phosphine 55 as ligand.
Using 55 as chiral ligands, the authors further evaluated their efficiency in the Pd-catalyzed asymmetric Heck cross-coupling reaction using 2,3-dihydrofuran and cyclohex-1-en-1-yl trifluoromethanesulfonate as substrates. Similar to the trend observed for the alkylation reaction, 55a and 55b offered the cross-coupled product in good yields of 73% and 91% as well as high enantioselectivity of 78% and 93% ee, respectively. However, 55c and 55d provided moderate stereoselectivity. In another study by Mathey[22], the Pd-catalyzed asymmetric Heck cross-coupling of PhOTf and 2,3-dihydrofuran using bis-bicyclic phosphine 46 as a chiral ligand could provide the chiral product in 70% yield with 95% ee.
By employing the skeletally similar P-chiral phosphine-sulfinamide compounds 55e and 55e’ (Figure 13A), obtained via column chromatography separation, as ligands, Li/Duan/Mathey in 2019 described a copper-catalyzed 1,3-dipolar cycloaddition of alkylidene succinimides 56 and imines 57 for the asymmetric synthesis of [4.4.0]-spiropyrrolines 58 and 58’, respectively[28] (Figure 13B). The reaction displayed a broad substrate scope and provided very high yields with excellent enantioselectivity. Almost at the same time, the authors also achieved an Ag-catalyzed 1,3-dipolar cycloaddition with 55e’ as ligand for efficient synthesis of [5.4.0]-spiropyrroline derivatives[29]. In 2022, the same group disclosed a Pd-catalyzed Tsuji-Trost-type reaction of carbonate 59 and benzooxathiazine 60 using 55f as chiral ligand[30] (Figure 13C). Benzooxathiazines 61 tethered with a chiral tetrahydro-2H-pyran ring with two vicinal stereocenters were synthesized. Except that a low enantioselctivity (32% ee) was obtained for a substrate bearing an alkyl tBu group in 60, high yields and enantioselectivity were obtained for a broad array of aryl substituted substrates.
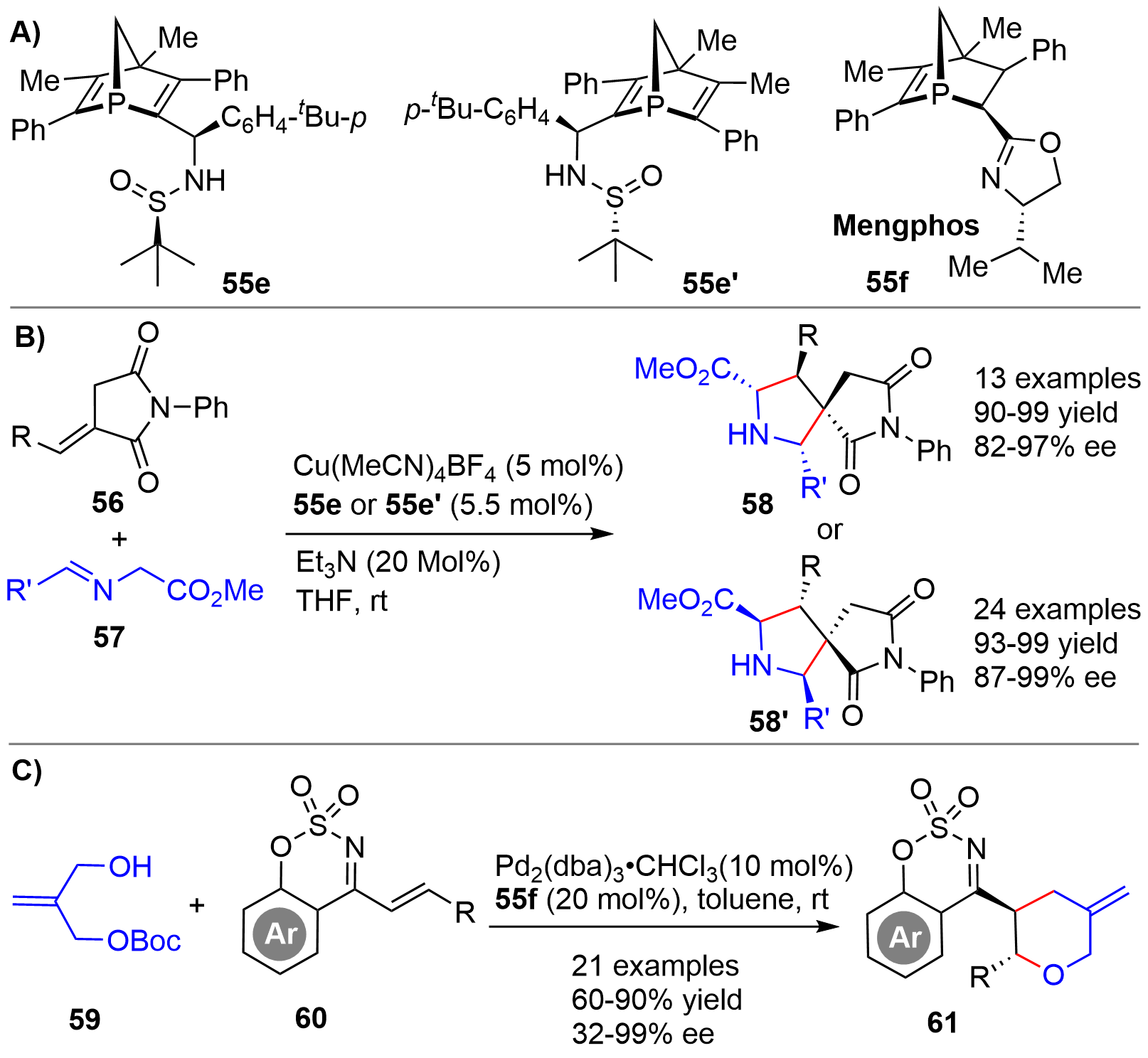{kind=link}
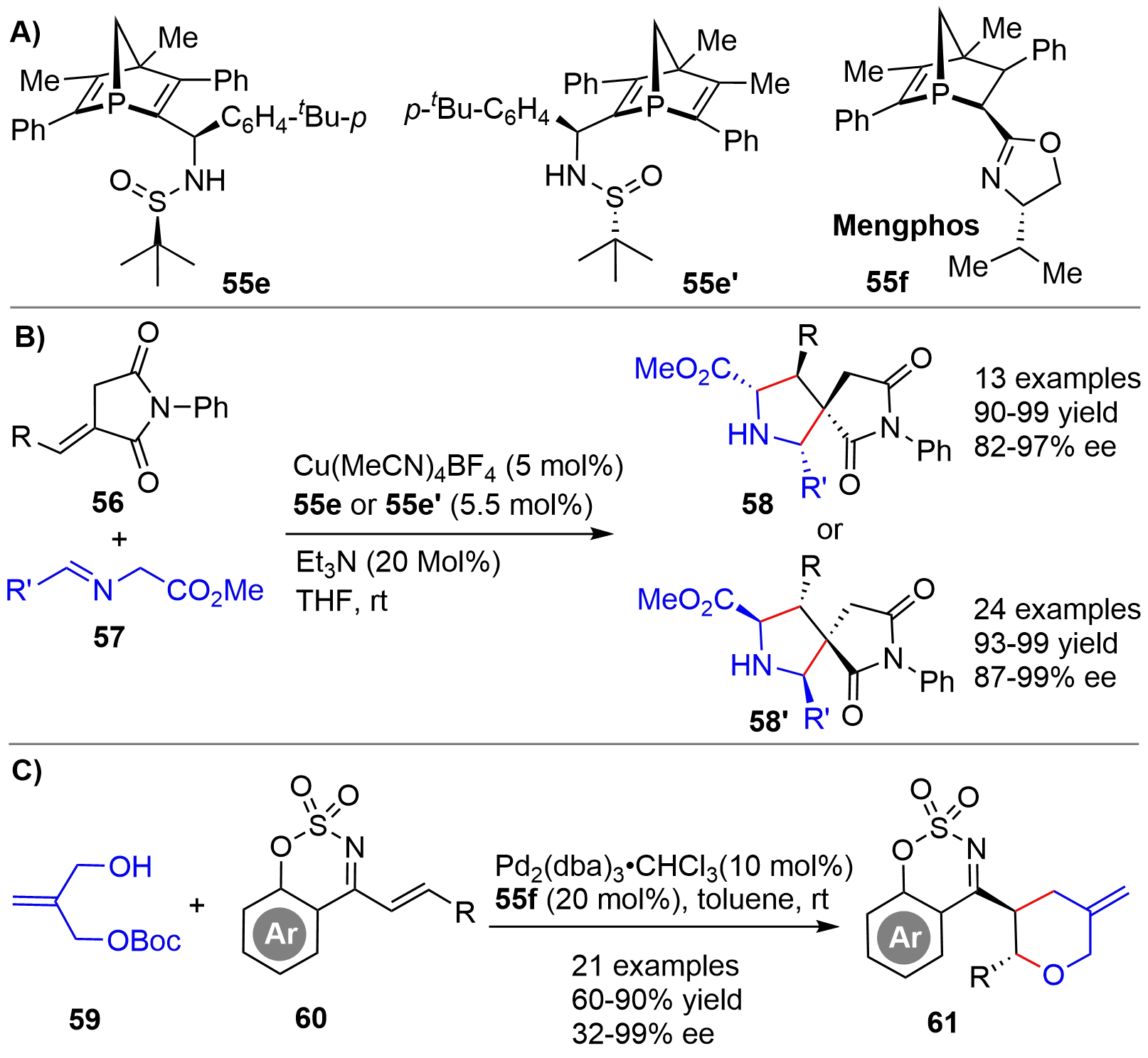
Figure 13. Structures of chiral bridged-cyclic phosphorus ligands (A), and Cu-catalyzed asymmetric 1,3-dipolar (B) and Pd-catalyzed Tsuji-Trost-type (C) cycloaddition.
3.2 Application as organocatalysts
3.2.1 Asymmetric [m+n] annulation with allenes
Hinted by Lu’s pioneering finding that phosphines could catalyze the [3+2] annulation reaction of 2,3-butadienoates or 2-butynoates with electron-deficient olefins for the synthesis of cyclopentenes[31], the Zhang’s group disclosed the first asymmetric [3+2] cycloaddition reaction of allenes with olefins using chiral phosphines as organocatalysts in 1997[14]. A screen of various catalysts showed that the bridged-bicyclic phosphines 20a and 20b exhibited much better catalytic performance than 62, 63, and BINAP as seen from the ratio of regioisomers of the products 64a:64’a and enantioselectivity (Figure 14A). Particularly, the enantioselectivity with [2.2.1] bridged-bicyclic 20a was much higher than that with the five-membered phosphine 63, which, once again, demonstrated the significance of conformational rigidity. In addition, 20a and 20b also showed high regioselectivity as well as enantioselectivity for other substrates (64b and 64c). It was proposed that the reaction proceeded through a concerted mechanism. Namely, the “bottom” face of the allylic anion was effectively blocked by the R groups of the rigid catalyst 20, which forced the olefins to approach from the “top” face with an endo orientation as shown in TS1 (Figure 14B). Consequently, products 64 were formed regio- and stereoselectively via Int1.
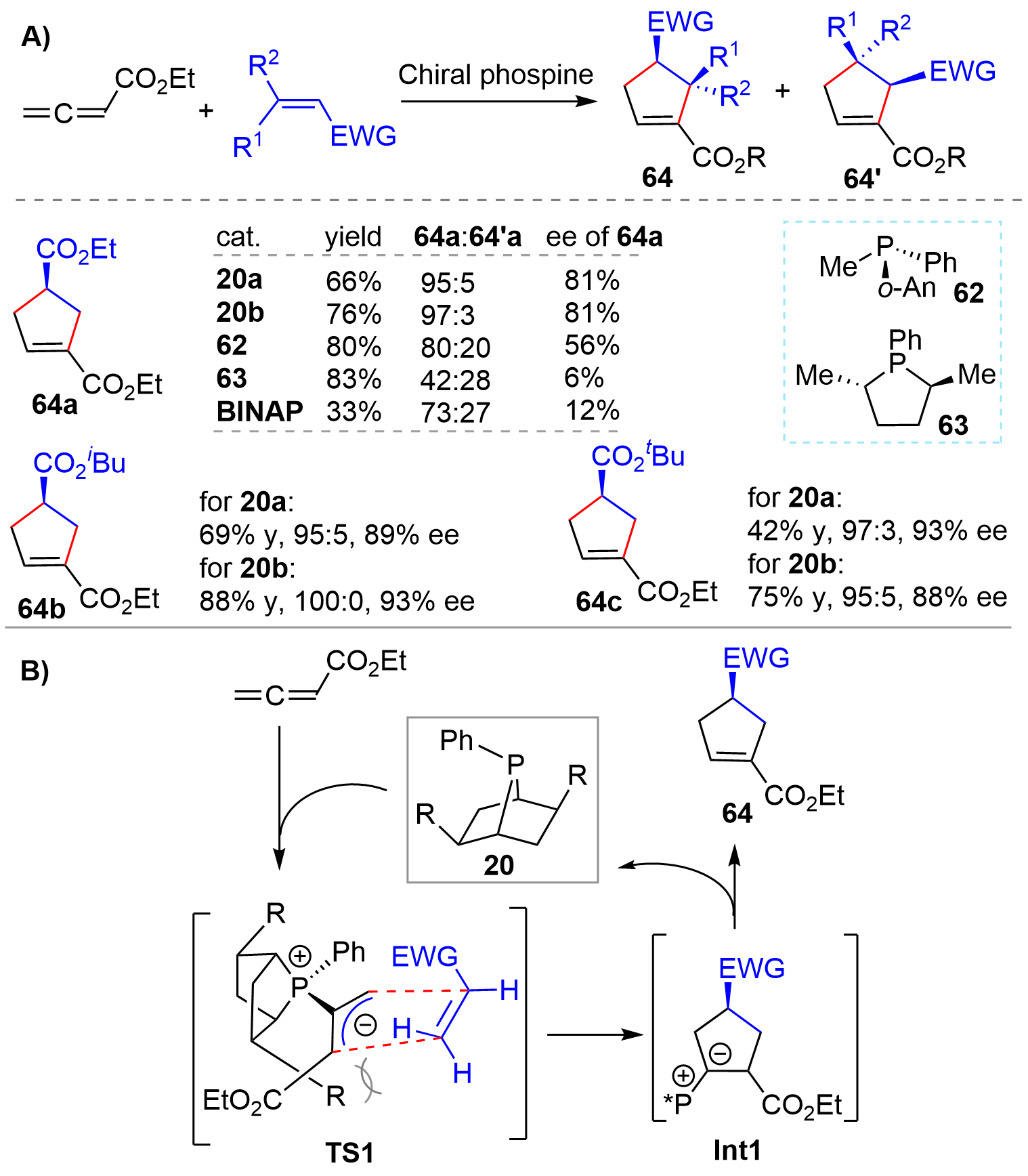{kind=link}
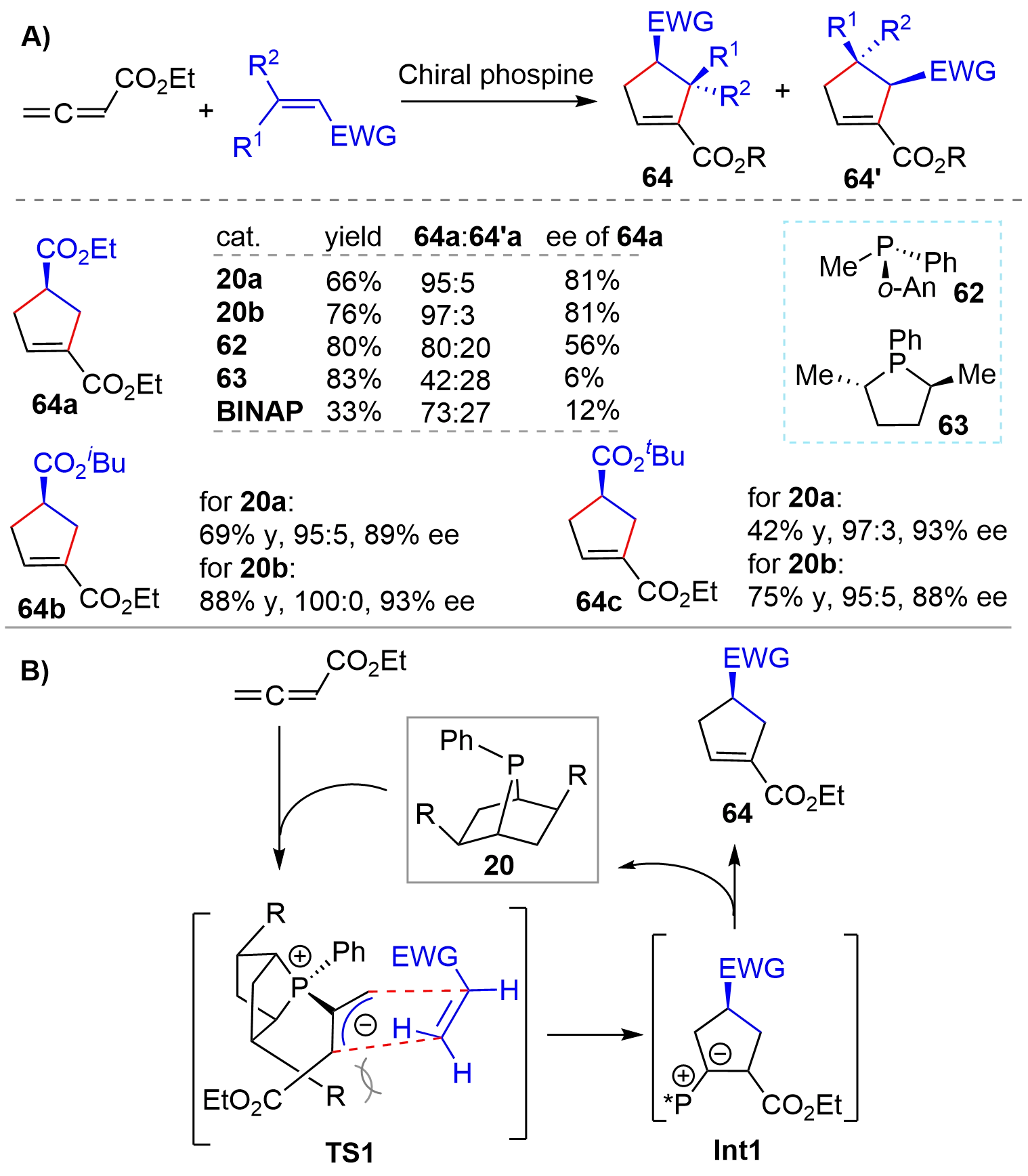
Figure 14. Chiral phosphine-catalyzed asymmetric [3+2] cycloaddition of allene and olefin.
In 2014, the Kwon’s group reported a [3+2] cycloaddition of allenes with imines[16]. Using the aza-[2.2.1]-bicyclic chiral phosphines exo-28 and endo-28’ (Figure 4) as organocatalysts, the authors successfully established a set of efficient conditions for the asymmetric synthesis of pyrrolines with two stereogenic centers (Figure 15). To compare with the earlier reports for asymmetric synthesis of pyrrolines[32,33], the bridged-bicyclic phosphines were compatible with a broader range of substrates and, in general, afforded high yields with excellent ee, although small R groups on allenes (65i and 65j) and electron-rich aryl groups on sulfonyl (65c and 65d) resulted in a marked decrease of enantioselectivity for catalyst 28. Interestingly, the diastereoisomeric 28’ not only provided the chiral products with opposite enantiomers but also with significantly improved enantioselectivity as shown from the results of 65c vs. ent-65c and 65d vs. ent-65d. Consequently, both (+)- and (-)-pyrrolines could be synthesized from the pseudoenantiomeric phosphine catalysts in high yields with excellent enantioselectivity. Based on DFT calculations, it was suggested that the stereoselection was governed mainly by the TS2 and TS2’, respectively, for the two catalysts. Both transition states were stabilized by two common factors including a hydrogen bond between the N-sulfonyl oxygen atom on the imine and a hydrogen atom on the bicyclic system of the catalyst, and a Coulombic interaction between the allenoate C=O oxygen atom and the phosphorus atom. Later in 2018, Kwon demonstrated that phosphines with structures featuring 31 derived from (R)-(-)-carvone (Figure 4) were also powerful catalysts for the [3+2] annulation of allenes and imines, affording pyrrolines with broad structural diversity, high yields, and excellent enantioselectivity[17].
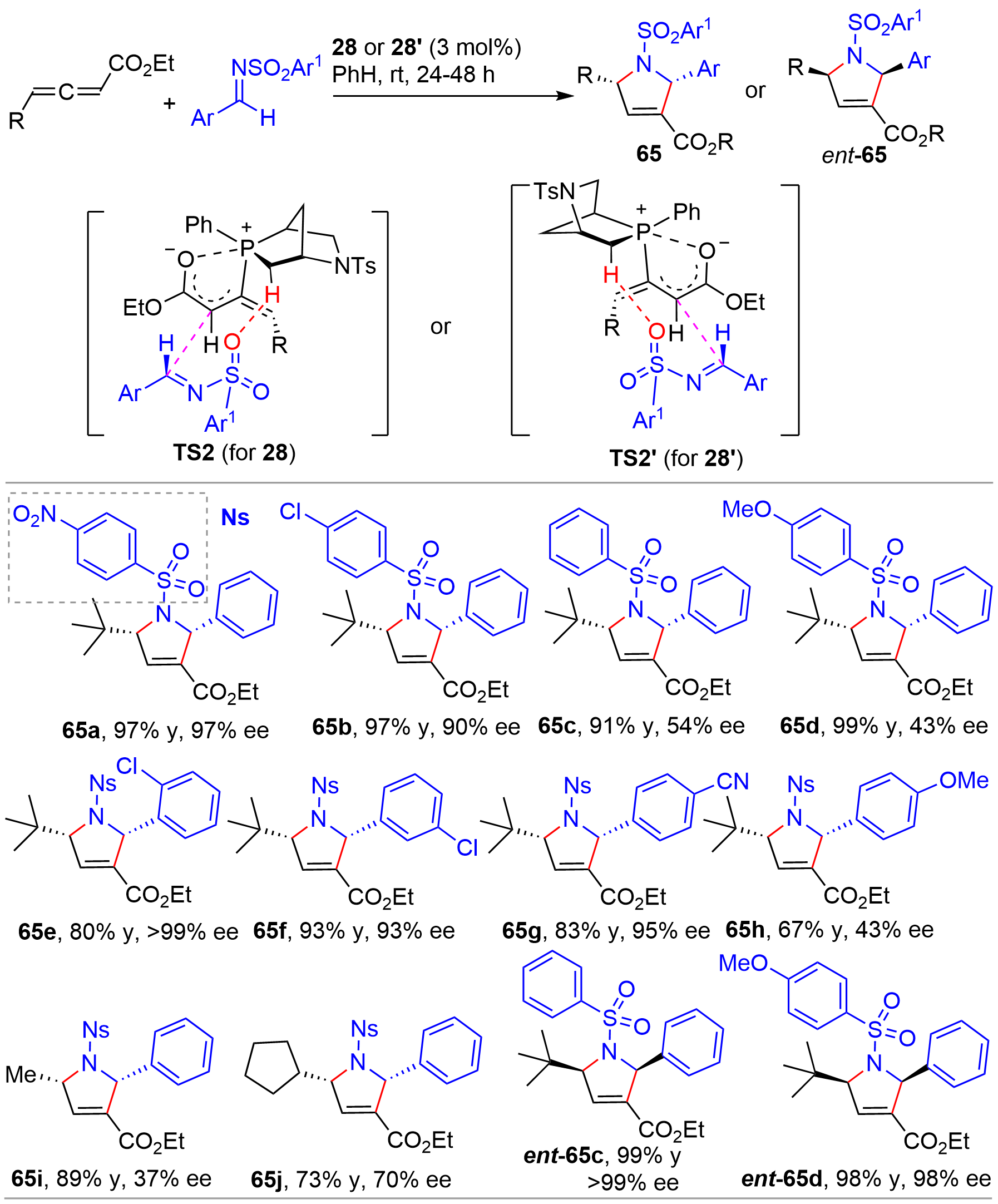{kind=link}
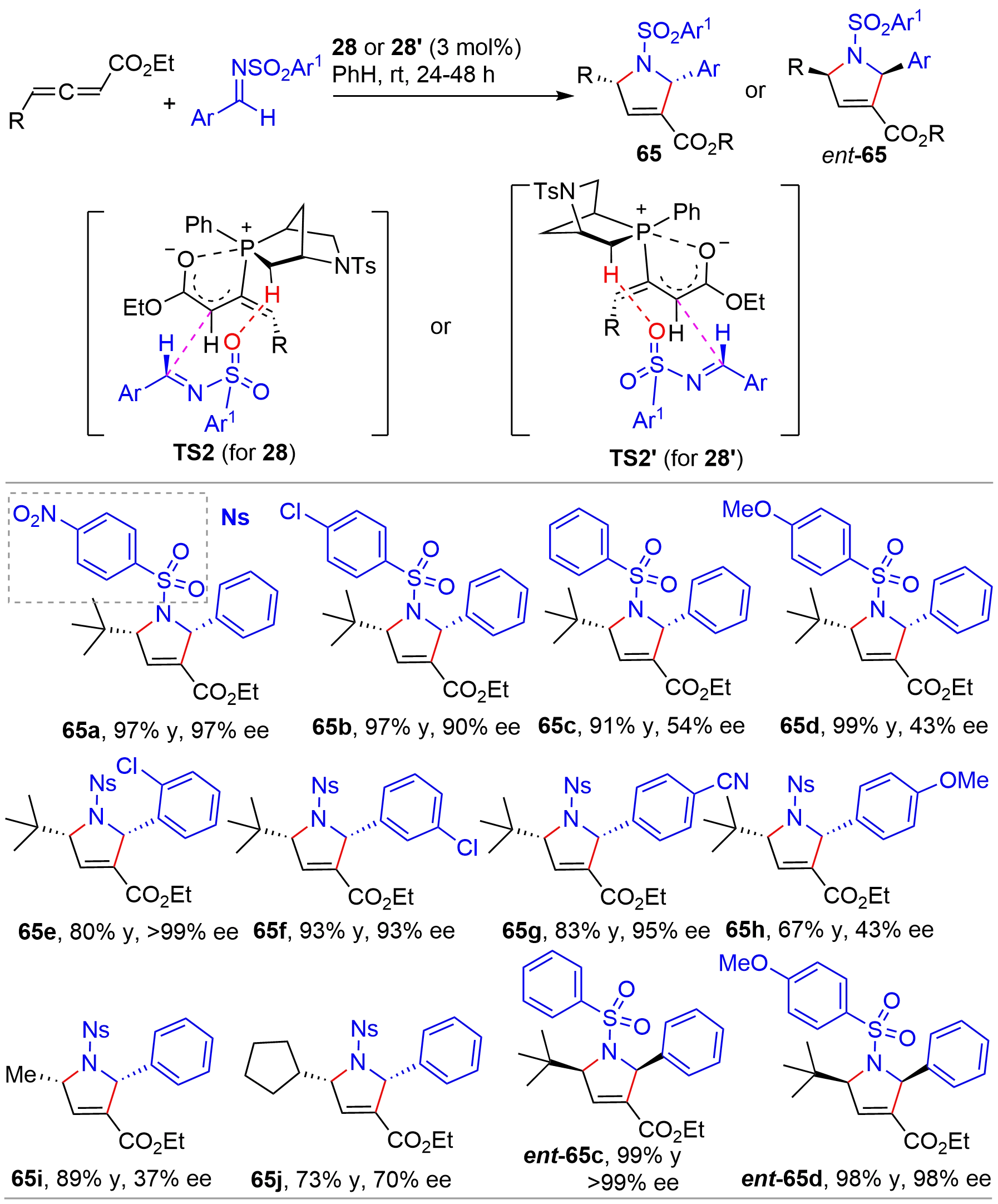
Figure 15. Chiral phosphine-catalyzed asymmetric [3+2] cycloaddition of allenes and imines.
The practical applications of the novel method were culminated by the efficient enantioselective syntheses of two natural products (+)-ibophyllidine[34] and (-)-actinophyllic acid[35]. As illustrated in Figure 16, asymmetric [3+2]-annulation of indolyl imine 66 and allene 67 catalyzed by 28’ afforded the cycloaddition product in excellent yield and ee, which upon hydrogenation gave 68 bearing three cis-stereocenters. From this key intermediate, a concise enantioselective synthesis of (+)-ibophyllidine could be achieved. Similarly, the asymmetric reaction of 66 and 69 using Kwon’s chiral phosphine 28’a as catalyst delivered 70 in high yield and excellent stereoselectivity, which was then used to accomplish the enantioselective total synthesis of (-)-actinophyllic acid.
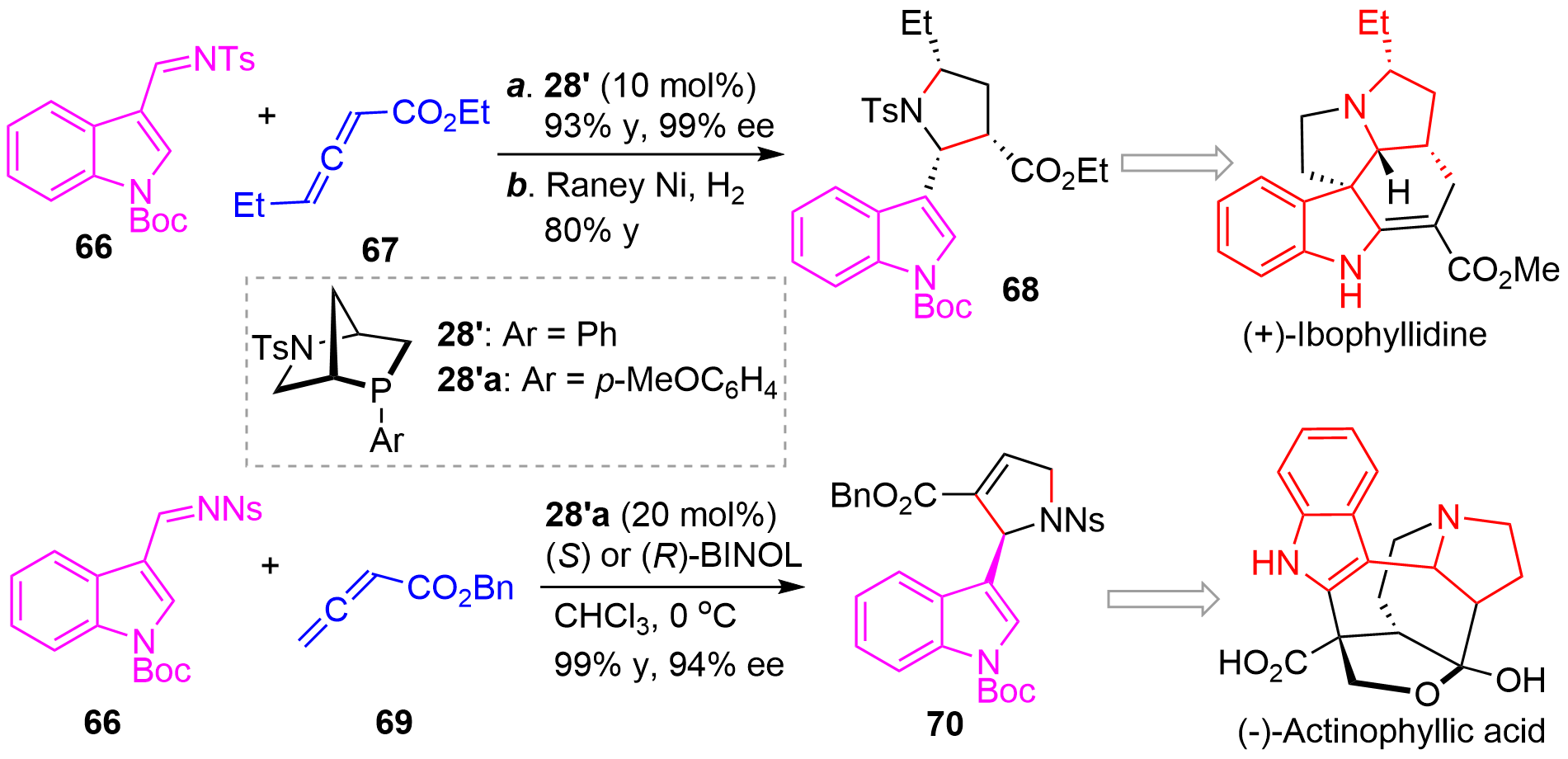{kind=link}
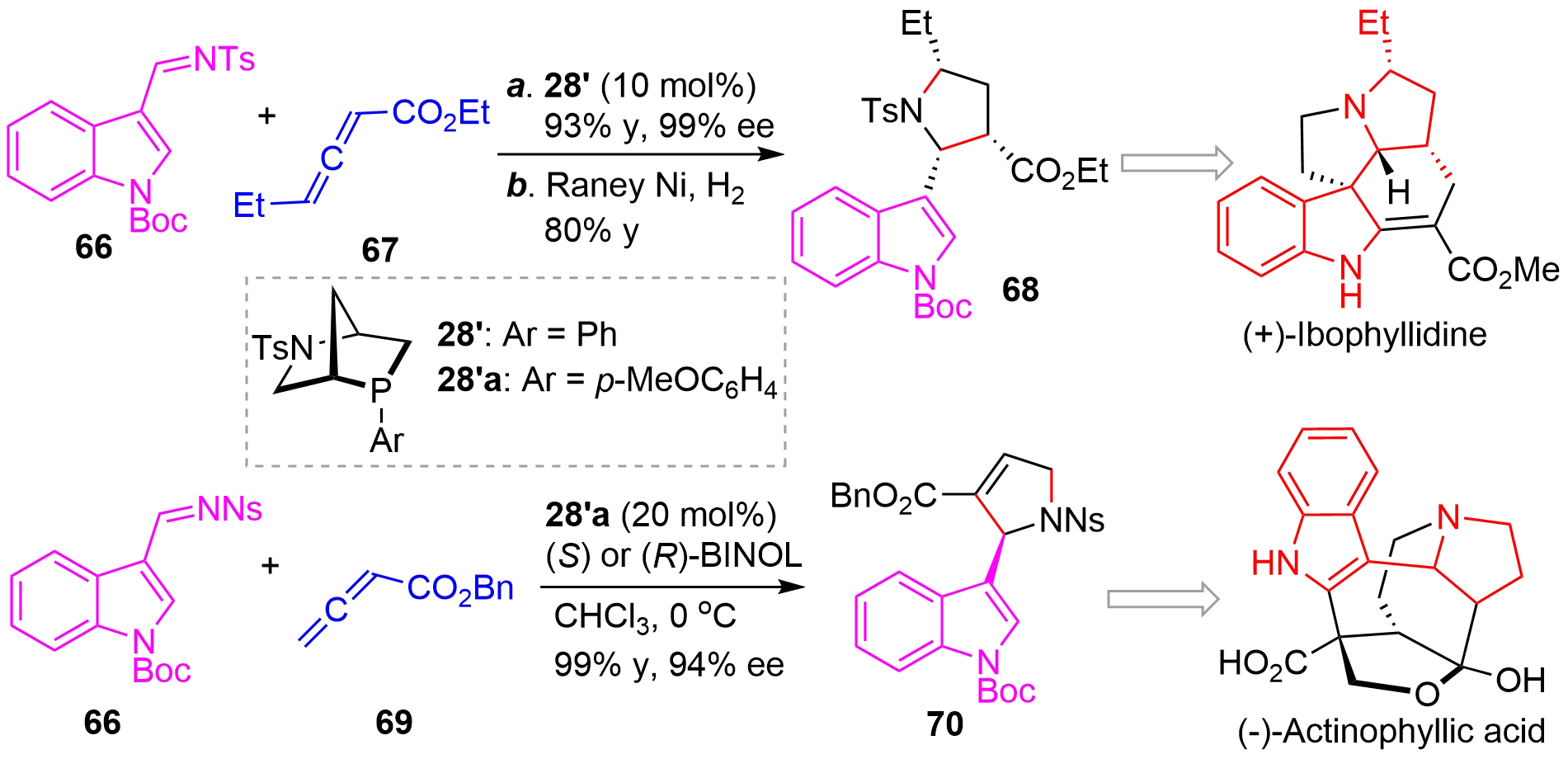
Figure 16. Application of the asymmetric [3+2] cycloaddition in the enantioselective synthesis of natural products.
Following Zhang’s and Kwon’s pioneering works[14,16], various other types of asymmetric catalytic annulation of allenes using bridged-cyclic chiral phosphorus as organocatalysts were reported[36-40] (Figure 17). In 2015, a successful demonstration of an asymmetric -addition/[4+4]cycloaddition domino reaction was reported by Tong using Kwon’s catalyst[36], delivering 2-oxabicyclo[3.3.1]nonanes 71 (Figure 17A). In 2016, Guo and coworkers reported a [4+3] annulation for the construction of quinazoline-based tricyclic heterocycles 72[37] (Figure 17B) and a [2+4] annulation for synthesizing functionalized 6,7-dihydro-5H-pyrano[2,3-d]thiazole derivatives 73[38] (Figure 17C). The Kwon’s group, following their extensive investigations on asymmetric [3+2] annulation for the synthesis of five-membered pyrrolidines, achieved the asymmetric synthesis of six-membered guvacine derivatives 74 in 2018 through a [4+2] annulation[39] (Figure 16D). Alternatively, by also applying a [4+2] annulation strategy, the team of Li/Duan could create the tricyclic spirooxindoles 75 and ent-75 in 2021[40] (Figure 17E). Notably, all the above [m+n] annulation reactions showed high yields and enantioselectivity.
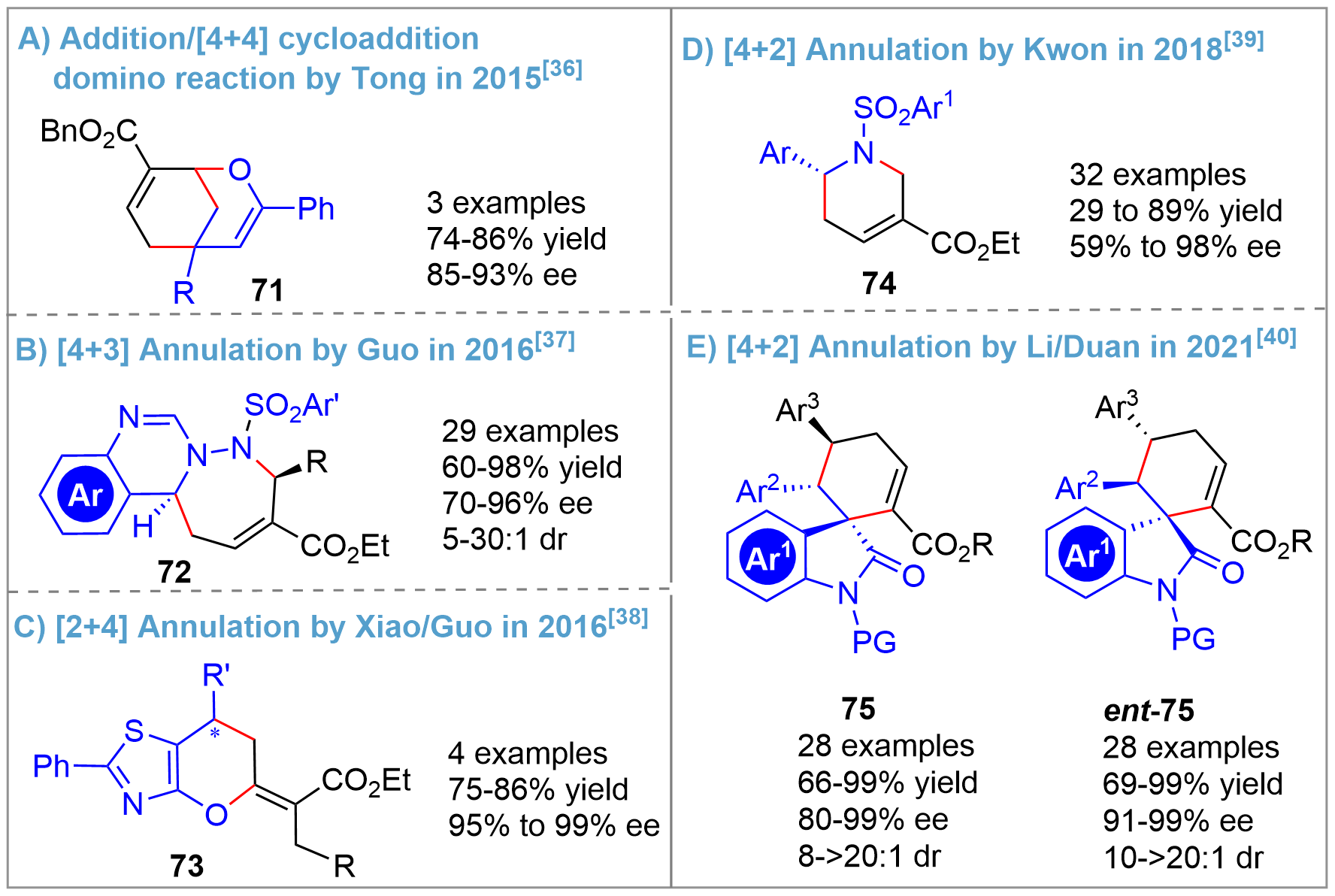{kind=link}
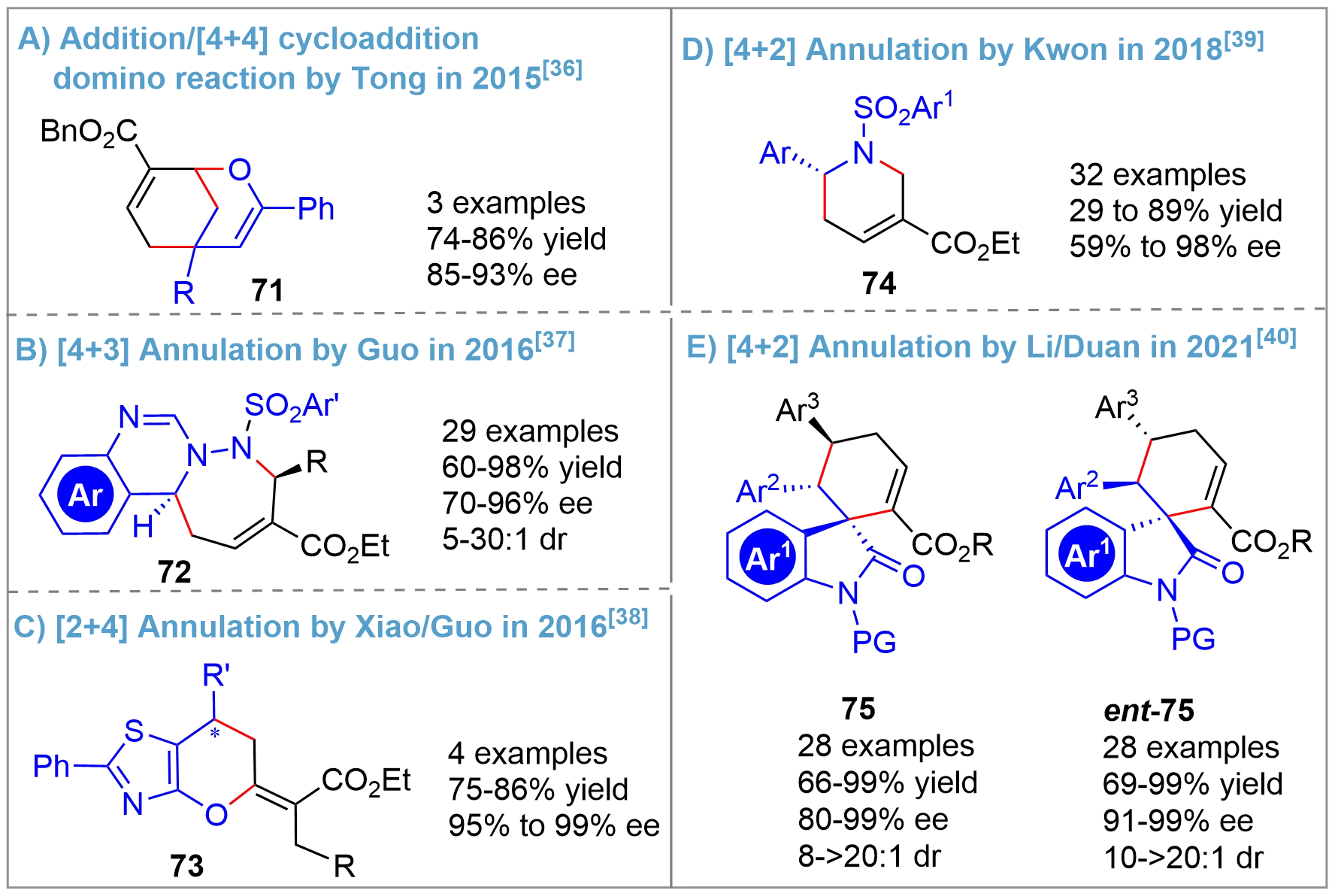
Figure 17. Other asymmetric annulation reactions of allenes catalyzed by bridged-cyclic P-stereogenic phosphines.
3.2.2 Asymmetric Morita–Baylis–Hillman-type (MBH) reaction
The asymmetric MBH reaction is an important method for the synthesis of chiral building blocks with dense functionalities[41,42]. In this regard, the bridged-cyclic chiral phosphines were also demonstrated to be powerful organocatalysts for asymmetric MBH-type reactions. In 2016, Ouyang/Chen disclosed an asymmetric MBH-type reaction of isatin-derived carbonates 76 and 2-alkylidene-1H-indene-1,3(2H)-dione derivatives 77 using the catalyst 28, affording the pentacyclic frameworks 78 bearing two vicinal all-carbon spirocenters in high yields with excellent enantio- and diastereo-selectivity[43] (Figure 18A). Shortly after, the Guo’s team reported a reaction of carbonates 79 and azomethine imine derivatives 80 using 28’ as a chiral phosphine catalyst[44] (Figure 18B). Generally, excellent yields as well as enantioselectivity for the multi-N-containing heterocycles 81 were obtained from a broad range of substrates. Recently, the He’s group reported the asymmetric synthesis of a densely substituted cyclopentene with two chiral centers[45] (Figure 18C). In the presence of chiral phosphine catalyst 28’, the reaction of allylic acetates 82 and polar dienes 83 proceeded smoothly to afford the cyclic products 84 in good yields with moderately high to high enantioselectivity.
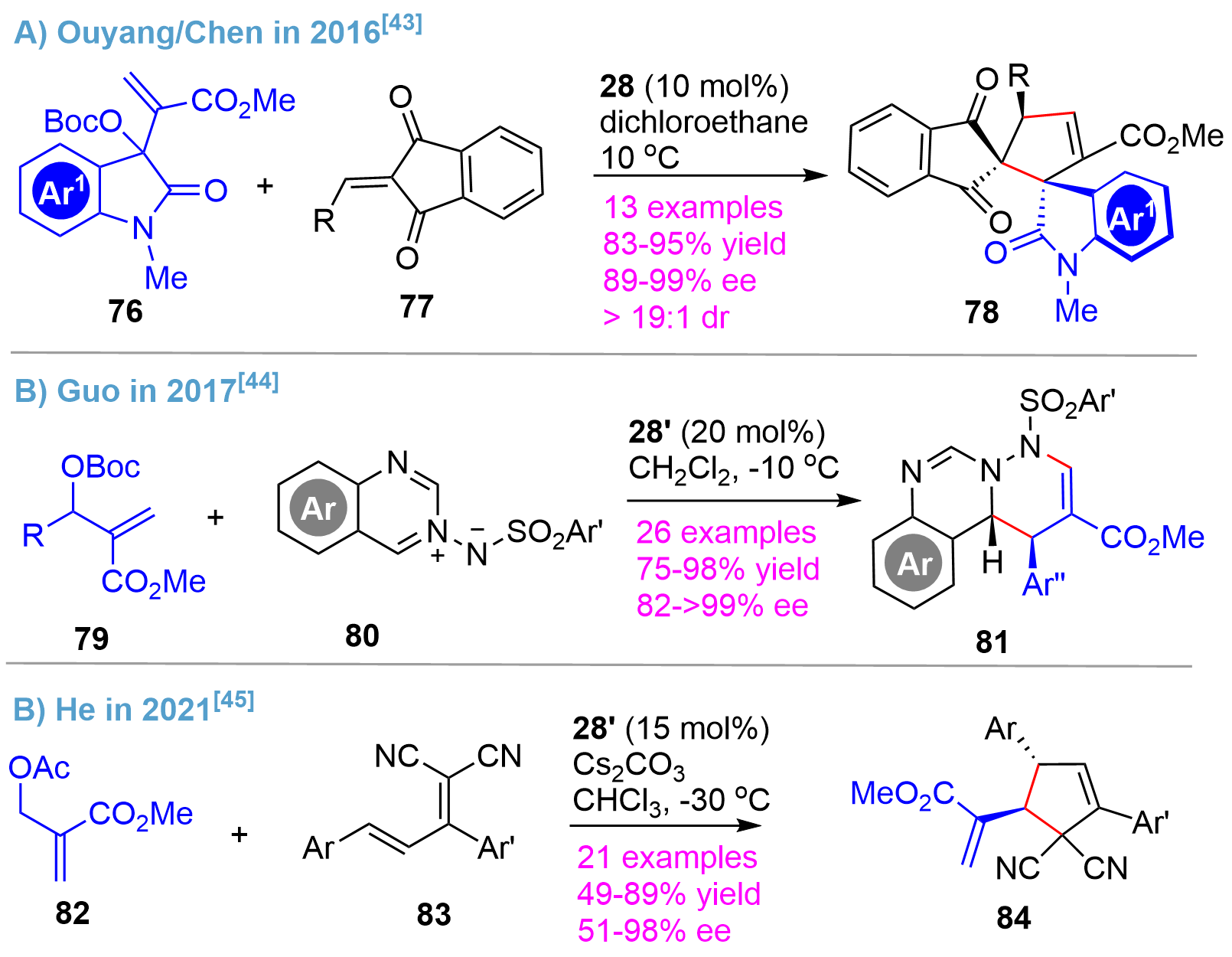{kind=link}
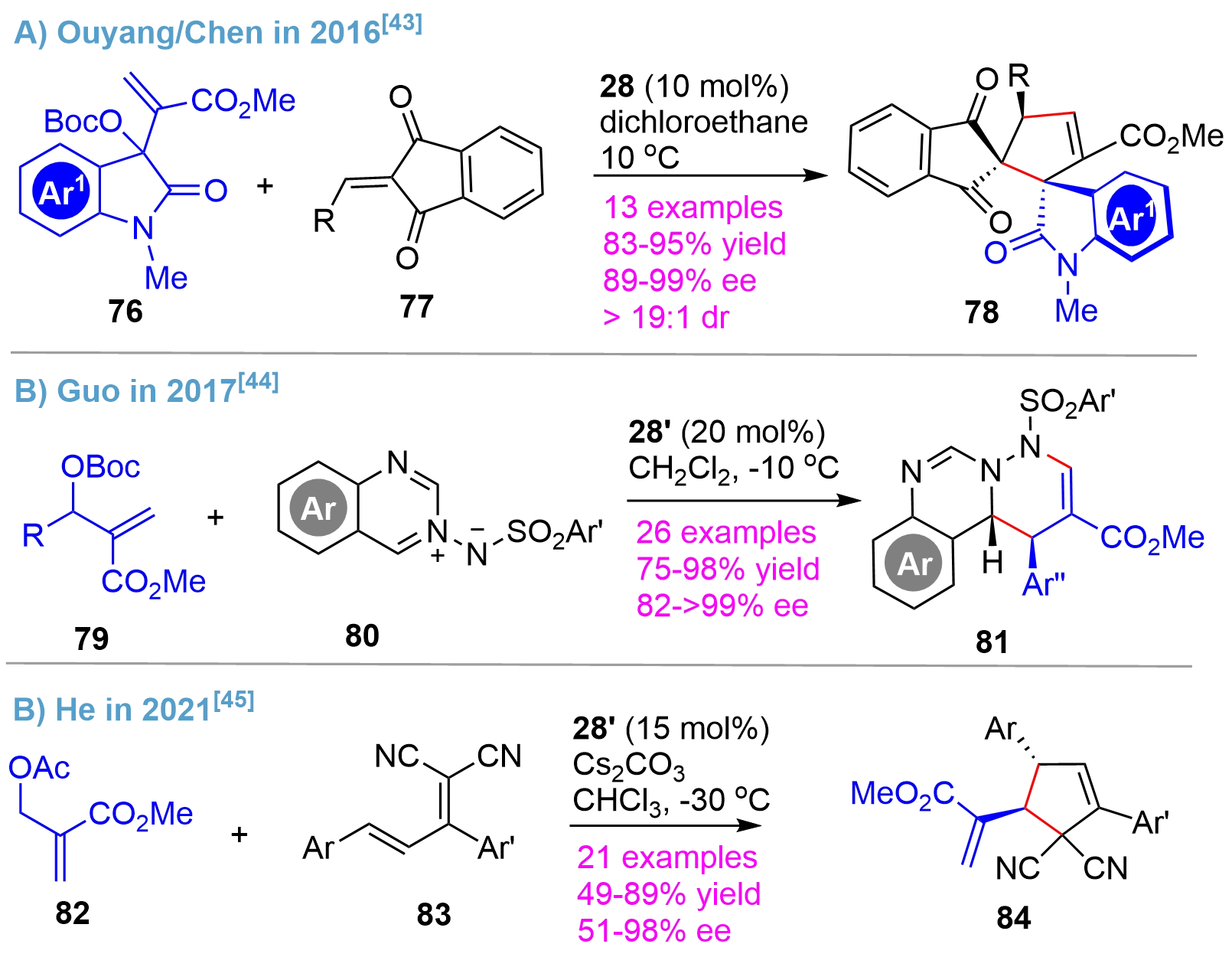
Figure 18. Asymmetric MBH-type reactions catalyzed by bridged-cyclic P-stereogenic phosphines. MBH-type: Morita–Baylis–Hillman-type.
3.2.3 Asymmetric Staudinger–aza-Wittig reaction
Chiral heterocyclic amines possessing chiral quaternary carbon centers are very common structural elements in biologically active compounds and natural products[46,47]. However, catalytic asymmetric synthesis of such scaffolds, especially of the angular all-carbon quaternary centers, remains a great challenge. The reported examples often proceeded with low efficiency and required a stoichiometric amount of chiral phosphines[48]. In 2019, Kwon and his colleagues reported for the first time a catalytic desymmetric asymmetric intramolecular Staudinger–aza-Wittig reaction of diketone azides 85 or 86 that could achieve N-heterocyclic imines 87 or 88, respectively, using their bridged-cyclic P-stereogenic phosphines as catalysts[49] (Figure 19A). High efficiency of the catalysts 28 and 28’ was exemplified by the broad scope of substrates, high yields, and enantioselectivity via an in situ P(III)/P(V)=O redox strategy, whereas earlier reports required stoichiometric chiral phosphines, limited substrate compatibility, and significantly lower yields and enantioselectivity. The high catalytic activity of the [2.2.1]-bridged cyclic phosphine should be attributed to the faster reducibility from P(V)=O to P(III) resulting from a suitable C–P–C angle of the bridged cyclic motif compared with the non-bridged cycles and the more electron rich alkyl substituents connected with the phosphorus atom compared with the triaryl-substituted counterpart[50].
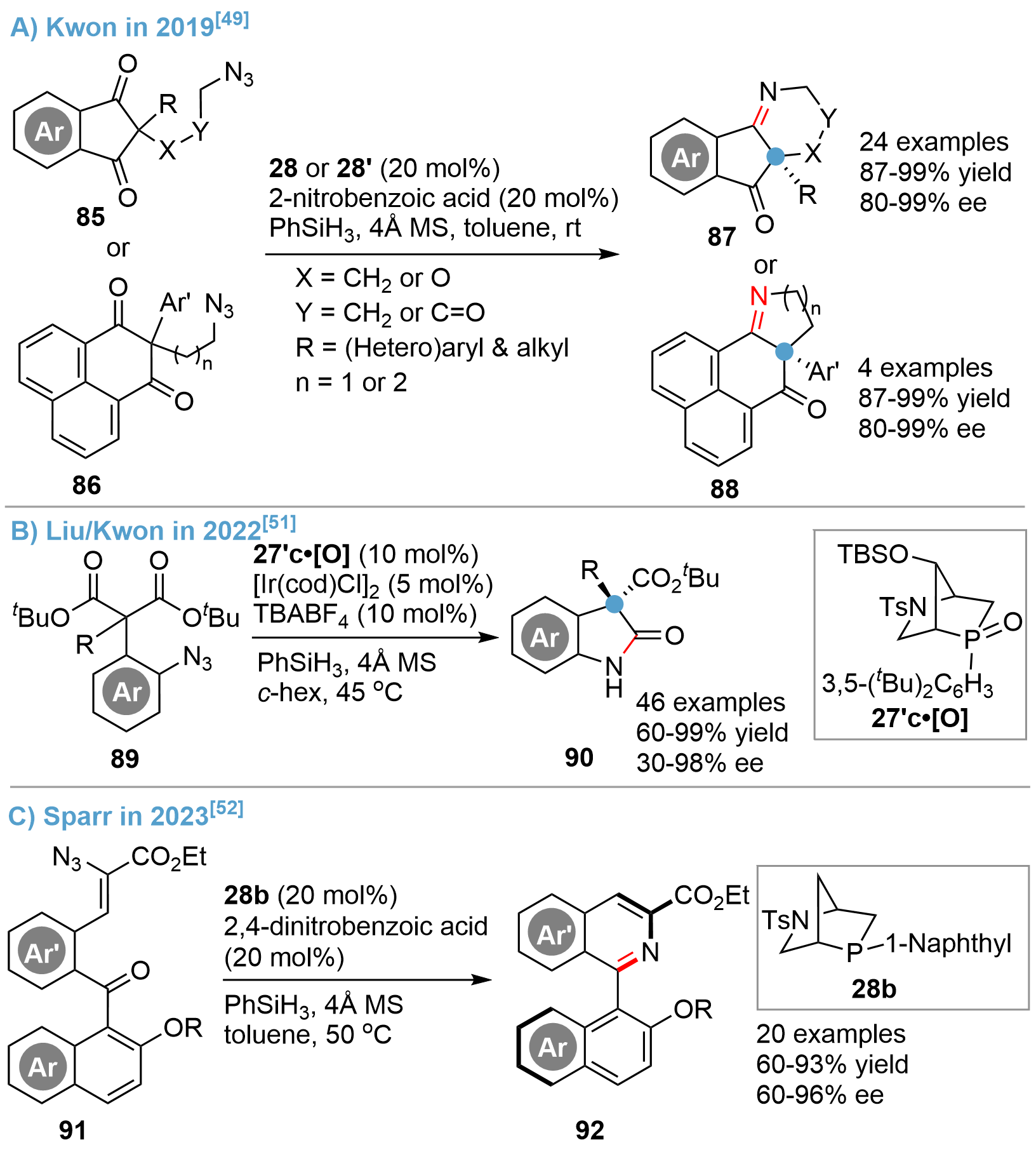{kind=link}
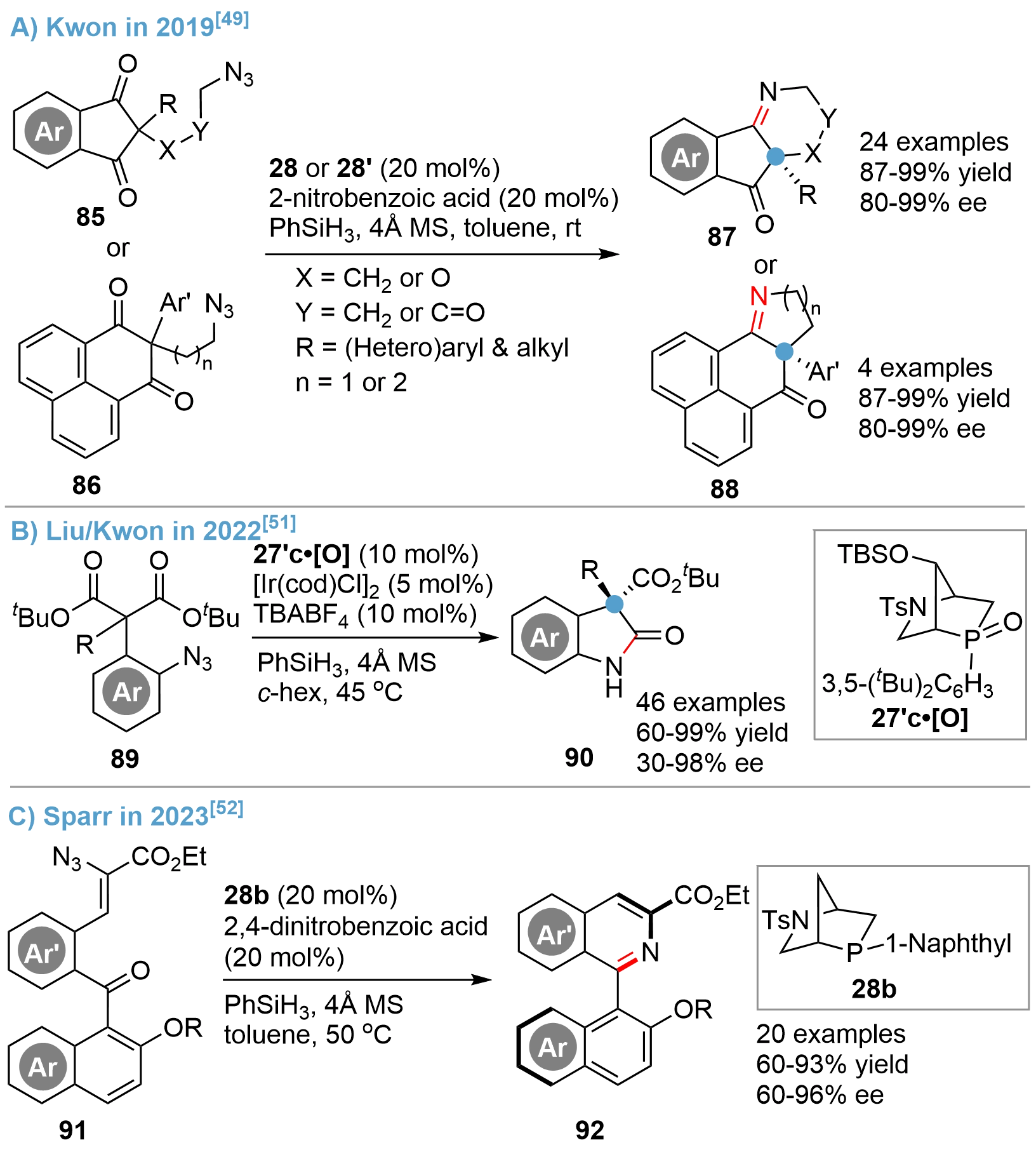
Figure 19. Asymmetric Staudinger-aza-Wittig reaction catalyzed by bridged-cyclic P-stereogenic phosphines.
Following this first report, Kwon further extended the protocol for the synthesis of oxindole derivatives containing a C3-quaternary carbon center via a catalytic desymmetrization of (o-azidoaryl)malonate esters 89[51] (Figure 19B). The reaction proceeded efficiently to give a broad range of oxindoles 90 with good functional group tolerance, wide substrate scope, and excellent enantioselectivity. Moreover, successful applications of the protocol were demonstrated by the syntheses of several alkaloids. A theoretical study suggested that the Ir(I) additive may serve as a Lewis acid to activate the phosphine oxide catalyst and the ester group in the substrate.
In a 2023 publication by Sparr[52], an asymmetric Staudinger–aza-Wittig reaction of diaryl ketone azides 91 for the atroposelective synthesis of isoquinoline-based heterocycles 92 was reported. A diversity of atropisomers were obtained in high yields with good enantioselectivity by using the bridged-cyclic phosphine 28b as catalyst (Figure 19C). The products could be transformed into a multitude of heteroarenes, which might be potentially used as ligands, medicinally relevant compounds, and functional molecular scaffolds.
3.2.4 Asymmetric Wittig reaction
On the basis of their results on the asymmetric Staudinger–aza-Wittig reaction for atroposelective synthesis of isoquinoline-based heterocycles[52], Sparr developed an ingenious intramolecular. Wittig reaction for atroposelective synthesis of biaryl atropisomers using the substrates 93 bearing biaryl carbonyl and MBH carbonate functionalities[53] (Figure 20A). The reaction proceeded efficiently via a P(III)/P(V) = O redox cycle using Kwon’s bridged-cyclic phosphine 28a as catalyst, giving 94 in high yields and enantioselectivity. A broad substrate scope was exemplified by varying Ar/Ar’ and R substitutions. Ir(I) was found to be an efficient Lewis acid, which, upon anion exchange with nBu4NBF4, to activate the substrates.
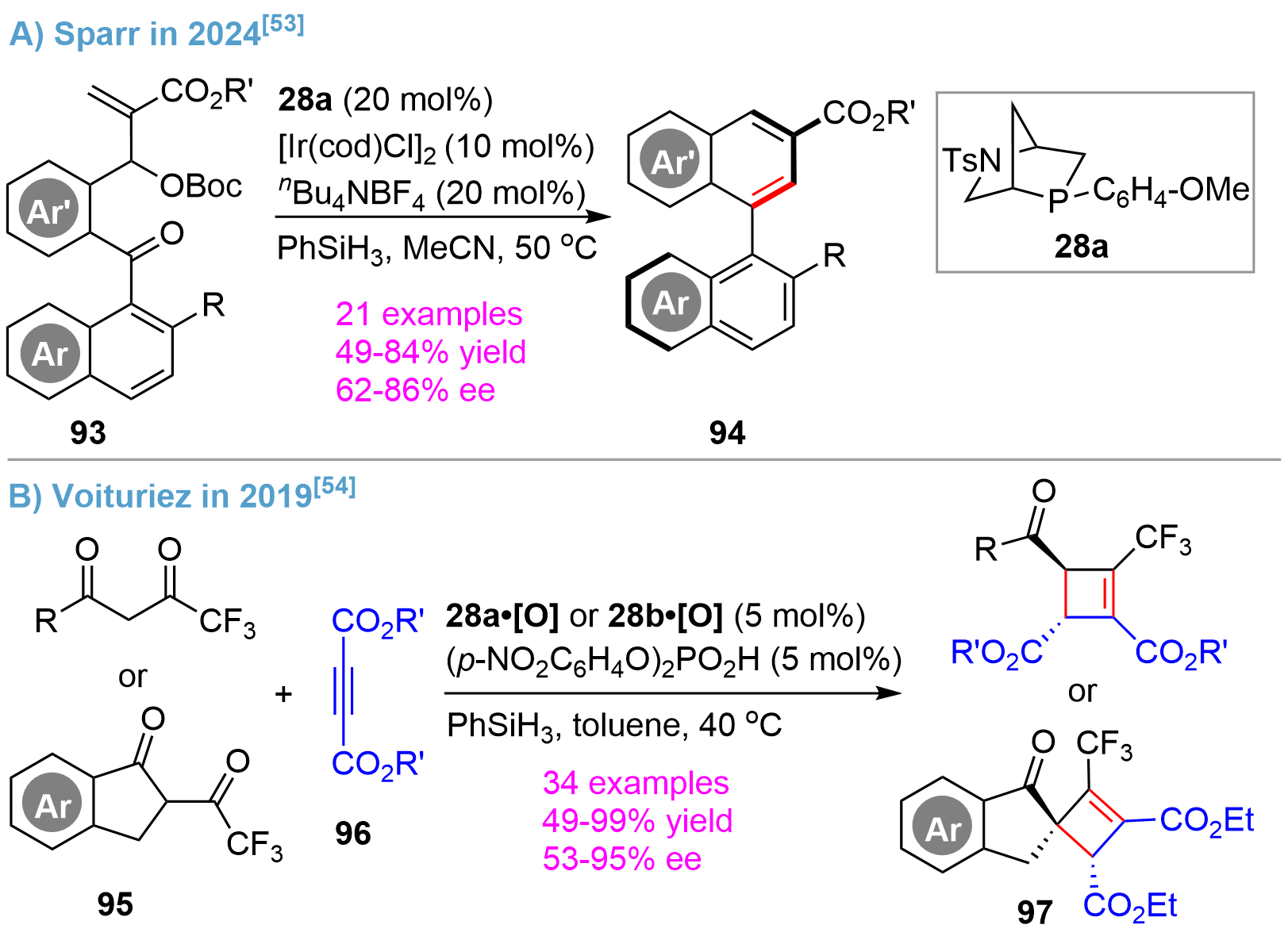{kind=link}
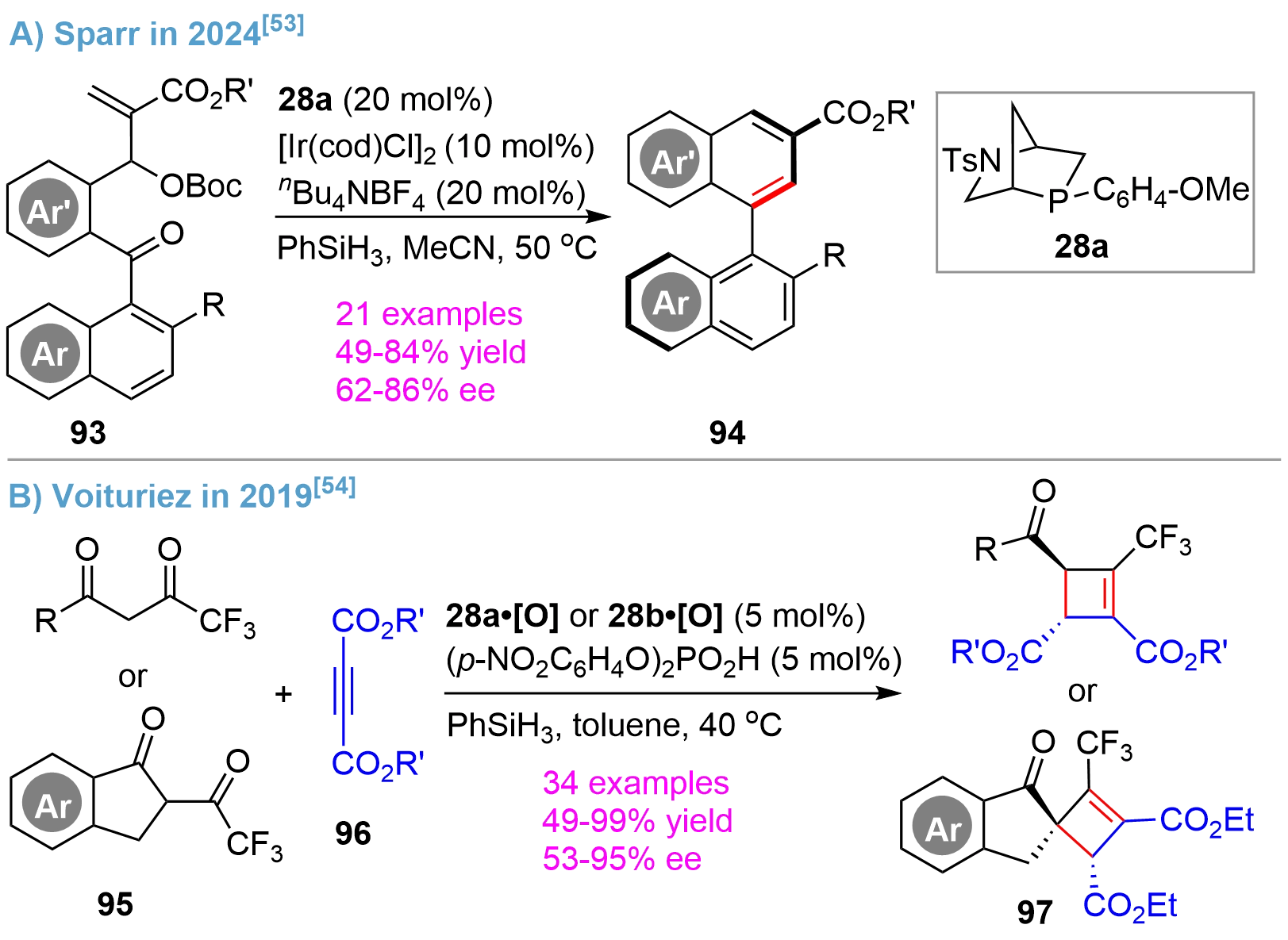
Figure 20. Asymmetric Wittig reaction catalyzed by bridged-cyclic P-stereogenic phosphines.
In an earlier report in 2019 by Voituriez[54], the tetrasubstituted chiral cyclobutenes 97 could be synthesized through the Michael addition/Wittig cascade from 1,3-diones 95 and acetylenedicarboxylates 96 (Figure 20B). By assaying various chiral phosphines and phosphine oxides, it was found that the Kown’s bridged-cyclic phosphine oxide 28a•[O] and 28b•[O] were highly efficient. Good yields and excellent enantioselectivity were obtained for a rich range of substrates except for a small alkyl group (R = Me) substituted dione, which provided a moderate selectivity of 53% ee. The results represented the first successful example of a chiral phosphine-catalyzed asymmetric Wittig reaction in terms of the yield, enantioselectivity, and substrate compatibility, since various non-bridged cyclic monophosphines and diphosphines showed much worse outcomes as shown in this and other studies[55].
4. Conclusion
In summary, this review summarized the advances in the synthesis of chiral bridged-cyclic phosphorus compounds and their applications as ligands or organocatalysts in asymmetric catalysis. While significant progress has been made in both aspects, the investigations are rather much less than those into the acyclic and non-bridged-cyclic backbone chiral or P-stereogenic phosphorus compounds. The key barriers should result from the lack of efficient methods for the synthesis of chiral bridged-cyclic phosphorus compounds with structural diversity. Especially, the catalytic asymmetric methods are still absent. Currently, most of the investigations are focused on Kwon’s catalysts derived from chiral 4-hydroxyproline though it still needs a somewhat tedious process based on synthetic step-count and the use of pungent and flammable PhPH2. In addition, limited availability of suitable phosphorus starting materials is also an additional concern. For instance, both phospholes and phosphaalkenes used for D-A reaction are difficult to synthesize. However, due to the significant advantages of bridged-cyclic chiral phosphorus compounds in asymmetric catalysis as summarized in this review, it is believed that more efforts in the future will be devoted to this challenging yet highly promising field.
Authors contribution
The author contributed solely to the article.
Conflicts of interest
The author declares no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
This work is supported by the National Natural Science Foundation of China (22471261).
Copyright
© The Author(s) 2026.
References
-
1. Imamoto T. P-stereogenic phosphorus ligands in asymmetric catalysis. Chem Rev. 2024;124(14):8657-8739.[DOI]
-
2. Imamoto T. Correction to “P-Stereogenic Phosphorus Ligands in Asymmetric Catalysis”. Chem Rev. 2024;124(21):12382-12389.[DOI]
-
3. Yang H, Yu H, Stolarzewicz IA, Tang W. Enantioselective transformations in the synthesis of therapeutic agents. Chem Rev. 2023;123(15):9397-9446.[DOI]
-
4. Wan F, Tang W. Phosphorus ligands from the Zhang lab: Design, asymmetric hydrogenation, and industrial applications. Chin J Chem. 2021;39(4):954-968.[DOI]
-
5. Cao Z, He D, Tang W. Catalysis and synthesis enabled by P-chiral dihydrobenzooxaphosphole ligands. Org Process Res Dev. 2024;28(4):949-977.[DOI]
-
6. Parmar D, Sugiono E, Raja S, Rueping M. Complete field guide to asymmetric BINOL-phosphate derived Brønsted acid and metal catalysis: History and classification by mode of activation; Brønsted acidity, hydrogen bonding, ion pairing, and metal phosphates. Chem Rev. 2014;114(18):9047-9153.[DOI]
-
7. Pietrusiewicz KM, Zablocka M. Preparation of scalemic P-chiral phosphines and their derivatives. Chem Rev. 1994;94(5):1375-1411.[DOI]
-
8. De Vaumas R, Marinetti A, Ricard L, Mathey F. Use of prochiral phosphaalkene complexes in the synthesis of optically active phosphines. J Am Chem Soc. 1992;114(1):261-266.[DOI]
-
9. Mattmann E, Mercier F, Ricard L, Mathey F. Synthesis of new tricyclic phosphines and phosphinites by intramolecular Diels-Alder reactions of trivalent phospholes. J Org Chem. 2002;67(15):5422-5425.[DOI]
-
13. Chen Z, Jiang Q, Zhu G, Xiao D, Cao P, Guo C, et al. Syntheses of novel chiral monophosphines, 2,5-dialkyl-7-phenyl-7-phosphabicyclo-[2.2.1]heptanes, and their application in highly enantioselective Pd-catalyzed allylic alkylations. J Org Chem. 1997;62(13):4521-4523.[DOI]
-
14. Zhu G, Chen Z, Jiang Q, Xiao D, Cao P, Zhang X. Asymmetric [3 + 2] cycloaddition of 2,3-butadienoates with electron-deficient olefins catalyzed by novel chiral 2,5-dialkyl-7-phenyl-7-phosphabicyclo[2.2.1]heptanes. J Am Chem Soc. 1997;119(16):3836-3837.[DOI]
-
16. Henry CE, Xu Q, Fan YC, Martin TJ, Belding L, Dudding T, et al. Hydroxyproline-derived pseudoenantiomeric [2.2.1] bicyclic phosphines: Asymmetric synthesis of (+)- and (–)-pyrrolines. J Am Chem Soc. 2014;136(34):11890-11893.[DOI]
-
18. Leung PH, Loh SK, Mok KF, White AJP, Williams DJ. A simple route to a novel enantiomerically pure P-chiral phosphine ligand containing a tertiary amide functional group. Chem Commun. 1996(5);591-592.[DOI]
-
20. Robin F, Mercier F, Ricard L, Mathey F, Spagnol M. BIPNOR: A new, efficient bisphosphine having two chiral, nonracemizable, bridgehead phosphorus centers for use in asymmetric catalysis. Chem Eur J. 1997;3(8):1365-1369.[DOI]
-
21. Lelièvre S, Mercier F, Ricard L, Mathey F. Enantiopure 1-phosphanorbornadiene-2-carboxaldehydes. Tetrahedron Asymmetry. 2000;11(22):4601-4608.[DOI]
-
22. Siutkowski M, Mercier F, Ricard L, Mathey F. C2-BIPNOR: An easily accessible homologue of BIPNOR for asymmetric catalysis. Organometallics. 2006;25(10):2585-2589.[DOI]
-
23. Breit B, Fuchs E. Chiral phosphabarrelene ligands: Synthesis and evaluation in rhodium-catalyzed asymmetric hydrogenation. Synthesis. 2006;2006(13):2121-2128.[DOI]
-
24. Jiang Q, Xiao D, Zhang Z, Cao P, Zhang X. Highly enantioselective hydrogenation of cyclic enol acetates catalyzed by a Rh HYPERLINK "https://onlinelibrary.wiley.com/doi/abs/10.1002/(SICI)1521-3773(19990215)38:4%3c516::AID-ANIE516%3e3.0.CO;2-B" –PennPhos complex. Angew Chem Int Ed. 1999;38(4):516-518.
-
26. Trost BM, Van Vranken DL. Asymmetric transition metal-catalyzed allylic alkylations. Chem Rev. 1996;96(1):395-422.[DOI]
-
28. Gan Z, Zhi M, Han R, Li EQ, Duan Z, Mathey F. P-stereogenic phosphines directed copper(I)-catalyzed enantioselective 1,3-dipolar cycloadditions. Org Lett. 2019;21(8):2782-2785.[DOI]
-
30. Meng Y, Wang Q, Yao X, Wei D, Liu YG, Li EQ, et al. Rigid P-chiral phosphorus ligands for highly selective palladium-catalyzed (4+2) and (4+4) annulations. Org Lett. 2022;24(50):9205-9209.[DOI]
-
31. Zhang C, Lu X. Phosphine-catalyzed cycloaddition of 2, 3-butadienoates or 2-butynoates with electron-deficient olefins. a novel [3+2] annulation approach to cyclopentenes. J Org Chem. 1995;60(9):2906-2908.[DOI]
-
35. Cai L, Zhang K, Kwon O. Catalytic asymmetric total synthesis of (−)-actinophyllic acid. J Am Chem Soc. 2016;138(10):3298-3301.[DOI]
-
37. Yuan C, Zhou L, Xia M, Sun Z, Wang D, Guo H. Phosphine-catalyzed enantioselective [4+3] annulation of allenoates with C, N-cyclic azomethine imines: Synthesis of quinazoline-based tricyclic heterocycles. Org Lett. 2016;18(21):5644-5647.[DOI]
-
39. Xu Q, Dupper NJ, Smaligo AJ, Fan YC, Cai L, Wang Z, et al. Catalytic enantioselective synthesis of guvacine derivatives through [4+2] annulations of imines with α-methylallenoates. Org Lett. 2018;20(19):6089-6093.[DOI]
-
44. Zhou L, Yuan C, Zhang C, Zhang L, Gao Z, Wang C, et al. Enantioselective synthesis of quinazoline-based heterocycles through phosphine-catalyzed asymmetric [3+3] annulation of Morita–Baylis–Hillman carbonates with azomethine imines. Adv Synth Catal. 2017;359(13):2316-2321.[DOI]
-
45. Li H, He Z. Chiral phosphine-catalyzed asymmetric [4+1] annulation of polar dienes with allylic derivatives: Enantioselective synthesis of substituted cyclopentenes. Tetrahedron Lett. 2021;67:152863.[DOI]
-
46. Xie C, Chen G, Feng CG, Lin GQ, Hong R. Recent advances in asymmetric P(III)/P(V)=O redox catalysis. Tetrahedron Lett. 2024;152:155337.[DOI]
-
51. Xie C, Kim J, Mai BK, Cao S, Ye R, Wang XY, et al. Enantioselective synthesis of quaternary oxindoles: Desymmetrizing Staudinger−Aza-Wittig reaction enabled by a Bespoke HypPhos oxide catalyst. J Am Chem Soc. 2022;144(46):21318-21327.[DOI]
-
52. Moser D, Jana K, Sparr C. Atroposelective PIII/PV=O redox catalysis for the isoquinoline-forming Staudinger–aza-Wittig reaction. Angew Chem Int Ed. 2023;62(39):e202309053.[DOI]
-
53. Jana K, Zhao Z, Musies J, Sparr C. Atroposelective arene-forming Wittig reaction by phosphorus PIII/PV=O redox catalysis. Angew Chem Int Ed. 2024;63(37):e202408159.[DOI]
-
54. Lorton C, Castanheiro T, Voituriez A. Catalytic and asymmetric process via PIII/PV=O redox cycling: Access to (trifluoromethyl)cyclobutenes via a Michael addition/Wittig olefination reaction. J Am Chem Soc. 2019;141(26):10142-10147.[DOI]
-
55. Werner T, Hoffmann M, Deshmukh S. First enantioselective catalytic Wittig reaction. Eur J Org Chem. 2014;2014(30):6630-6633.[DOI]
Copyright
© The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Publisher’s Note
Science Exploration remains a neutral stance on jurisdictional claims in published
maps
and institutional affiliations. The views expressed in this article are solely those
of
the author(s) and do not reflect the opinions of the Editors or the publisher.
Share And Cite



Science Exploration Style
Han FS. Advances in the synthesis of chiral bridged-cyclic phosphorus compounds and the applications in asymmetric catalysis. Chiral Chem. 2026;2:202623. https://doi.org/10.70401/cc.2026.0030
Tips
Copy completed.
Submit a Manuscript
Author Instructions
Cite this Article
Article Metrics
0
View
0
Download
Cited
Article Updates

- Abstract
- Keywords
- 1. Introduction
- 2. Synthesis of Bridged-Cyclic Chiral Phosphorus Compounds
- 3. Application of Bridged-Cyclic Chiral Phosphorus Compounds in Asymmetric Catalysis
- 4. Conclusion
- Authors contribution
- Conflicts of interest
- Ethical approval
- Consent to participate
- Consent for publication
- Availability of data and materials
- Funding
- References
- Copyright
Science Exploration Style
Han FS. Advances in the synthesis of chiral bridged-cyclic phosphorus compounds and the applications in asymmetric catalysis. Chiral Chem. 2026;2:202623. https://doi.org/10.70401/cc.2026.0030
copy
Share Link
copy