The gut-brain axis in Parkinson’s disease: A macrophage perspective
Viktoras Konstantellos
1,2,3

,
Pieter Van De Walle
2
,
Sebastiaan De Schepper
1,2,*
*Correspondence to:
Sebastiaan De Schepper, Laboratory for Gut-Immune-Brain Axis (GIBA) Research, VIB Center for Molecular Neurology, VIB, 2610 Antwerp, Belgium.
E-mail: Sebastiaan.deschepper@uantwerpen.be
Myeloid Cells. 2026;1:202606. 10.70401/mc.2026.0008
Received: March 31, 2026Accepted: July 03, 2026Published: July 06, 2026
Abstract
Parkinson’s disease (PD) is defined by the progressive accumulation of misfolded α-synuclein (αSyn) across the gut–brain axis, yet the cellular systems that normally contain this burden are only beginning to be understood. Among them, tissue-resident macrophages (RTM) occupy strategically positioned niches and functionally interact with their microenvironment. In this Perspective, we examine how muscularis externa macrophages in the enteric nervous system, parenchymal microglia, and border-associated macrophages in the central nervous system respond to αSyn and shape disease progression. We argue that these RTM serve as early sentinels of synucleinopathy by taking up extracellular αSyn and attempting to degrade it through lysosomal pathways. However, with ageing and in the context of genetic or environmental stress, this protective function may break down. As degradative capacity declines, RTM may enter dysfunctional states marked by cellular stress, incomplete processing of αSyn, propagation of pathogenic species, and enhanced antigen presentation. In this way, RTM may influence not only the local handling of αSyn, but also the emergence of adaptive immune responses and neuroinflammation across the enteric nervous system (ENS)–central nervous system (CNS) axis. We further discuss how anatomical niche, ontogeny, and αSyn conformational diversity may shape these responses, and how restoring macrophage proteostatic function could offer therapeutic opportunities to limit disease progression.
Keywords
Parkinson’s disease, tissue-resident macrophages, neuroimmunology, gut-brain axis
1. Introduction
Parkinson’s disease (PD), first described as “shaking palsy” by James Parkinson in 1817[1], is a synucleinopathy characterized by the misfolding and aggregation of α-synuclein (αSyn) into Lewy bodies and Lewy neurites, accompanied by the neurodegeneration of dopaminergic neurons in the basal ganglia of the central nervous system (CNS)[2,3] (A full list of abbreviated molecules and terms is provided in Table 1). In addition to the classical motor symptoms of tremor, bradykinesia, and rigidity, PD is characterised by a spectrum of prodromal manifestations; for example, constipation, which could reflect enteric nervous system (ENS) dysfunction[4,5]. In 2003, Braak and colleagues proposed a pathological staging scheme in which αSyn aggregation follows a caudo-rostral progression; from the dorsal motor nucleus of the vagus in the brainstem, through pontine and midbrain nuclei to limbic and neocortical regions[6]. The early involvement of the vagal system, together with the observation of αSyn aggregation in the submucosal and myenteric plexuses of the ENS of patients, led to the hypothesis that αSyn pathology could originate in the intestine, from where it could spread, via the vagus nerve, to the CNS, potentially in a cell-to-cell transsynaptic manner[5]. This concept is supported by animal studies showing gut-to-brain transmission of αSyn that is attenuated by vagotomy[7-9], and by human epidemiological data suggesting a reduced PD risk following truncal vagotomy[10], although this association remains debated. However, accumulating evidence supports substantial clinical and pathological heterogeneity in PD. Proposed disease trajectories include “body-first” PD, in which αSyn pathology may arise in peripheral autonomic or enteric structures before spreading to the CNS, and “brain-first” PD, in which pathology may originate within the CNS and subsequently involve peripheral autonomic pathways[11]. Consistently, early brain-first cases may show little or no peripheral Lewy pathology, indicating that ENS involvement and gut-to-brain propagation are not universal features of PD. Although strict dichotomisation of patients into “body-first” and “brain-first” categories remains challenging and controversial[12], the ENS–CNS axis may nevertheless represent an important route of αSyn propagation in a subset of patients.
Table 1. Key molecules and pathways cited in the text.
| Molecule/pathway | Function |
| αSyn; s129p αSyn | Core pathological protein and phosphorylated pathology-associated form used as a readout. |
| Aβ; tau | Comparator proteopathic proteins discussed in relation to age-related clearance burden. |
| GBA1; VPS35; GALC; SMPD1; CTSD; CTSB; GRN; LAMP1; p62 | Endolysosomal/autophagy and lysosomal cargo-handling pathways relevant to αSyn processing. |
| PINK1; PRKN; NADH-ubiquinone oxidoreductase; ROS | Mitochondrial quality-control and oxidative-stress-related genes and molecules linked to macrophage vulnerability. |
| LRRK2; Rab10 | Pathway implicated in lysosomal stress and αSyn release from macrophage-lineage cells. |
| MerTK; TLR2; TLR4; LAG3; FAM171A2 | Candidate αSyn uptake or sensing receptors with context-dependent evidence. |
| NF-κB; TNF; JAK-STAT; HSPA1A; HSPA1B; HSPA5; HSPA9 | Inflammatory and cellular-stress programmes induced by αSyn exposure. |
| C1q; complement | Engulfment-related pathway implicated in αSyn handling and synaptic/protein-associated clearance. |
| MHC-II; HLA-DQB1; HLA-DRB5; HLA-DRB1*15:01 | Antigen-presentation molecules linking αSyn processing to adaptive immune recognition. |
| TGFβ1; CCR2; CD163; CD206; LYVE1; CD22; CD68 | Macrophage subset, trafficking, scavenger, phagocytic and regulatory markers discussed across ENS and CNS niches. |
αSyn: α-synuclein; s129p αSyn: serine-129-phosphorylated α-synuclein; Aβ: amyloid-beta; ROS: reactive oxygen species; NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells; TNF: tumor necrosis factor; JAK-STAT: Janus kinase-signal transducer and activator of transcription; MHC-II: major histocompatibility complex class II; ENS: enteric nervous system; CNS: central nervous system.
Alongside the growing recognition of the gut-brain axis in PD, the immune system has also emerged as an important contributor to its pathogenesis[13,14]. Initial postmortem studies in the late 1980s reported the presence of reactive microglia, the major tissue-resident macrophages (RTM) of the CNS, in areas of gliosis adjacent to Lewy body pathology and degenerating neurons in the substantia nigra[15]. Subsequent positron emission tomography scan studies using translocator protein (TSPO) ligands suggested increased microglial activity early in the disease[16,17], although the specificity of TSPO as a marker and the concept of an “activated” microglial state in general remain debated[18]. While most PD cases appear to be sporadic and influenced by a complex interplay of genetic and environmental factors, studies have identified genes of the endolysosomal and autophagic pathways as risk factors, including glucocerebrosidase 1 (GBA1), vacuolar protein sorting-associated protein 35 (VPS35), galactocerebrosidase (GALC), sphingomyelin phosphodiesterase 1 (SMPD1), and cathepsin D (CTSD) among others[4,19]. Other risk genes include PINK1 and parkin RBR E3 ubiquitin protein ligase (PRKN), which function in mitochondrial quality control, and the multifunctional kinase leucine-rich repeat kinase 2 (LRRK2), the most frequent genetic cause of familial PD[4]. Of note, these genes are expressed at moderate to high levels in RTM, in some cases at higher levels than in neurons, suggesting that PD-associated pathways are active in these cells[20,21]. It also suggests that the interactions of RTM with αSyn, for example through uptake and lysosomal clearance, may contribute to PD pathogenesis.
In this Perspective, we explore the concept that RTM across the ENS–CNS interface are not mere bystanders in synucleinopathy, but active participants and potential modulators of disease. We frame impaired RTM-mediated αSyn clearance as a plausible vulnerability that may amplify and accelerate existing pathology. We discuss how the interaction of αSyn with RTM could modulate its aggregation, and how their engagement with the adaptive immune system may link synucleinopathy to disease progression. Although a myriad of RTM are present along the ENS–CNS axis, we focus on muscularis externa macrophages (ME-Macs) in the ENS, and on parenchymal microglia and border-associated macrophages in the CNS, given the mounting experimental evidence of their importance in PD pathogenesis.
The mechanistic evidence discussed here comes primarily from rodent models of synucleinopathy and in vitro systems involving genetic or pharmacological manipulation, while available human tissue evidence is largely correlative. We therefore distinguish model-system observations from the broader pathogenic framework proposed for PD.
2. Tissue-Resident Macrophages in the ENS and CNS: Mechanisms of Neuronal Clearance
Across the ENS and CNS, RTM occupy functionally analogous niches and maintain long-term relationships with neuronal tissues. In the ENS, ME-Macs represent the dominant RTM population and are primarily associated with the myenteric plexus, the major ganglion network governing intestinal motility, secretion, and blood flow[22]. They differ transcriptionally and regionally from submucosal RTM that are localized in the submucosal plexus and regulate vasculature permeability and epithelial homeostasis[23,24]. In the CNS, microglia are the main phagocyte population residing in the parenchyma. A second macrophage population, collectively termed border-associated macrophages (BAMs), occupies CNS-body interfaces at the brain border regions. These include the perivascular spaces, meninges (dura mater, arachnoid, and pia mater), and the choroid plexus (apical epiplexus Kolmer cells and stromal BAMs)[25,26]. Despite these anatomical differences, both systems are populated by long-lived macrophages of embryonic origin. In mice, microglia derive from yolk-sac progenitors from E7.25-E7.5 onwards, seed the brain by E9.5, and are maintained largely by self-renewal, although niche disruption or severe inflammation can permit limited engraftment by monocyte-derived macrophages[27,28]. ME-Macs are also established during embryogenesis and are partially self-maintaining, but, in contrast to microglia, they undergo substantial age-dependent replacement by C-C motif chemokine receptor 2 (CCR2)+ monocytes, reaching approximately 70% renewal by eight months of age[24]. BAMs receive a similar monocytic input to ME-Macs, depending on their anatomical location; for example, dural macrophages are 80% replenished in the first year of murine life[29].
Both ME-Macs and microglia closely interact with neurons and contribute to neural circuit homeostasis, in part through clearance functions. During murine early postnatal development, ME-Macs localize to enteric ganglia and dynamically extend processes towards neurons, engulfing synapses and other neuronal material to refine ENS circuits[30]. This functional interaction with neurons persists into adulthood, where ME-Macs are proposed to continuously phagocytose dying myenteric neurons and neuronal debris in the healthy mouse gut, consistent with their ongoing role in ENS maintenance and potentially neurogenesis[31]. Similarly, in the murine brain, microglia shape developing neuronal circuits through complement C1q-mediated synaptic engulfment of less-active synapses[32]. Beyond synaptic engulfment, microglia phagocytose apoptotic newborn oligodendrocytes in neurogenic regions[33], whereas in the developing cortex, interferon-responsive microglia can also engulf whole neurons, linking microglial phagocytosis to neuronal sculpting during circuit maturation[34]. In adulthood, murine microglia continuously engulf myelin to prevent hypermyelination[35] and, in zebrafish, clear cellular remnants after traumatic brain injury via efferocytosis[36]. TAM receptor signalling, particularly through Mer and Axl, allows microglia to phagocytose apoptotic cells during adult neurogenesis, whereas loss of these receptors on microglia results in the accumulation of apoptotic cells in the subventricular zone[37]. In conclusion, microglia and ME-Macs continuously balance clearance and remodelling of surrounding neurons to maintain neuronal homeostasis, at least in mice. Whether human RTM exhibit similar roles in neuronal clearance remains to be elucidated.
With ageing, however, protein clearance appears to decline in these RTM[38]. A recent proteomic study on nascent proteomes showed prolonged half-lives of synaptic proteins in aged murine microglia, particularly in the hippocampus and hypothalamus, with many of these proteins also displaying increased aggregation propensity[39]. This suggests that protein aggregates may directly impair the clearing capacity of microglia, or alternatively, that protein aggregation arises from a progressive failure of degradative pathways in microglia, potentially related to lifelong exposure to microbial signals and misfolded proteins such as amyloid-beta (Aβ), tau, and αSyn. In line with this, as myelin fragmentation increases with age, microglia accumulate insoluble myelin aggregates and exhibit increased lysosomal volume together with the upregulation of antigen-presentation molecules[38]. In the ENS, murine ME-Macs adopt an age-associated CLEC7A+CD11c+ phenotype and show reduced phagocytic activity[40]. At the same time, αSyn, a physiological regulator of enteric neurotransmission[41], progressively accumulates in enteric neuronal axons and terminal varicosities with age, and is detected within ME-Macs[40,42]. This suggests that the clearance capacity of ME-Macs might become gradually outpaced by the rising burden of neuronal αSyn pathology.
At CNS–peripheral interfaces, BAMs restrict the entry and accumulation of peripheral signals, pathogens, and cellular debris[25,26]. They express scavenger receptors such as CD163 and CD206 and are enriched in antigen-presentation pathways compared to microglia. In highly permeable regions, including the area postrema and choroid plexus, CD163+ macrophages rapidly capture circulating serum proteins, thereby limiting their access to the parenchyma[43,44]. BAMs also continuously sample antigens present in the cerebrospinal fluid (CSF): after intra-ventricular or intra-cisterna magna tracer delivery, labelled antigens are detected in epiplexus macrophages and in meningeal BAMs across the pia and subarachnoid space[45,46]. Along venous and sinus-associated drainage routes, CD163+LYVE1+ macrophages further sample CSF contents while supporting CSF efflux through extracellular matrix remodelling[45]. Because CSF flow declines with ageing and in Alzheimer’s disease[47,48], impaired clearance at these interfaces may promote retention of disease-associated proteins. Given that Aβ accumulates in meningeal and vascular deposits[49] and misfolded αSyn is detectable in the CSF of PD patients[50], BAMs are strategically positioned to capture such CNS-derived proteins and potentially shape their clearance and immune presentation[51].
3. Tissue-Resident Macrophages into the Spotlight in Parkinson’s Disease
We propose that declining RTM clearance capacity with age may increase susceptibility to synucleinopathy. PD, whose strongest risk factor is ageing[4], is characterized by the accumulation of misfolded αSyn in the ENS and CNS. In this Perspective, we suggest that RTM across these compartments respond to extracellular αSyn in broadly similar ways (Figure 1). Following uptake, RTM may undergo one or more of three pathological trajectories: 1) induction of cellular stress due to incomplete digestion; 2) propagation of pathology through transport or release of partially processed αSyn species; and/or 3) antigen presentation following processing of αSyn-derived peptides. These RTM outcomes should be viewed as parallel, potentially reversible states that can coexist rather than as a fixed progression from protection to dysfunction. Their balance is shaped by the tissue niche, RTM ontogeny, host genetics, environmental exposures, αSyn conformer, and disease stage.
{kind=link}
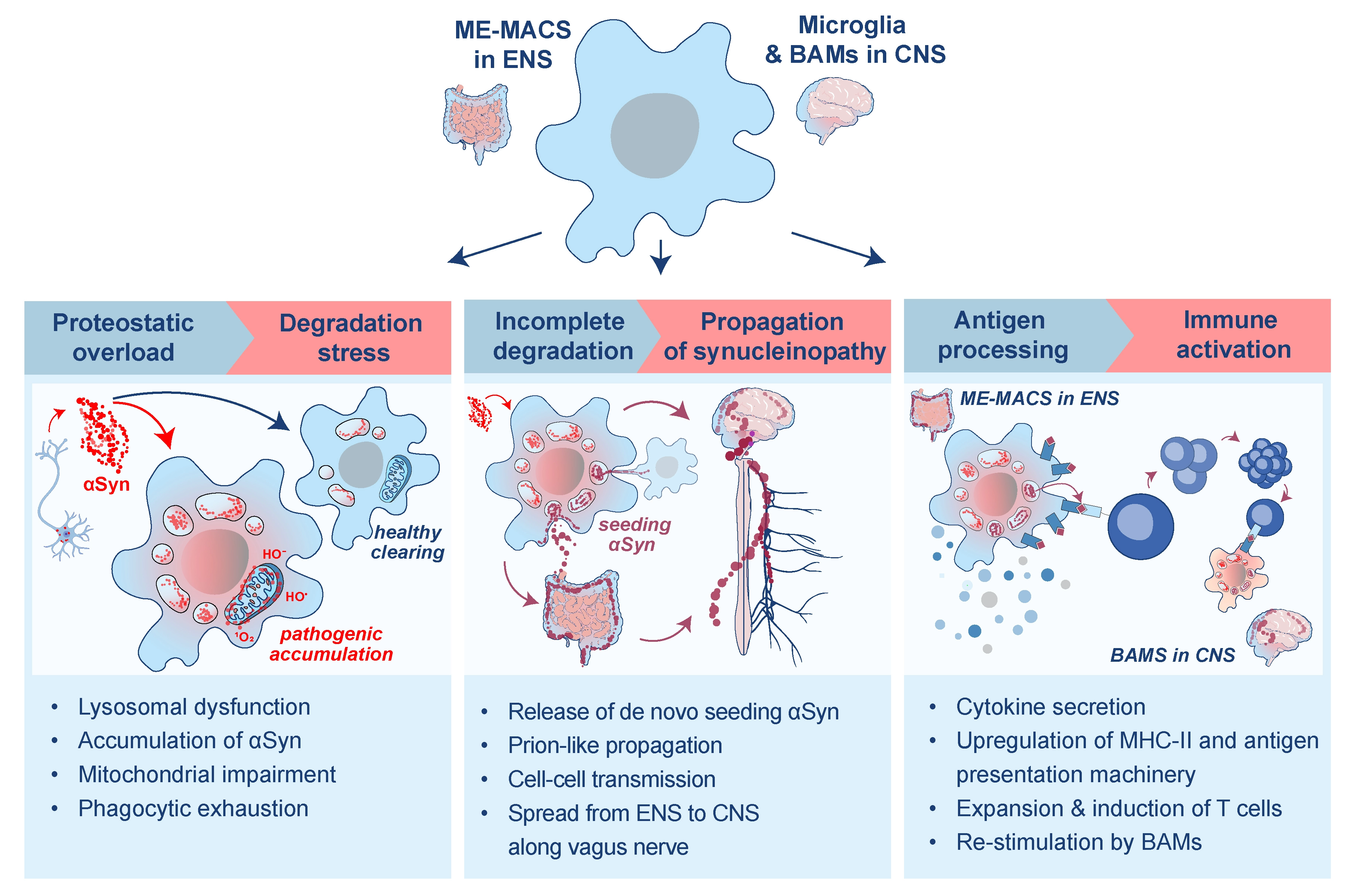
Figure 1. Pathological trajectories of tissue-resident macrophages following αSyn uptake. The schematic depicts parallel/coexisting outcomes after αSyn uptake rather than a required deterministic sequence. ME-Macs: muscularis externa macrophages; ENS: enteric nervous system; αSyn: α-synuclein; CNS: central nervous system; BAMs: border-associated macrophages.
In interpreting the evidence reviewed below, we distinguish between three levels of inference: findings from direct experimental manipulation in animal models, including genetic deletion and cell-type-specific depletion models; mechanistic data from in vitro or ex vivo systems, which illuminate molecular pathways but may not fully recapitulate in vivo conditions; and observations from human postmortem tissue or patient cohorts, which are largely correlative with disease state.
4. Cellular Stress Responses
While neurons can internalize and degrade extracellular αSyn, in vitro studies indicate that microglia have a greater capacity to do so[48]. This is consistent with a division of labour in which neurons may rely in part on RTM, as professional phagocytes, to limit extracellular proteopathic burden[49]. However, the relative contributions of these cell types in vivo remain to be established. In the homeostatic murine brain, microglia sequester extracellular αSyn, accumulate it in ubiquitin-positive puncta, and degrade it through p62- and TLR4-dependent autophagy[52]. In microglia-specific Atg7-deficient mice expressing human αSyn, disruption of autophagy caused αSyn accumulation and increased insoluble αSyn pathology in whole-brain lysates, demonstrating that intact microglial degradative pathways are required to limit αSyn buildup through efficient clearance[52].
Uptake of αSyn aggregates partly occurs through receptor-mediated pathways, although the dominant receptor depends on both cell type and αSyn conformer. In neurons, early studies implicated lymphocyte-activation gene 3 (LAG3) in αSyn uptake[53], but this has been challenged by more recent evidence showing minimal neuronal expression and no survival benefit of LAG3 deletion in A53T αSyn mice[54]. Attention has therefore shifted towards alternative receptors, including FAM171A2, which was recently identified as a neuronal receptor for fibrillar, but not monomeric, αSyn in mice and humans[55]. Consistent with this, adeno-associated virus (AAV)-driven FAM171A2 overexpression increased serine-129-phosphorylated αSyn, a widely used marker of pathological αSyn[56,57], in striatal and substantia nigral neurons of mice[55]. Notably, in vitro experiments support alternative mechanisms of αSyn transport in neurons, including axonal transport[58], transfer via tubular structures that connect donor and recipient cells[59], or exosome-mediated uptake of αSyn[60], although their contribution in vivo and relevance to humans remain to be investigated.
In microglia, mechanistic studies in primary human cells and induced pluripotent stem cell (iPSC)-derived models indicate that mer tyrosine kinase (MerTK) is a major contributor to αSyn fibril internalisation[61]. In parallel, pattern-recognition pathways influence both uptake and inflammatory sensing: TLR2 binds aggregated αSyn in a conformation-dependent manner, whereas TLR4 links extracellular αSyn exposure to nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) activation in microglia[62-64]. In murine primary microglia, exposure to extracellular αSyn activates inflammatory programmes including NF-κB, tumor necrosis factor (TNF), and Janus kinase (JAK)-signal transducer and activator of transcription (STAT) signalling, together with heat-shock proteins such as HSPA1A, HSPA1B, HSPA5, and HSPA9, consistent with a cellular stress response to internalized αSyn aggregates[65,66]. These findings support the idea that pathological αSyn triggers innate receptor signalling and inflammasome-linked cascades rather than the immunologically silent pathways related to efferocytosis[67]. Instead, the response resembles activation by pathogen-associated stimuli such as lipopolysaccharide (LPS)[68]. In line with this, fibrillar αSyn induces high levels of reactive oxygen species (ROS) in microglia in vitro[69,65]. Similarly, selective overexpression of human A53T αSyn in microglia in vivo drives accumulation of serine-129-phosphorylated α-synuclein (s129p αSyn), increased ROS production, and upregulation of CD22[70]. As CD22 has also been identified as a negative regulator of phagocytosis in aged microglia, this may point to a negative feedback response to intrinsic αSyn burden[71]. In line with impaired homeostatic function, microglia exposed to fibrillar αSyn acquire an amoeboid morphology with retracted processes[52,62,63], yet simultaneously upregulate pathways linked to microtubule assembly and polymerisation[65]. These coinciding features suggest attempted but ineffective engagement of the phagocytic machinery, reminiscent of “frustrated phagocytosis” described during the incomplete clearance of persistent particles, such as microplastics[65,72,73]. Nevertheless, because these models artificially overload microglia with fibrillar αSyn, they may overestimate the extent to which αSyn uptake drives inflammatory activation under physiological disease conditions.
A similar pattern appears to emerge in the ENS. In the 3KL transgenic mouse model, which expresses aggregation-prone human αSyn carrying three lysine substitutions[74], ME-Macs show increased lysosomal lysosome-associated membrane protein 1 (LAMP1) volume together with the appearance of s129p+ αSyn within lysosomes[75]. Proteomic profiling further revealed upregulation of lysosomal and phagosome-associated proteins, including GRN, cathepsin B (CTSB) and CD68. Simultaneously, the mitochondrial nicotinamide adenine dinucleotide (reduced form) (NADH)-ubiquinone oxidoreductase complex was downregulated in ME-Macs, pointing to concurrent activation of degradative pathways and mitochondrial vulnerability[75]. We further found that injection of brain-derived fibrillar αSyn from PD postmortem donors into the muscularis externa of wildtype mice increased enteric s129p pathology and promoted its uptake by ME-Macs. Similar findings were reported by Mackie et al. using intestinal injection of endotoxin-free, Escherichia coli-derived murine αSyn preformed fibrils[76]. In that study, ME-Macs initially upregulated complement-associated engulfment pathways, and C1q-dependent clearance transiently constrained enteric synucleinopathy. However, this protective response appeared to decline over time, suggesting a state in which sustained αSyn uptake exceeds degradative capacity and progressively impairs phagocytic function[76]. Mechanistically, the involvement of C1q aligns with a broader theme in neurodegeneration, whereby complement tags synaptic or protein-associated targets for removal by microglia[77]. Together, these observations suggest that ME-Macs, like microglia, initially mount a degradative response to extracellular αSyn but may be progressively overwhelmed and can exhibit features associated with endolysosomal stress and impaired phagocytic function as pathological burden accumulates. This response is likely heterogeneous across RTM populations: some subsets may retain efficient αSyn clearance and degradation, whereas others may fail to resolve αSyn burden and transition toward cellular stress and inflammatory activation. The mechanisms governing this functional divergence remain to be elucidated.
5. Propagation of Synucleinopathy
When lysosomal degradation is incomplete, the sequestration of αSyn may generate an intracellular reservoir of pathogenic species, while lysosomal stress or rupture could allow seeding-competent material to escape into the cytosol and further influence αSyn aggregation[78,79]. In a catecholaminergic neuronal cell line, de novo aggregation between exogenous fibrillar and endogenous αSyn largely colocalized with LAMP1+ lysosomes, indicating that the lysosome may present a favourable environment to facilitate aggregation[79]. By contrast, studies in macrophage-like RAW264.7 cells show that exogenous fibrillar αSyn initially localizes to lysosomes but is subsequently exported in exosomes through LRRK2–Rab10 signalling, indicating that undigested αSyn can be released back into the extracellular space[80]. Retained αSyn can also be transferred between neighbouring microglia through F-actin-dependent intercellular “nanotubes”, although this has so far only been shown in primary microglia and interpreted as a cooperative clearance mechanism rather than direct evidence of pathological propagation[65]. Consistent with the idea that RTM in the ENS and CNS could modulate αSyn propagation, we found that depletion of ME-Macs via anti-CSF1R blockade prior to administration of patient-derived αSyn fibrils reduced local enteric neuronal s129p+ αSyn pathology and limited its spread along the ENS[75]. Whether this propagation occurs trans-synaptically via the vagus nerve, as suggested by previous studies[8], remains to be determined. By analogy, microglial depletion following the injection of murine αSyn preformed fibrils (PFF) in the striatum of mice reduced the presence of s129p+ αSyn in the striatum and amygdala, and attenuated dopaminergic neuron loss in the striatum and substantia nigra[62]. Another study similarly found that microglial depletion was beneficial in an rAAV-hSYN mouse model, in which neuronal overexpression of human αSyn in the substantia nigra induces progressive dopaminergic neuron loss and motor deficits[81]. Together, these findings from rodent models suggest that RTM can shift from a transiently protective role towards one that facilitates the persistence and dissemination of pathogenic αSyn species. Whether this transition occurs in (a fraction of) human PD patients, at which stage, and to what extent it is reversible, remains open questions.
Notably, distinct αSyn polymorphs differentially engage receptors such as TLR2 and elicit divergent transcriptional and cytokine programs in human iPSC-derived microglia[62]. The uptake of αSyn is size- and species-dependent, with smaller prefibrillar or oligomeric assemblies entering cells more efficiently than longer fibrils and potentially exerting distinct downstream effects[82]. Mackie et al. observed that ME-Mac depletion increased neuronal pathology following murine αSyn PFF administration, in contrast to our findings with human fibrils, suggesting that both neuronal and macrophage responses are shaped by strain-specific properties[76]. More broadly, this fits with the concept that αSyn strain identity emerges from the interplay between the seed and the host-cell environment, raising the possibility that RTM might actively shape the fate, conformation, and downstream toxicity of internalized αSyn assemblies[83]. These apparently divergent depletion and uptake studies are important because they argue against a single unified macrophage function in synucleinopathy. Depending on the route of administration, timing of depletion, tissue niche and readout, macrophages or microglia may appear protective and/or pathogenic. Whether targeting macrophage uptake or survival would be beneficial in human PD remains to be determined.
6. Antigen Presentation
We hypothesise that antigen-presenting cells, including RTM, may provide a mechanistic link between αSyn processing and the adaptive immune activation observed in PD. In humans, this is supported by a genome-wide association study (GWAS) identifying human leukocyte antigen (HLA) class II alleles, including HLA-DQB1 and HLA-DRB5, as genetic risk factors for PD[84]. Experimental evidence comes from a landmark study by Sulzer et al., which showed that T cells from PD patients recognise defined αSyn epitopes, with immunoreactivity concentrated in discrete regions of the protein[85]. Notably, an N-terminal epitope within the Y39 region (aa32-46; KTKEGVLYVGSKTKE) binds with high affinity to PD-associated HLA class II alleles, including the abovementioned HLA-DRB1*15:01 and HLA-DRB5*01:01. A second antigenic region localises to the C-terminus and includes epitopes surrounding s129, linking immune recognition to a disease-relevant post-translational modification state[85]. Importantly, αSyn-specific T cell responses are enriched in prodromal and early-stage PD and can be detected before motor symptom onset, indicating that adaptive immune activation may contribute to early disease mechanisms rather than simply reflecting inflammatory responses to central neurodegeneration[86]. Consistently, work by Garretti et al. showed that, in HLA-DRB1*15:01 humanized mice, immunization with the αSyn-derived epitope αSyn[32-46] triggers intestinal inflammation, enteric neurodegeneration, delayed gastrointestinal transit, and weight loss, supporting a potential role for αSyn-specific adaptive immunity in prodromal enteric PD-like pathology[87]. Because single-nucleotide substitutions in HLA-DQ and HLA-DR can alter peptide binding affinity and major histocompatibility complex class II (MHC-II) complex stability[88], this raises the possibility that RTM and other antigen-presenting cells might generate and present disease-relevant αSyn-derived epitopes in the context of synucleinopathy.
In mice, enteric injection of PD patient-derived αSyn fibrils induced surface MHC-II upregulation on ME-Macs following αSyn engulfment and coincided with local T-cell expansion. Compared with neurologically healthy control-derived extracts, mice injected with PD-derived αSyn fibrils showed increased numbers of CD3+ T cells, predominantly CD4+ T cells, within myenteric ganglia[75]. These expanded T cells subsequently migrated from the ENS to the dura mater and brain during disease progression, whereas targeted depletion of ME-Macs attenuated T cell expansion and ameliorated neurodegeneration and motor deficits. Importantly, ME-Macs may also promote T cell expansion through cytokine signalling. Single-cell RNA-seq analysis of ME-Macs and T cells in αSyn mouse models identified distinct macrophage subsets, including CCR2+ and CD163+ populations, that express high levels of transforming growth factor beta 1 (TGFβ1), together with corresponding receptor expression on gut CD4+ T cells[75]. Moreover, deletion of TGFβ1 in ME-Macs abrogated αSyn-induced T cell expansion in the ENS, demonstrating a functional requirement for macrophage-derived TGFβ1 in this process. Together, these data position ME-Macs as local immune instructors that couple αSyn handling to T cell expansion and differentiation within the ENS.
In the murine CNS, antigen presentation in αSyn-driven pathology appears to be organized primarily at the brain borders rather than within the parenchyma. Although murine and human microglia are able to express MHC-II[15,89], microglia-specific MHC-II deletion does not significantly alter peripheral immune cell infiltration in an AAV2-SYN induced mouse model of nigral human αSyn overexpression[90]. Instead, BAMs appear to act as the dominant antigen-presenting cells: αSyn induces their expansion into a disease-activated state marked by the upregulation of antigen-presentation, chemotactic, and matrix-remodelling programmes, and BAM depletion markedly reduces CD4+ T cell recruitment and neuroinflammation[90]. Together, these findings from experimental models support a model in which BAMs at vascular and meningeal interfaces may orchestrate T cell entry and restimulation, whereas parenchymal microglia primarily execute downstream inflammatory and phagocytic responses. Perivascular BAMs and T cells are also found in close proximity in the human PD midbrain, although the directionality and functional significance of this spatial relationship in human disease remain to be determined[75].
7. Outlook: From Clearance Failure to Therapeutic Opportunities
We hypothesize that RTM dysfunction may constitute an underlying vulnerability factor in PD, one that could contribute to and amplify αSyn pathology rather than being solely a secondary response to neuronal loss. With ageing, genetic predisposition, environmental insults, or their combined effects, αSyn uptake by RTM may shift the distribution of RTM states: some cells may retain efficient degradative clearance, whereas others may accumulate incompletely processed or immunogenic αSyn cargo. Familial PD genetics support this idea, as risk variants are enriched in pathways regulating endolysosomal and mitochondrial homeostasis, processes that are also relevant to RTM[19]. Environmental exposures may further exacerbate RTM stress, including pesticides such as paraquat and rotenone, which impair lysosomal and mitochondrial function, as well as indigestible contaminants such as microplastics[73,91]. This is supported by epidemiological studies linking environmental toxins to increased risk of developing PD[92]. It will be important to experimentally define the thresholds that distinguish age-associated decline, reversible stress responses, and loss of phagocytic performance within αSyn-challenged RTM compartments.
An important consideration is that pathological αSyn is unlikely to behave as a single ligand. Instead, increasing evidence suggests that misfolded αSyn comprises a structurally heterogeneous family of assemblies with distinct biochemical and immunological properties[62,83]. Conformation-specific epitopes can be differentially recognised by antibodies[93], and distinct αSyn conformations have been shown to trigger divergent inflammatory programmes in macrophages[62]. Although some studies implicate specific TLRs in the recognition of different αSyn structures, the precise epitopes that drive pro-inflammatory responses remain unclear. Future work is therefore needed to determine how αSyn strain diversity might impact RTM phenotypes, and in turn, how the RTM response could shape αSyn strain identity[83] across the ENS and CNS. An additional consideration regarding αSyn strain identity is that fibril-injection systems likely do not fully recapitulate idiopathic PD. Although studies injecting fibrils commonly perform endotoxin testing to rule out adjuvant-like immunological effects, pre-formed aggregates may activate RTMs differently compared to endogenously derived aggregates. Therefore, incorporating genetic mouse models with fibril injection systems will be beneficial to future studies.
It remains to be determined to what extent RTM responses are shaped by cell-intrinsic programming, for example ontogeny, in addition to local tissue cues and αSyn burden. Although microglia and ME-Macs are largely self-maintained in homeostasis, monocyte-derived macrophages can enter the CNS under pathological conditions and may remain transcriptionally and functionally distinct from embryonically derived resident cells[28,94]. In neurodegenerative settings, these recruited populations often show increased expression of inflammatory cytokines, chemokines, and antigen-presentation machinery compared with resident microglia[94]. In an AAV2-SYN mouse model of PD, in which full-length human αSyn is virally overexpressed in dopaminergic neurons of the substantia nigra pars compacta, peripheral monocytes infiltrate the ventral midbrain and contribute to dopaminergic neurodegeneration[95]. In the ENS, monocyte-derived macrophage subsets with stronger antigen-presentation signatures appear to show enhanced αSyn uptake relative to more trophic populations[75]. In line with these findings, monocytes from individuals at prodromal disease stages display heightened cytokine responsiveness compared with controls[96]. Together, these observations suggest that RTM ontogeny may critically shape responses to αSyn pathology. Furthermore, although core microglia programmes are conserved across species[97], primate- and human-specific states are not fully captured in mice, highlighting the need for validation in human tissues.
Beyond ontogeny and tissue-specific signals, the microbiota may further modulate RTM activation thresholds in synucleinopathy. This is supported by germ-free and antibiotic-treatment studies showing that microbial signals regulate microglial maturation, morphology, and inflammatory responsiveness, with short-chain fatty acids such as acetate, propionate, and butyrate acting as microbiota-derived metabolites that can restore several microglial defects[98]. In αSyn-overexpressing mice, microbiota depletion attenuates microglial activation, αSyn pathology, and motor deficits, whereas short-chain fatty acid supplementation and transfer of PD patient-derived microbiota worsen disease-associated phenotypes[99]. These data suggest that microbial metabolites and pattern-recognition receptor ligands can modulate how resident macrophages respond to αSyn. A key unresolved question is whether such signals act similarly on ME-Macs, microglia, and BAMs, or whether each compartment interprets microbiota-derived cues according to its local niche and ontogeny.
Finally, any RTM-based therapeutic strategy will need to account for the functional heterogeneity of these cells across subsets and disease stages, rather than treating macrophages as a single uniform target. In the ENS, ME-Macs may act as early regulators of αSyn handling and as coordinators of adaptive immune activation, suggesting that enhancing αSyn clearance, preserving lysosomal competence, or limiting inappropriate antigen presentation in the gut could represent upstream intervention strategies. In the CNS, by contrast, promoting efficient but non-inflammatory clearance by microglia or BAMs may become more relevant once brain αSyn accumulation is established[100]. This implies that targeting uptake alone is unlikely to be sufficient and may even prove counterproductive unless coupled to reinforcement of degradative capacity. Approaches that enhance lysosomal biogenesis, stabilize lysosomal membranes, improve hydrolase activity, or restore autophagic flux are especially attractive, as they address a convergent vulnerability linking intracellular stress, seed persistence, and antigen generation. More broadly, reprogramming RTM towards states marked by enhanced proteostatic resilience, restrained inflammatory output, and efficient, non-propagative cargo disposal may offer a more plausible translational strategy than indiscriminate depletion.
Authors contribution
Konstantellos V: Conceptualisation, writing-original draft, writing-review & editing.
Van De Walle P: Writing-original draft, visualisation, writing-review & editing.
De Schepper S: Conceptualisation, writing-original draft, supervision.
Conflicts of interest
The authors declare no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
Sebastiaan De Schepper receives funding from the University of Antwerp/BOF, European Research Council (ERC StG, Nr 101221591) and FWO (FWO Odysseus II, Nr G0ARS25N).
Copyright
© The Author(s) 2026.
References
-
1. Parkinson J. An essay on the shaking palsy. J Neuropsychiatry Clin Neurosci. 2002;14(2):441-445.[DOI]
-
2. Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M, et al. α-synuclein in lewy bodies. Nature. 1997;388(6645):839-840.[DOI]
-
3. Calabresi P, Mechelli A, Natale G, Volpicelli-Daley L, Di Lazzaro G, Ghiglieri V, et al. Alpha-synuclein in Parkinson’s disease and other synucleinopathies: From overt neurodegeneration back to early synaptic dysfunction. Cell Death Dis. 2023;14(3):176.[DOI]
-
4. Kalia LV, Lang AE. Parkinson’s disease. Lancet. 2015;386(9996):896-912.[DOI]
-
5. Fasano A, Visanji NP, Liu LWC, Lang AE, Pfeiffer RF. Gastrointestinal dysfunction in Parkinson’s disease. Lancet Neurol. 2015;14(6):625-639.[DOI]
-
6. Braak H, Del Tredici K, Rüb U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24(2):197-211.[DOI]
-
7. Challis C, Hori A, Sampson TR, Yoo BB, Challis RC, Hamilton AM, et al. Gut-seeded α-synuclein fibrils promote gut dysfunction and brain pathology specifically in aged mice. Nat Neurosci. 2020;23(3):327-336.[DOI]
-
8. Kim S, Kwon SH, Kam TI, Panicker N, Karuppagounder SS, Lee S, et al. Transneuronal propagation of pathologic α-synuclein from the gut to the brain models Parkinson’s disease. Neuron. 2019;103(4):627-641.e7.[DOI]
-
9. Van Den Berge N, Ferreira N, Gram H, Mikkelsen TW, Alstrup AKO, Casadei N, et al. Evidence for bidirectional and trans-synaptic parasympathetic and sympathetic propagation of alpha-synuclein in rats. Acta Neuropathol. 2019;138(4):535-550.[DOI]
-
10. Svensson E, Horváth-Puhó E, Thomsen RW, Djurhuus JC, Pedersen L, Borghammer P, et al. Vagotomy and subsequent risk of Parkinson’s disease. Ann Neurol. 2015;78(4):522-529.[DOI]
-
11. Borghammer P, van Den Berge N. Brain-first versus gut-first Parkinson’s disease: A hypothesis. J Parkinsons Dis. 2019;9(s2):S281-S295.[DOI]
-
12. Fearon C, Lang AE, Espay AJ. The logic and pitfalls of Parkinson’s disease as “brain-first” versus “body-first” subtypes. Mov Disord. 2021;36(3):594-598.[DOI]
-
13. Tansey MG, Wallings RL, Houser MC, Herrick MK, Keating CE, Joers V, et al. Inflammation and immune dysfunction in Parkinson disease. Nat Rev Immunol. 2022;22(11):657-673.[DOI]
-
14. Harms AS, Yang YT, Tansey MG. Central and peripheral innate and adaptive immunity in Parkinson’s disease. Sci Transl Med. 2023;15(721):eadk3225.[DOI]
-
15. McGeer PL, Itagaki S, Boyes BE, McGeer EG. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology. 1988;38(8):1285-1291.[DOI]
-
16. Gerhard A, Pavese N, Hotton G, Turkheimer F, Es M, Hammers A, et al. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiol Dis. 2006;21(2):404-412.[DOI]
-
17. Bartels AL, Willemsen ATM, Doorduin J, De Vries EFJ, Dierckx RA, Leenders KL, et al. [11C]-PK11195 PET: Quantification of neuroinflammation and a monitor of anti-inflammatory treatment in Parkinson’s disease? Park Relat Disord. 2010;16(1):57-59.[DOI]
-
18. Tronel C, Largeau B, Santiago Ribeiro MJ, Guilloteau D, Dupont AC, Arlicot N, et al. Molecular targets for PET imaging of activated microglia: The current situation and future expectations. Int J Mol Sci. 2017;18(4):802.[DOI]
-
19. Robak LA, Jansen IE, van Rooij J, Uitterlinden AG, Kraaij R, Jankovic J, et al. Excessive burden of lysosomal storage disorder gene variants in Parkinson’s disease. Brain. 2017;140(12):3191-3203.[DOI]
-
20. Heng TSP, Painter MW, Elpek K, Lukacs-Kornek V, Mauermann N, Turley SJ, et al. The Immunological Genome Project: Networks of gene expression in immune cells. Nat Immunol. 2008;9(10):1091-1094.[DOI]
-
21. Ghoochani A, Heiby JC, Rawat ES, Medoh UN, Di Fraia D, Dong W, et al. Cell-type resolved protein atlas of brain lysosomes identifies SLC45A1-associated disease as a lysosomal disorder. Cell. 2026;189(3):765-782.e31.[DOI]
-
22. Stakenborg N, Viola MF, Boeckxstaens G. Intestinal neuron-associated macrophages in health and disease. Nat Immunol. 2025;26(7):1004-1013.[DOI]
-
23. Gabanyi I, Muller PA, Feighery L, Oliveira TY, Costa-Pinto FA, Mucida D, et al. Neuro-immune interactions drive tissue programming in intestinal macrophages. Cell. 2016;164(3):378-391.[DOI]
-
24. DSchepper S, Verheijden S, Aguilera-Lizarraga J, Viola MF, Boesmans W, Stakenborg N, et al. Self-maintaining gut macrophages are essential for intestinal homeostasis. Cell. 2019;176(3):676.[DOI]
-
25. Munro DAD, Movahedi K, Priller J. Macrophage compartmentalization in the brain and cerebrospinal fluid system. Sci Immunol. 2022;7(69):eabk0391.[DOI]
-
26. Vara-Pérez M, Movahedi K. Border-associated macrophages as gatekeepers of brain homeostasis and immunity. Immunity. 2025;58(5):1085-1100.[DOI]
-
27. Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330(6005):841-845.[DOI]
-
28. Du S, Ou F, Drieu A, Xu EZ, Cheng Y, Storck SE, et al. Brain-engrafted monocyte-derived macrophages from blood and skull-bone marrow exhibit distinct properties. Neuron. 2026;114(11):1986-2005.e9.[DOI]
-
29. Amann L, Fell A, Monaco G, Sankowski R, Wu HZQ, Jordão MJC, et al. Extrasinusoidal macrophages are a distinct subset of immunologically active dural macrophages. Sci Immunol. 2024;9(102):eadh1129.[DOI]
-
30. Viola MF, Chavero-Pieres M, Modave E, Delfini M, Stakenborg N, Estévez MC, et al. Dedicated macrophages organize and maintain the enteric nervous system. Nature. 2023;618(7966):818-826.[DOI]
-
31. Kulkarni S, Micci MA, Leser J, Shin C, Tang SC, Fu YY, et al. Adult enteric nervous system in health is maintained by a dynamic balance between neuronal apoptosis and neurogenesis. Proc Natl Acad Sci U S A. 2017;114(18):E3709-E3718.[DOI]
-
32. Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, et al. Synaptic pruning by microglia is necessary for normal brain development. Science. 2011;333(6048):1456-1458.[DOI]
-
33. Li Q, Cheng Z, Zhou L, Darmanis S, Neff NF, Okamoto J, et al. Developmental heterogeneity of microglia and brain myeloid cells revealed by deep single-cell RNA sequencing. Neuron. 2019;101(2):207-223.e10.[DOI]
-
34. Escoubas CC, Dorman LC, Nguyen PT, Lagares-Linares C, Nakajo H, Anderson SR, et al. Type-I-interferon-responsive microglia shape cortical development and behavior. Cell. 2024;187(8):1936-1954.e24.[DOI]
-
35. McNamara NB, Munro DAD, Bestard-Cuche N, Uyeda A, Bogie JFJ, Hoffmann A, et al. Microglia regulate central nervous system myelin growth and integrity. Nature. 2023;613(7942):120-129.[DOI]
-
36. Herzog C, Pons Garcia L, Keatinge M, Greenald D, Moritz C, Peri F, et al. Rapid clearance of cellular debris by microglia limits secondary neuronal cell death after brain injury in vivo. Development. 2019;146(9):dev174698.[DOI]
-
37. Fourgeaud L, Través PG, Tufail Y, Leal-Bailey H, Lew ED, Burrola PG, et al. TAM receptors regulate multiple features of microglial physiology. Nature. 2016;532(7598):240-244.[DOI]
-
38. Safaiyan S, Kannaiyan N, Snaidero N, Brioschi S, Biber K, Yona S, et al. Age-related myelin degradation burdens the clearance function of microglia during aging. Nat Neurosci. 2016;19(8):995-998.[DOI]
-
39. Guldner IH, Wagner VP, Moran-Losada P, Shi SM, Golub SW, Hevler JF, et al. Ageing promotes microglial accumulation of slow-degrading synaptic proteins. Nature. 2026;650(8103):930-941.[DOI]
-
40. Bishop ES, Namkoong H, Aurelian L, McCarthy M, Nallagatla P, Zhou W, et al. Age-dependent microglial disease phenotype results in functional decline in gut macrophages. Gastro Hep Adv. 2023;2(2):261-276.[DOI]
-
41. Swaminathan M, Fung C, Finkelstein DI, Bornstein JC, Foong JPP. α-synuclein regulates development and function of cholinergic enteric neurons in the mouse colon. Neuroscience. 2019;423:76-85.[DOI]
-
42. Phillips RJ, Billingsley CN, Powley TL. Macrophages are unsuccessful in clearing aggregated alpha-synuclein from the gastrointestinal tract of healthy aged Fischer 344 rats. Anat Rec. 2013;296(4):654-669.[DOI]
-
43. Willis CL, Garwood CJ, Ray DE. A size selective vascular barrier in the rat area postrema formed by perivascular macrophages and the extracellular matrix. Neuroscience. 2007;150(2):498-509.[DOI]
-
44. Du S, Nguyen KM, Ulezko Antonova A, Fachi JL, Rodrigues PF, Verdiani A, et al. Diversity and immune dynamics of choroid plexus macrophages are shaped by distinct developmental origins. Nat Neurosci. 2026;29(3):717-731.[DOI]
-
45. Drieu A, Du S, Storck SE, Rustenhoven J, Papadopoulos Z, Dykstra T, et al. Parenchymal border macrophages regulate the flow dynamics of the cerebrospinal fluid. Nature. 2022;611(7936):585-593.[DOI]
-
46. Smyth LCD, Xu D, Okar SV, Dykstra T, Rustenhoven J, Papadopoulos Z, et al. Identification of direct connections between the dura and the brain. Nature. 2024;627(8002):165-173.[DOI]
-
47. Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, et al. Structural and functional features of central nervous system lymphatic vessels. Nature. 2015;523(7560):337-341.[DOI]
-
48. Da Mesquita S, Louveau A, Vaccari A, Smirnov I, Cornelison RC, Kingsmore KM, et al. Functional aspects of meningeal lymphatics in ageing and Alzheimer’s disease. Nature. 2018;560(7717):185-191.[DOI]
-
49. Joachim CL, Duffy LK, Morris JH, Selkoe DJ. Protein chemical and immunocytochemical studies of meningovascular β-amyloid protein in Alzheimer’s disease and normal aging. Brain Res. 1988;474(1):100-111.[DOI]
-
50. Shahnawaz M, Tokuda T, Waragai M, Mendez N, Ishii R, Trenkwalder C, et al. Development of a biochemical diagnosis of Parkinson disease by detection of α-synuclein misfolded aggregates in cerebrospinal fluid. JAMA Neurol. 2017;74(2):163-172.[DOI]
-
51. Kim MW, Gao W, Lichti CF, Gu X, Dykstra T, Cao J, et al. Endogenous self-peptides guard immune privilege of the central nervous system. Nature. 2025;637(8044):176-183.[DOI]
-
52. Choi I, Zhang Y, Seegobin SP, Pruvost M, Wang Q, Purtell K, et al. Microglia clear neuron-released α-synuclein via selective autophagy and prevent neurodegeneration. Nat Commun. 2020;11(1):1386.[DOI]
-
53. Mao X, Ou MT, Karuppagounder SS, Kam TI, Yin X, Xiong Y, et al. Pathological α-synuclein transmission initiated by binding lymphocyte-activation gene 3. Science. 2016;353(6307):aah3374.[DOI]
-
54. Emmenegger M, de Cecco E, Hruska-Plochan M, Eninger T, Schneider MM, Barth M, et al. LAG3 is not expressed in human and murine neurons and does not modulate α-synucleinopathies. EMBO Mol Med. 2021;13(9):e14745.[DOI]
-
55. Wu KM, Xu QH, Liu YQ, Feng YW, Han SD, Zhang YR, et al. Neuronal FAM171A2 mediates α-synuclein fibril uptake and drives Parkinson’s disease. Science. 2025;387(6736):892-900.[DOI]
-
56. Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS, et al. α-Synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol. 2002;4(2):160-164.[DOI]
-
57. Gorbatyuk OS, Li S, Sullivan LF, Chen W, Kondrikova G, Manfredsson FP, et al. The phosphorylation state of Ser-129 in human α-synuclein determines neurodegeneration in a rat model of Parkinson disease. Proc Natl Acad Sci U S A. 2008;105(2):763-768.[DOI]
-
58. Freundt EC, Maynard N, Clancy EK, Roy S, Bousset L, Sourigues Y, et al. Neuron-to-neuron transmission of α-synuclein fibrils through axonal transport. Ann Neurol. 2012;72(4):517-524.[DOI]
-
59. Abounit S, Bousset L, Loria F, Zhu S, de Chaumont F, Pieri L, et al. Tunneling nanotubes spread fibrillar α-synuclein by intercellular trafficking of lysosomes. EMBO J. 2016;35(19):2120-2138.[DOI]
-
60. Danzer KM, Kranich LR, Ruf WP, Cagsal-Getkin O, Winslow AR, Zhu L, et al. Exosomal cell-to-cell transmission of alpha synuclein oligomers. Mol Neurodegener. 2012;7:42.[DOI]
-
61. Dorion MF, Yaqubi M, Senkevich K, Kieran NW, MacDonald A, Chen CXQ, et al. MerTK is a mediator of alpha-synuclein fibril uptake by human microglia. Brain. 2024;147(2):427-443.[DOI]
-
62. Chang K, Kim J, Iba M, Park JH, Beilina A, Syed ZA, et al. Differential microglial responses to structurally distinct alpha-synuclein polymorphs. Mol Brain. 2025;19(1):3.[DOI]
-
63. Doorn KJ, Moors T, Drukarch B, van de Berg WD, Lucassen PJ, van Dam AM, et al. Microglial phenotypes and toll-like receptor 2 in the substantia nigra and hippocampus of incidental Lewy body disease cases and Parkinson’s disease patients. Acta Neuropathol Commun. 2024;2(1):90.[DOI]
-
64. Kim C, Ho DH, Suk JE, You S, Michael S, Kang J, et al. Neuron-released oligomeric α-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat Commun. 2013;4:1562.[DOI]
-
65. Scheiblich H, Dansokho C, Mercan D, Schmidt SV, Bousset L, Wischhof L, et al. Microglia jointly degrade fibrillar alpha-synuclein cargo by distribution through tunneling nanotubes. Cell. 2021;184(20):5089-5106.e21.[DOI]
-
66. Hong H, Wang Y, Menard M, Buckley JA, Zhou L, Volpicelli-Daley L, et al. Suppression of the JAK/STAT pathway inhibits neuroinflammation in the line 61-PFF mouse model of Parkinson’s disease. J Neuroinflammation. 2024;21(1):216.[DOI]
-
67. A-Gonzalez N, Quintana JA, García-Silva S, Mazariegos M, González de la Aleja A, Nicolás-Ávila JA, et al. Phagocytosis imprints heterogeneity in tissue-resident macrophages. J Exp Med. 2017;214(5):1281-1296.[DOI]
-
68. He Y, Taylor N, Yao X, Bhattacharya A. Mouse primary microglia respond differently to LPS and poly (I:C) in vitro. Sci Rep. 2021;11(1):10447.[DOI]
-
69. Su X, Maguire-Zeiss KA, Giuliano R, Prifti L, Venkatesh K, Federoff HJ, et al. Synuclein activates microglia in a model of Parkinson’s disease. Neurobiol Aging. 2008;29(11):1690-1701.[DOI]
-
70. Bido S, Muggeo S, Massimino L, Marzi MJ, Giannelli SG, Melacini E, et al. Microglia-specific overexpression of α-synuclein leads to severe dopaminergic neurodegeneration by phagocytic exhaustion and oxidative toxicity. Nat Commun. 2021;12(1):6237.[DOI]
-
71. Wei P, Li J. Commentary: CD22 blockade restores homeostatic microglial phagocytosis in ageing brains. Front Immunol. 2019;10:1301.[DOI]
-
72. Masters TA, Pontes B, Viasnoff V, Li Y, Gauthier NC. Plasma membrane tension orchestrates membrane trafficking, cytoskeletal remodeling, and biochemical signaling during phagocytosis. Proc Natl Acad Sci U S A. 2013;(29):11875-11880.[DOI]
-
73. Codo AC, Romero-Pichardo JE, Wang Z, Aufiero MA, Lazarov T, Saitz Rojas W, et al. Polystyrene microplastic-induced pathophysiology is driven by disruption of efferocytosis. Immunity. 2026;59(3):618-636.e11.[DOI]
-
74. Nuber S, Rajsombath M, Minakaki G, Winkler J, Müller CP, Ericsson M, et al. Abrogating native α-synuclein tetramers in mice causes a L-DOPA-responsive motor syndrome closely resembling Parkinson’s disease. Neuron. 2018;100(1):75-90.e5.[DOI]
-
75. De Schepper S, Konstantellos V, Conway JA, Sokolova D, Zaccagnini L, Cowley MV, et al. Intestinal macrophages modulate synucleinopathy along the gut-brain axis. Nature. 2026;651(8104):174-184.[DOI]
-
76. Mackie PM, Koshy J, Bhogade M, Hammoor T, Hachmeister W, Lloyd GM, et al. Complement C1q-dependent engulfment of alpha-synuclein induces ENS-resident macrophage exhaustion and accelerates Parkinson’s-like gut pathology. bioRxiv [Preprint]. 2023.[DOI]
-
77. Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science. 2016;352(6286):712-716.[DOI]
-
78. Freeman D, Cedillos R, Choyke S, Lukic Z, McGuire K, Marvin S, et al. Alpha-synuclein induces lysosomal rupture and cathepsin dependent reactive oxygen species following endocytosis. PLoS One. 2013;8(4):e62143.[DOI]
-
79. Dilsizoglu Senol A, Samarani M, Syan S, Guardia CM, Nonaka T, Liv N, et al. α-Synuclein fibrils subvert lysosome structure and function for the propagation of protein misfolding between cells through tunneling nanotubes. PLoS Biol. 2021;19(7):e3001287.[DOI]
-
80. Abe T, Kuwahara T, Suenaga S, Sakurai M, Takatori S, Iwatsubo T, et al. Lysosomal stress drives the release of pathogenic α-synuclein from macrophage lineage cells via the LRRK2-Rab10 pathway. iScience. 2024;27(2):108893.[DOI]
-
81. Zhang Z, Niu K, Huang T, Guo J, Xarbat G, Gong X, et al. Microglia depletion reduces neurodegeneration and remodels extracellular matrix in a mouse Parkinson’s disease model triggered by α-synuclein overexpression. NPJ Parkinsons Dis. 2025;11(1):15.[DOI]
-
82. Cascella R, Chen SW, Bigi A, Camino JD, Xu CK, Dobson CM, et al. The release of toxic oligomers from α-synuclein fibrils induces dysfunction in neuronal cells. Nat Commun. 2021;12(1):1814.[DOI]
-
83. Peng C, Gathagan RJ, Covell DJ, Medellin C, Stieber A, Robinson JL, et al. Cellular milieu imparts distinct pathological α-synuclein strains in α-synucleinopathies. Nature. 2018;557(7706):558-563.[DOI]
-
84. Yu E, Ambati A, Andersen MS, Krohn L, Estiar MA, Saini P, et al. Fine mapping of the HLA locus in Parkinson’s disease in Europeans. NPJ Parkinsons Dis. 2021;7(1):84.[DOI]
-
85. Sulzer D, Alcalay RN, Garretti F, Cote L, Kanter E, Agin-Liebes J, et al. T cells from patients with Parkinson’s disease recognize α-synuclein peptides. Nature. 2017;546(7660):656-661.[DOI]
-
86. Lindestam Arlehamn CS, Dhanwani R, Pham J, Kuan R, Frazier A, Rezende Dutra J, et al. α-Synuclein-specific T cell reactivity is associated with preclinical and early Parkinson’s disease. Nat Commun. 2020;11(1):1875.[DOI]
-
87. Garretti F, Monahan C, Sloan N, Bergen J, Shahriar S, Kim SW, et al. Interaction of an α-synuclein epitope with HLA-DRB1* 15:01 triggers enteric features in mice reminiscent of prodromal Parkinson’s disease. Neuron. 2023;111(21):3397-3413.e5.[DOI]
-
88. Abualrous ET, Stolzenberg S, Sticht J, Wieczorek M, Roske Y, Günther M, et al. MHC-II dynamics are maintained in HLA-DR allotypes to ensure catalyzed peptide exchange. Nat Chem Biol. 2023;19(10):1196-1204.[DOI]
-
89. Harms AS, Cao S, Rowse AL, Thome AD, Li X, Mangieri LR, et al. MHCII is required for α-synuclein-induced activation of microglia, CD4 T cell proliferation, and dopaminergic neurodegeneration. J Neurosci. 2013;33(23):9592-9600.[DOI]
-
90. Schonhoff AM, Figge DA, Williams GP, Jurkuvenaite A, Gallups NJ, Childers GM, et al. Border-associated macrophages mediate the neuroinflammatory response in an alpha-synuclein model of Parkinson disease. Nat Commun. 2023;14(1):3754.[DOI]
-
91. Amaral L, Martins M, Côrte-Real M, Outeiro TF, Chaves SR, Rego A, et al. The neurotoxicity of pesticides: Implications for Parkinson’s disease. Chemosphere. 2025;377:144348.[DOI]
-
92. Dorsey ER, Bloem BR. Parkinson’s disease is predominantly an environmental disease. J Parkinsons Dis. 2024;14(3):451-456.[DOI]
-
93. Liekniņa I, Reimer L, Panteļejevs T, Lends A, Jaudzems K, El-Turabi A, et al. Structural basis of epitope recognition by anti-alpha-synuclein antibodies MJFR14-6-4-2. NPJ Parkinsons Dis. 2024;10(1):206.[DOI]
-
94. Silvin A, Uderhardt S, Piot C, Da Mesquita S, Yang K, Geirsdottir L, et al. Dual ontogeny of disease-associated microglia and disease inflammatory macrophages in aging and neurodegeneration. Immunity. 2022;55(8):1448-1465.e6.[DOI]
-
95. Harms AS, Thome AD, Yan Z, Schonhoff AM, Williams GP, Li X, et al. Peripheral monocyte entry is required for alpha-Synuclein induced inflammation and Neurodegeneration in a model of Parkinson disease. Exp Neurol. 2018;300:179-187.[DOI]
-
96. Mark JR, Titus AM, Staley HA, Alvarez S, Mahn S, McFarland NR, et al. Peripheral immune cell response to stimulation stratifies Parkinson’s disease progression from prodromal to clinical stages. Commun Biol. 2025;8(1):716.[DOI]
-
97. Geirsdottir L, David E, Keren-Shaul H, Weiner A, Bohlen SC, Neuber J, et al. Cross-species single-cell analysis reveals divergence of the primate microglia program. Cell. 2019;179(7):1609-1622.e16.[DOI]
-
98. Erny D, Hrabě de Angelis AL, Jaitin D, Wieghofer P, Staszewski O, David E, et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat Neurosci. 2015;18(7):965-977.[DOI]
-
99. Sampson TR, Debelius JW, Thron T, Janssen S, Shastri GG, Ilhan ZE, et al. Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson’s disease. Cell. 2016;167(6):1469-1480.e12.[DOI]
-
100. Albertini G, Zielonka M, Cuypers ML, Snellinx A, Xu C, Poovathingal S, et al. The Alzheimer’s therapeutic Lecanemab attenuates Aβ pathology by inducing an amyloid-clearing program in microglia. Nat Neurosci. 2026;29(1):100-110.[DOI]
Copyright
© The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Publisher’s Note
Science Exploration remains a neutral stance on jurisdictional claims in published
maps
and institutional affiliations. The views expressed in this article are solely those
of
the author(s) and do not reflect the opinions of the Editors or the publisher.
Share And Cite



Science Exploration Style
Konstantellos V, Van De Walle P, De Schepper S. The gut-brain axis in Parkinson’s disease: A macrophage perspective. 2026;1:202606. https://doi.org/10.70401/mc.2026.0008
Tips
Copy completed.
Submit a Manuscript
Author Instructions
Cite this Article
Article Metrics
0
View
0
Download
Cited
Article Updates

- Abstract
- Keywords
- 1. Introduction
- 2. Tissue-Resident Macrophages in the ENS and CNS: Mechanisms of Neuronal Clearance
- 3. Tissue-Resident Macrophages into the Spotlight in Parkinson’s Disease
- 4. Cellular Stress Responses
- 5. Propagation of Synucleinopathy
- 6. Antigen Presentation
- 7. Outlook: From Clearance Failure to Therapeutic Opportunities
- Authors contribution
- Conflicts of interest
- Ethical approval
- Consent to participate
- Consent for publication
- Availability of data and materials
- Funding
- References
- Copyright
Science Exploration Style
Konstantellos V, Van De Walle P, De Schepper S. The gut-brain axis in Parkinson’s disease: A macrophage perspective. 2026;1:202606. https://doi.org/10.70401/mc.2026.0008
copy
Share Link
copy