Delivering RNA-based therapeutics for pancreatic cancer treatment: Current progress and challenges
Haoyang Cong
1,#
,
Limin Chang
2,#
,
Yu Li
3
,
Wen-Bin Zou
4,*
,
Congcong Xu
1,2,5,*

*Correspondence to:
Wen-Bin Zou, Department of Gastroenterology, Shanghai Institute of Pancreatic Diseases, Changhai Hospital; National Key Laboratory of Immunity and Inflammation, Naval Medical University, Shanghai 200433, China.
E-mail: dr.wenbinzou@hotmail.com
Congcong Xu, International College of Pharmaceutical Innovation, Soochow University, Suzhou 215123, Jiangsu, China. E-mail: xucc@suda.edu.cn
Congcong Xu, International College of Pharmaceutical Innovation, Soochow University, Suzhou 215123, Jiangsu, China. E-mail: xucc@suda.edu.cn
BME Horiz. 2026;4:202609. 10.70401/bmeh.2026.0028
Received: January 24, 2026Accepted: May 20, 2026Published: May 20, 2026
This article belongs to the Special lssue Engineering RNA Delivery Technologies for Vaccine and Therapeutic Development
Abstract
Pancreatic cancer, particularly pancreatic ductal adenocarcinoma (PDAC), presents formidable treatment challenges owing to the intricacies of its anatomical structures and biological impediments. RNA-based therapeutics offer unprecedented potential for PDAC treatment. This review comprehensively summarizes the latest advances in PDAC-targeted nucleic acid delivery systems, including extracellular vesicles, polymer carriers, lipid nanoparticles, viral vectors, and antibody-drug conjugates. Key features of each delivery platform, such as their structural modifications and targeting mechanisms, are highlighted. Additionally, the review emphasizes their capacity to overcome PDAC-specific biological barriers (e.g., blood-pancreatic barrier, immunosuppressive tumor microenvironment, dense extracellular matrix). It also outlines current challenges and future directions in optimizing nucleic acid delivery for PDAC. Integrating the latest insights into PDAC biology, delivery system engineering, and nucleic acid drug development, this review provides a concise, up-to-date perspective to guide the design of effective translational strategies for PDAC therapy.
Keywords
Pancreatic cancer, RNA therapeutics, targeted delivery
1. Introduction
Pancreatic cancer broadly refers to all malignant tumors that originate from the epithelial tissue of pancreas, with the most common subtype being pancreatic ductal adenocarcinoma (PDAC). PDAC accounts for 80%-90% of all pancreatic cancers[1], and holds infamous distinction of being labeled the “king of cancers”. The deep anatomic location of pancreas contributes to subtle and non-specific early symptoms, inadequate surgical resection rates, and limited efficacy of chemotherapy, collectively posing significant challenges for developing efficacious therapeutic agents for pancreatic cancer[2]. RNA therapy is regarded as a powerful therapeutic method with its myriad biological functions, which has contributed to advances in treating various diseases such as cancers, viral infections, and inflammatory disorders in the last few years. Various therapeutic RNAs, such as small interfering RNA (siRNA), antisense oligonucleotide (ASO), messenger RNA (mRNA), have been used in gene therapy for several diseases. While these RNA-based drugs have led to successful therapeutic outcomes, there is still room for improvement in several aspects such as blood circulation time, stability, and RNA loading capacity. For example, RNA-based therapeutics are highly susceptible to degradation by serum nucleases in bloodstream[3]. Intravenously administered nanoparticles or delivery vehicles are readily taken up and cleared by the reticuloendothelial system, particularly in liver and spleen[4]. This significantly reduces the effective drug dose that reaches pancreas; After cellular uptake, RNA drugs are typically trapped within endosomes and ultimately transported to lysosomes where they are degraded by enzymes[5]. To exert their function, RNA-based drugs (especially siRNA/mRNA) must effectively escape from the endosomes with the help of a carrier and be released into the cytoplasm.
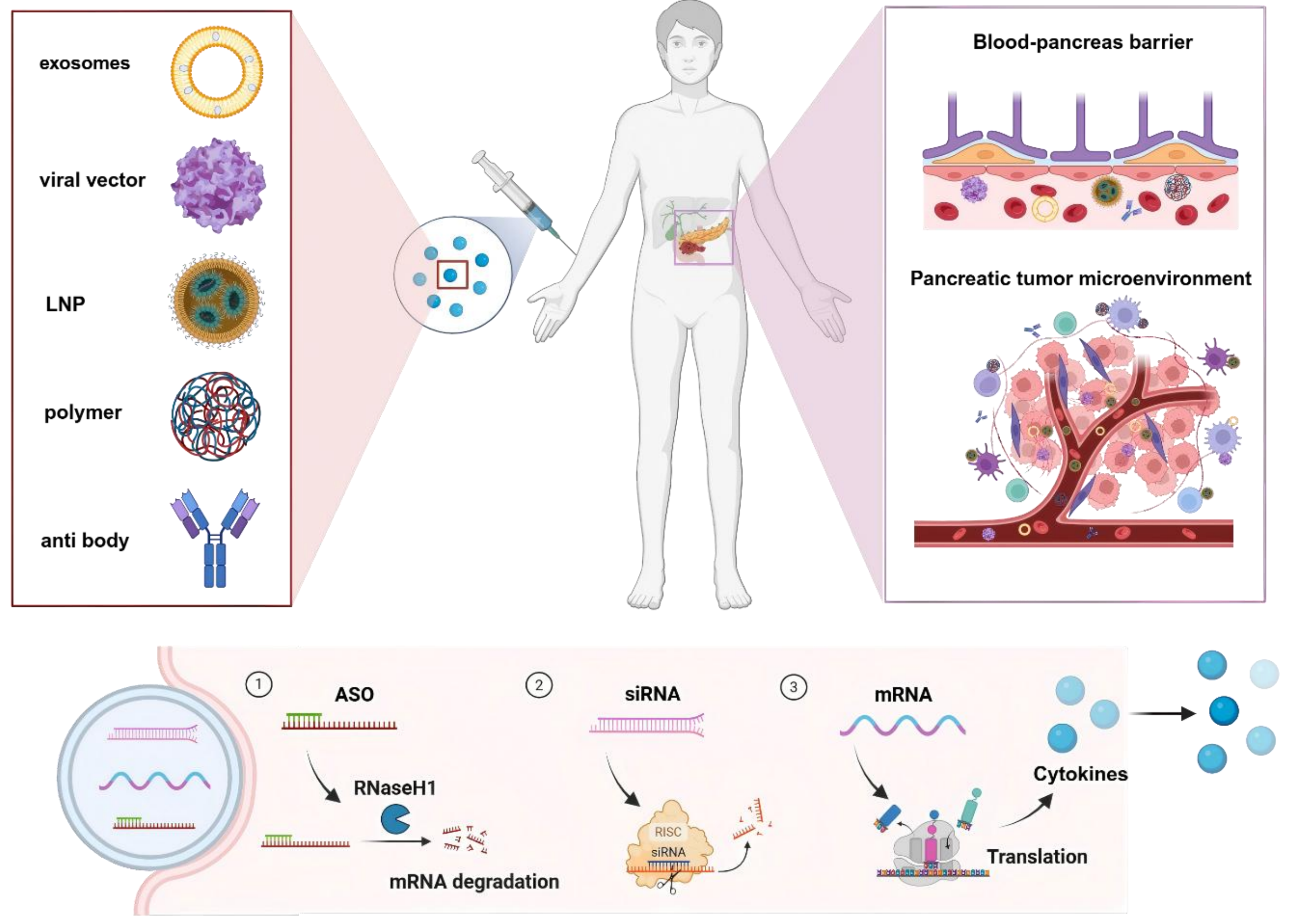
This article aims to systematically delineate the application of RNA-based therapeutics for pancreatic cancer, with a focus on three pivotal aspects: The challenges of pancreatic cancer treatment, existing strategies for pancreatic-targeted delivery strategies, and the latest advancements in the utilization of RNA-based therapeutics for the treatment of pancreatic cancer (Scheme 1).
{kind=link}
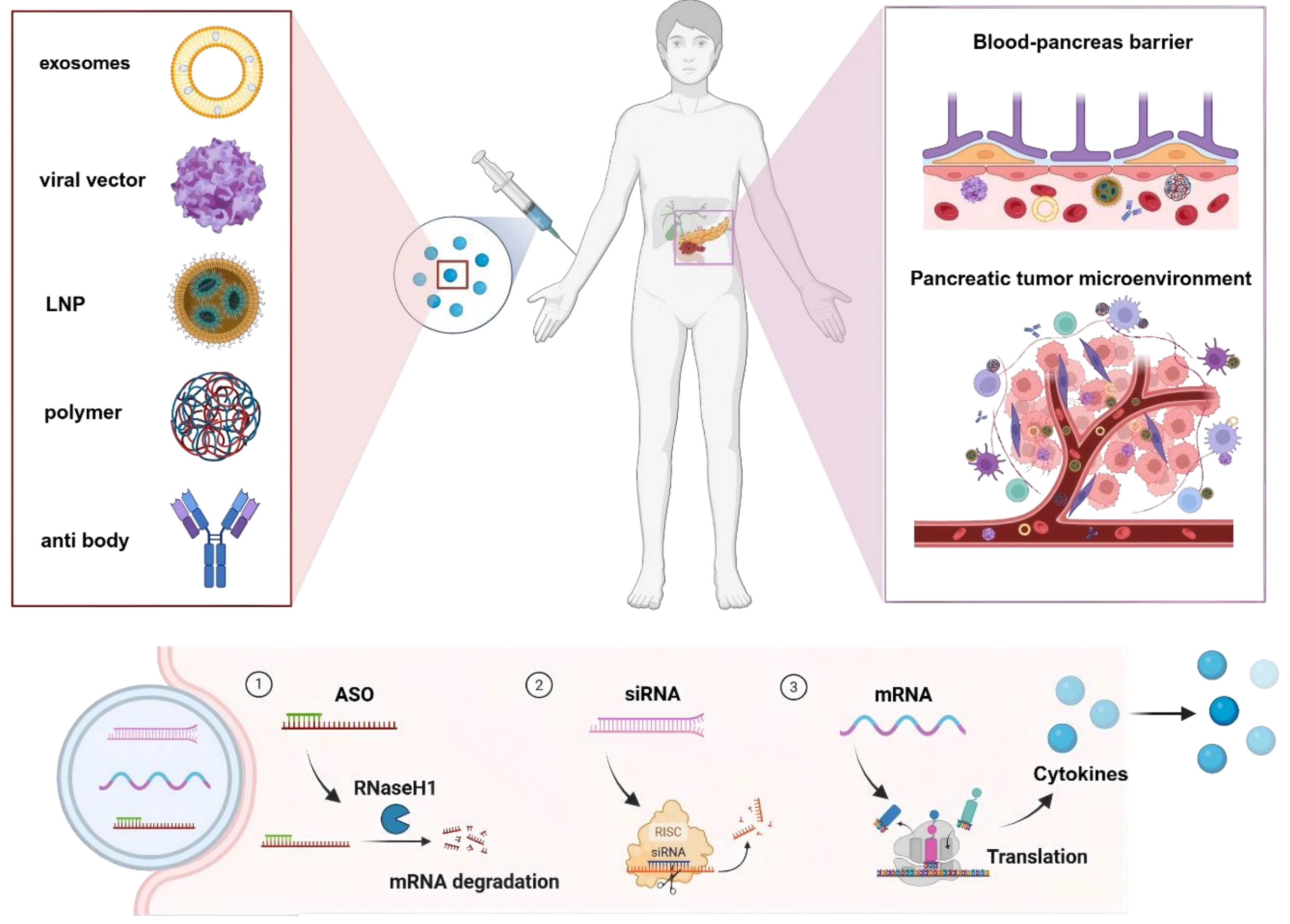
Scheme 1. Overview of delivering RNA-based therapeutics for pancreatic cancer treatment. Created in BioRender. Chang, L. (2026) https://BioRender.com/r5om53y. LNP: lipid nanoparticle; ASO: antisense oligonucleotide; siRNA: small interfering RNA; mRNA: messenger RNA.
2. Challenges of Pancreatic Cancer Treatment
This intricate convergence of anatomical constraints, pathophysiological aberrations, and immunosuppressive mechanisms orchestrates a robust defense that renders pancreatic tumors refractory to standard therapeutic modalities. The pancreas, with its unique lobulated architecture and stringent blood-pancreas barrier, presents a formidable fortress that limits drug accessibility. Within this tumor microenvironment (TME), tumor cells thrive amidst a densely desmoplastic stroma orchestrated by tumor-associated fibroblasts, while aberrant vascularization and a profoundly immunosuppressive microenvironment further impair therapeutic efficacy. This chapter systematically dissects these interconnected challenges, including the physical barricades of the blood-pancreas barrier and extracellular matrix, as well as the cellular conspirators of cancer-associated fibroblast (CAF).
2.1 Blood pancreas barrier (BPB)
The treatment of pancreatic cancer, particularly PDAC, faces a formidable obstacle in form of BPB. This highly selective physiological barrier severely restricts the penetration of both small and large molecules into pancreas, drastically limiting distribution and efficacy of chemotherapeutic drugs[6]. The BPB is a complex, multi-layered structure that includes pancreatic capillary endothelial cells with tight junctions, a continuous basement membrane, and a unique periacinal fibroblastic sheath, which is a prominent structure that physically separates capillaries from acinar cells. Furthermore, the acinar cell basement membrane and the tight junctions between acinar cells themselves add additional layers of defense. Consequently, this intricate barrier system presents a major challenge in developing effective therapeutics.
2.2 Pancreatic TME
TME is a complex ecosystem composed of both cellular and non-cellular components that surround tumor tissue. Its complex tissue structure creates a physiological barrier that prevents targeted drugs from reaching tumor sites. The following section will introduce the key components of the pancreatic TME.
2.2.1 Pancreatic tumor extracellular matrix (ECM)
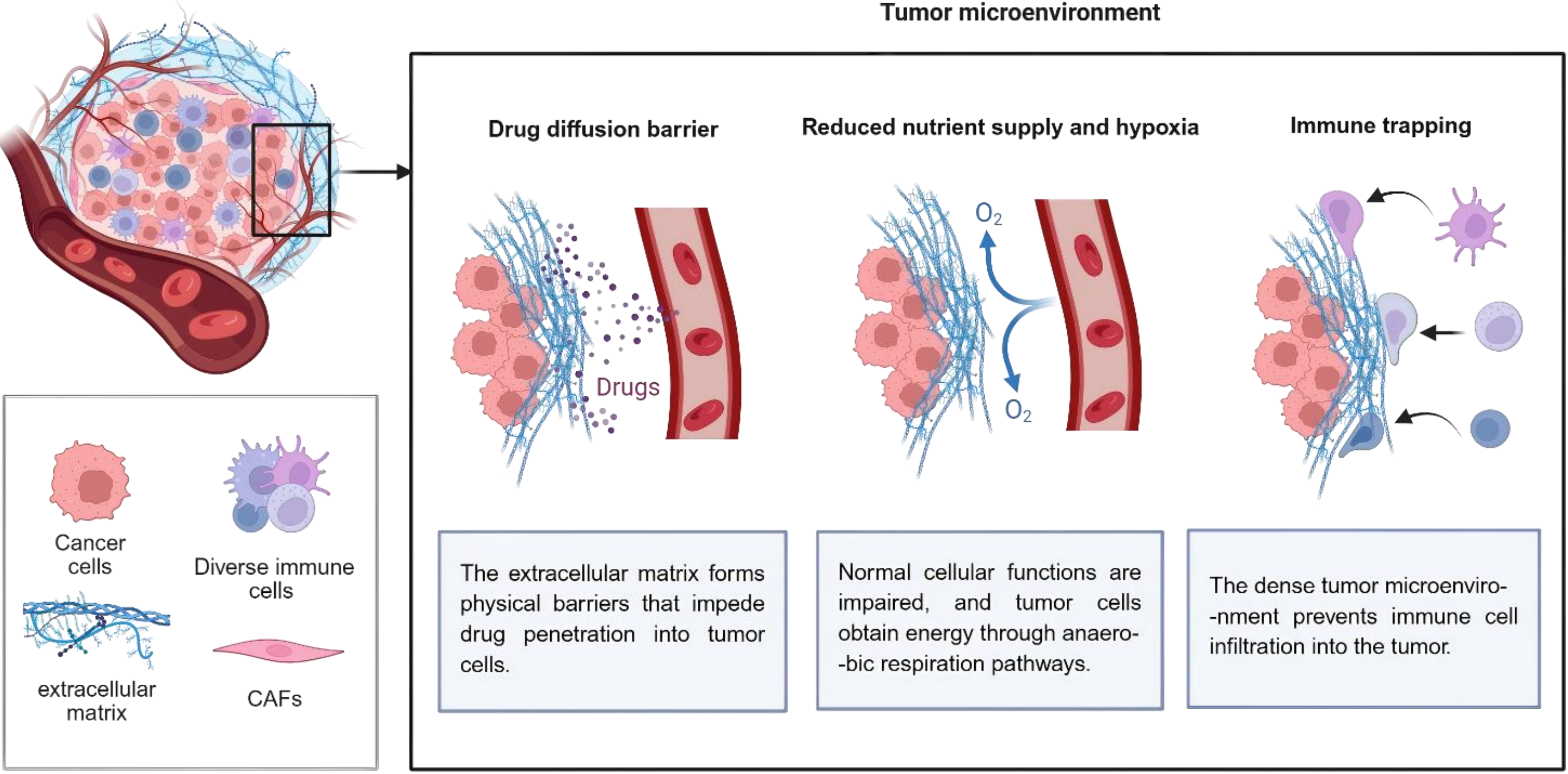
ECM, one of the sources of physiologic resistance of cancer cells to treatment, is regarded as a serious trouble for anti-cancer approaches such as gene and immune therapies. ECM stiffness is obstructive for effective uptake or delivery of drugs to the intratumoral area (Figure 1). High deposition of collagen proteins would do this possibly through binding to proteoglycans and further stabilizing these components of ECM[7]. Increased cancer stem cell (CSC) stemness and expansion of the stem cell niche in TME, mediated by ECM stiffness, hamper drug penetration to this niche[8]. It is hypothesized that ECM stiffness is associated with transformation of cancer cells toward CSCs that can survive under hypoxic conditions of the TME. These stem-like cells are also more resistant to cytotoxic drugs, and they have the potential to migrate and invade via surrounding tissues[9]. In addition, ECM stiffness stimulates hypoxic conditions within the TME. Hypoxic microenvironment triggers abnormal tumor vascular functions including increased permeability and disrupted vascular structure, which raise vascular rupture and leakage risks, and facilitate tumor cell intravasation and distant metastasis. Meanwhile, hypoxia hinders normal vascular maturation and results in immature neovessels, ultimately impairing blood perfusion and drug delivery efficiency in tumor tissues[10].
{kind=link}
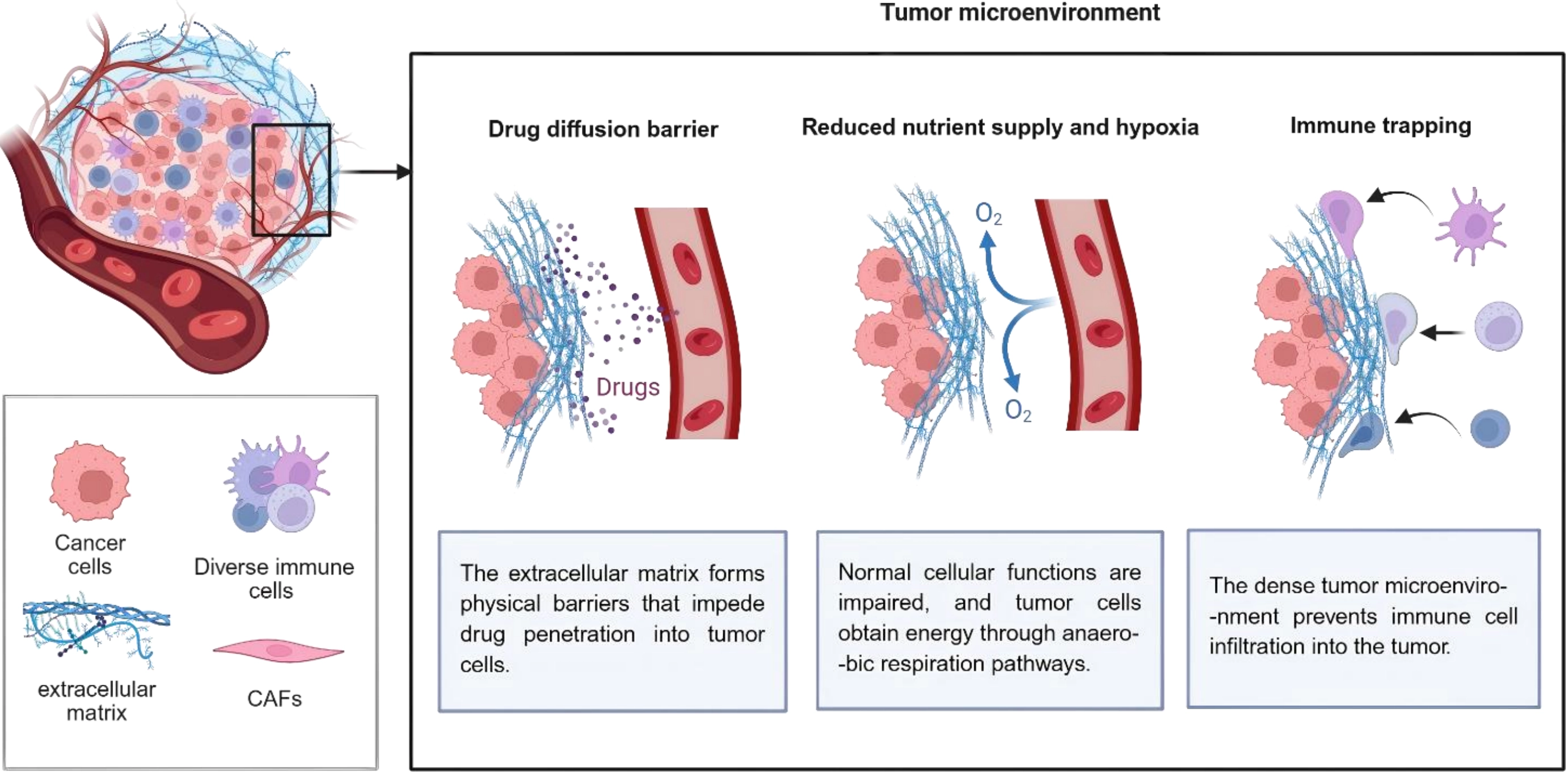
Figure 1. Tumor extracellular matrix reduces therapeutic efficiency in pancreatic tumors. Created in BioRender. Chang, L. (2026) https://BioRender.com/rsxzi9y. CAFs: cancer-associated fibroblasts.
2.2.2 CAFs
Fibroblasts represent the most abundant cell type in the TME and are largely responsible for the deposition of the extracellular matrix, driving characteristic physiological properties of PDAC. CAFs display substantial cellular heterogeneity that critically complicates tumor progression and metastatic dissemination. Multiple cellular origins, including tissue-resident fibroblasts, mesenchymal stem cells[11], and endothelial cells[12], can transdifferentiate into CAFs. CAFs characteristically upregulate α-smooth muscle actin, platelet-derived growth factor receptor α, and fibroblast activation protein, thereby directly fostering tumor cell invasion and proliferation[13]. Furthermore, CAFs comprise multiple distinct subtypes. Among them, myofibroblastic CAFs drive ECM remodeling and mechanical microenvironment modulation; inflammatory CAFs secrete immunosuppressive cytokines; and antigen-presenting CAFs (apCAFs) induce immune tolerance[14]. Notably, apCAFs are broadly distributed and can actively promote tumor progression in PDAC[15].
CAFs have the capacity to secrete a variety of ECM-related proteins, including collagen and hyaluronic acid, which form a physical and metabolic barrier, thereby reducing the clinical effect of chemotherapy and immunotherapy on PDAC, and promoting tumor growth and invasion[16]. CAFs have also been observed to secrete a variety of cytokines, which have been linked to the promotion of tumor invasion, metastasis and chemoresistance[17]. Furthermore, these cells have been shown to modify the phenotype of tumor tissue, ultimately resulting in the development of proliferative fibrosis within the connective tissue of the pancreas. Through their role in negative immune regulation, they can compromise the efficacy of immunotherapy. Strategically designed therapeutic interventions targeting the suppression of CAFs genesis or the disruption of CAFs functionality may effectively reverse their negative immunomodulatory effects within the tumor microenvironment, thereby potentiating the efficacy of immunotherapeutic approaches.
2.2.3 Hypervascularization of PDAC
Insufficient vascularization within pancreatic tumor tissue hinders drug delivery through the circulatory system. The presence of a dense extracellular matrix and elevated interstitial pressure has been demonstrated to result in the collapse of newly formed blood vessels. The glycolytic metabolism driven by KRAS mutations consumes local oxygen, further exacerbating vascular dysfunction[18].
2.2.4 Immunosuppressive properties of pancreatic tumors
Pancreatic cancer exhibits a profoundly immunosuppressive microenvironment characterized by a paucity of functional CD8+ cytotoxic T lymphocytes, restricted to the tumor periphery, scarce and dysfunctional dendritic cells, and extensive infiltration of Tregs, tumor-associated macrophages (TAMs), and myeloid-derived suppressor cells (MDSCs). Elevated transforming growth factor-β, interleukin (IL)-10, and vascular endothelial growth factor further consolidate this immunoinhibitory milieu, collectively rendering immunotherapy largely ineffective[19]. These multifaceted challenges necessitate innovative delivery platforms capable of simultaneous barrier penetration, stromal remodeling and immune activation, a promise that emerging nanotechnology seeks to fulfill.
3. Pancreatic Targeted Delivery Strategy
Exposed RNA nucleic acids (siRNA, mRNA and ASO) are easily and rapidly broken down by nucleases in plasma and tissues in the circulatory system, resulting in a loss of drug efficacy. At the same time, due to their electronegative nature, nucleic acid molecules find it difficult to enter target cells autonomously. Furthermore, the naked molecules accumulate easily in the liver, causing liver injury. Therefore, nucleic acid drug targeting of pancreatic organs must be achieved using a drug delivery system (Figure 2).
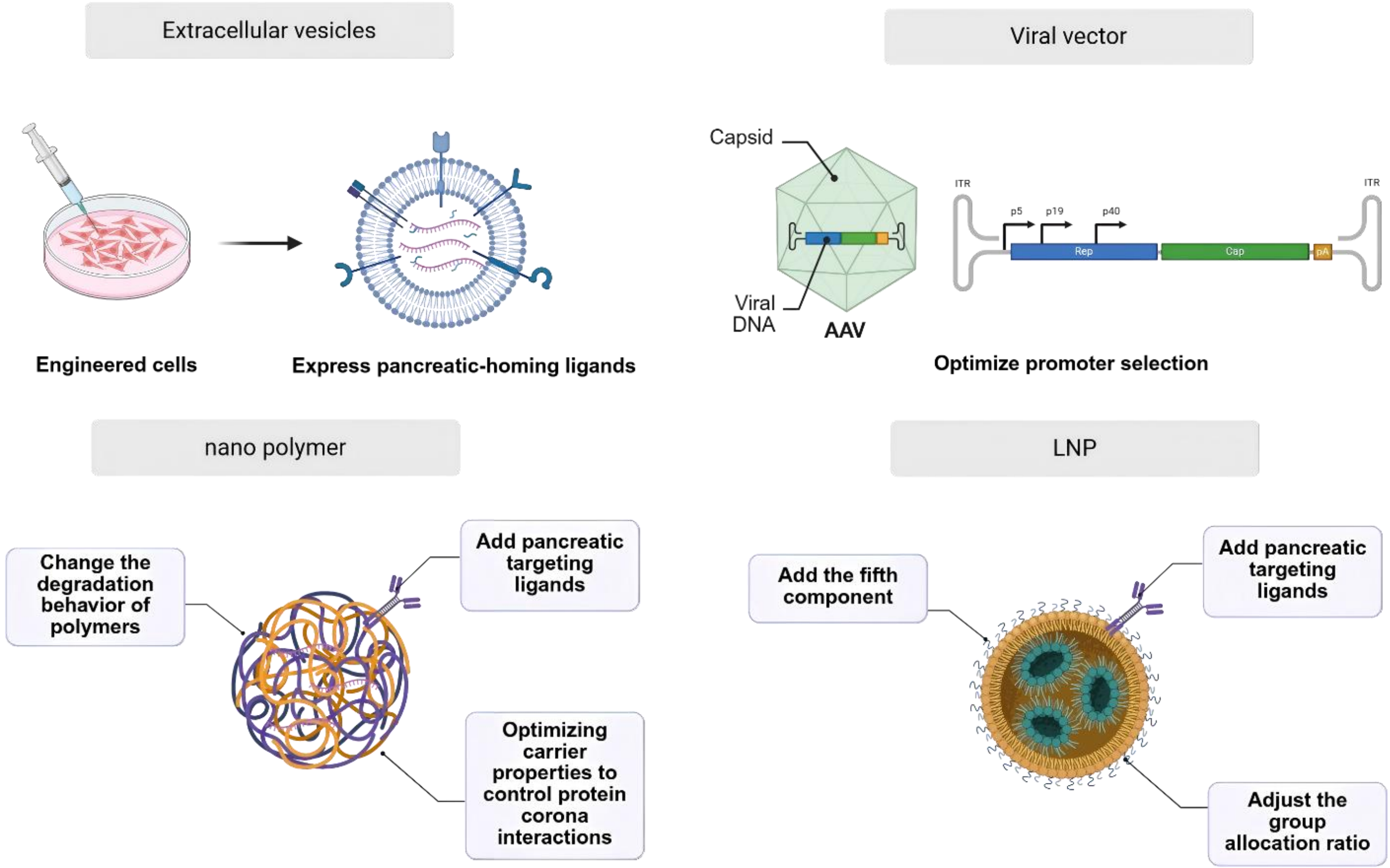{kind=link}
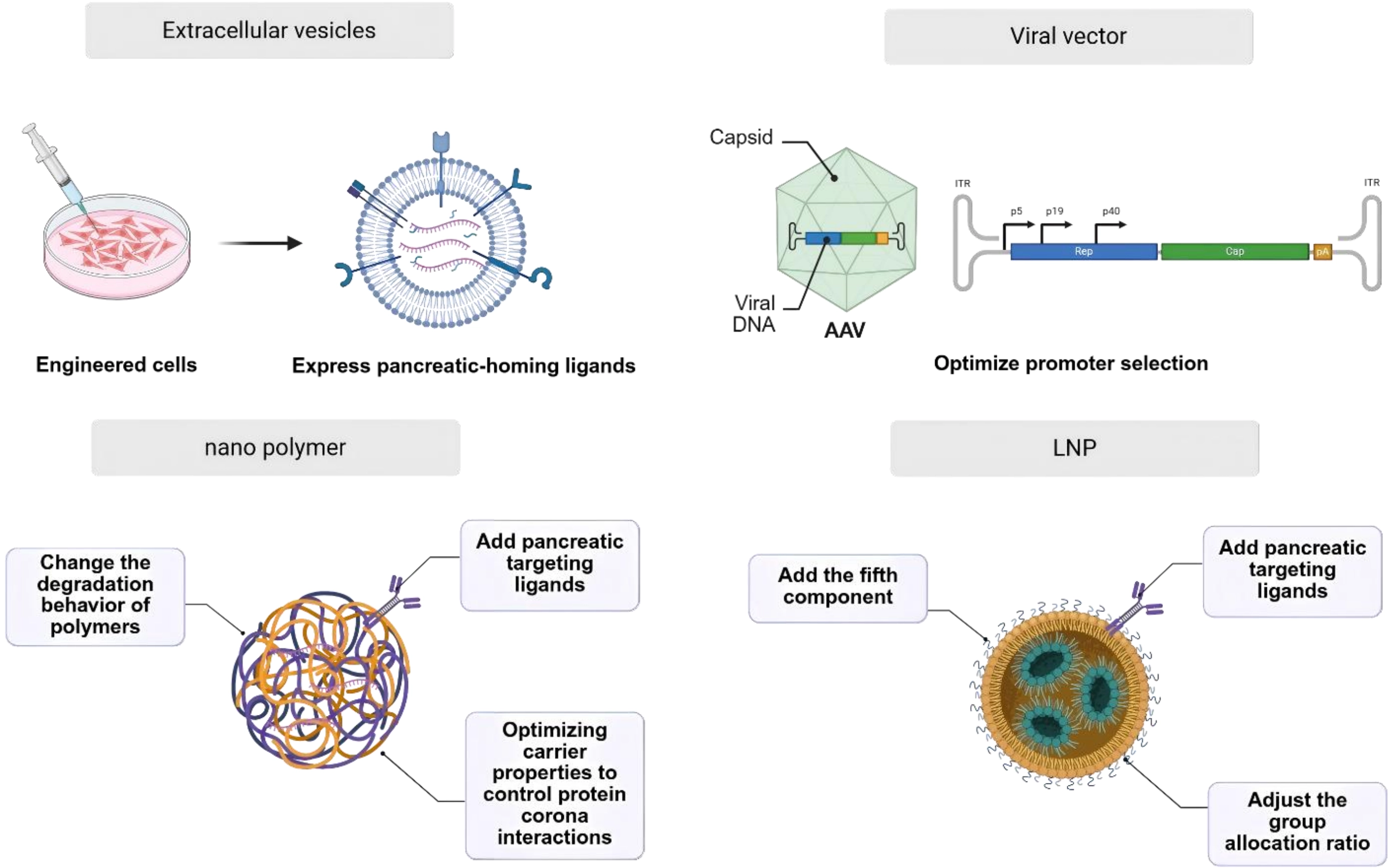
Figure 2. Delivery particle for pancreatic cancer therapy. Created in BioRender. Chang, L. (2026) https://BioRender.com/ue749o6. AAV: adeno-associated virus; LNP: lipid nanoparticle.
3.1 Exosomes delivery system
Exosomes are membrane-bound vesicles that are released into the extracellular matrix when intracellular multivesicular bodies fuse with the cell membrane. They can transport a variety of substances, including proteins, lipids, DNA and RNA. Using extracellular vesicles (EVs) as RNA-based drug delivery carriers targeted to the pancreas has the following advantages: 1. The surface of EVs can be equipped with specific membrane proteins, enabling them to actively target specific cells or tissues in the pancreas and pancreatic tumors[20]; 2. The extracted exosomes have a natural homing effect towards the pancreas[21]; 3. Derived from natural cells, exosomes can reduce immune rejection reactions and improve in vivo stability[22]; 4. They possess a transmembrane transport capacity and can cross the BPB.
The following table illustrates several methods for the realization of exosome delivery to the pancreas (Table 1). One method involves chemical modification of oxaliplatin prodrugs to the surface of exosome particles, thus enhancing their targeting ability towards pancreatic tumors[23]. Another method uses the CD47 or CD81 protein on the surface of exosome particles to inhibit phagocytosis by binding to SIRPα on monocytes and macrophages[24]. A further method involves the genetic engineering of exosome-producing source cells to secrete exosome particles that possess CD133 targeting properties, thus enabling the targeting of pancreatic tumors[27]. Finally, the co-displaying of Arg-Gly-Asp (a peptide capable of binding to αvβ3 integrin on the surface of tumor cells) on exosome particles enhances pancreatic tumor tropism[28].
Table 1. Exosome-based delivery to treat pancreatic cancer.
| Target | Source cells | Payload | Targeted delivery strategy | Ref |
| Pancreatic cancer | Bone marrow mesenchymal stem cells | PD-L1 siRNA | Exosomes modified with oxaliplatin properties | [23] |
| Pancreatic cancer | Normal fibroblast-like mesenchymal cells | Kras G12D siRNA | CD47 molecule protects exosomes from phagocytosis by monocytes and macrophages | [24] |
| Pancreatic cancer | HEK293T epithelial cells | CRISPR/Cas9 | CD81 molecule protects exosomes from being cleared by the immune system | [25] |
| Pancreatic cancer | Bone marrow mesenchymal stem cells | siKrasG12D-2 | CD47 & CD81 can avoid exosomes from being cleared by the immune system | [26] |
| Pancreatic cancer | Adipose-derived stem cells | PD-L1 siRNA | Genetically engineered exosomesCoupled with cd133 | [27] |
| Pancreatic cancer | HEK293T epithelial cells | Paclitaxel | Exosome coupled with CD47 and RGD | [28] |
SiRNA: small interfering RNA; RGD: Arg-Gly-Asp.
The following must be considered when transitioning exosomes into clinical practice: 1. The efficacy and safety of the cell line used for exosome production; 2. The stability of the exosomes following prolonged freeze-thaw cycles; 3. The potential in vivo toxicity associated with long-term administration[29]. To overcome these challenges, parental cells can be genetically engineered to express pancreatic-homing ligands, standardized large-scale production protocols can be developed, and exosome-mimetic nanovesicles can be constructed to improve targeting and controllability.
3.2 Pancreatic targeted polymeric nanodelivery systems
In recent years, there has been a notable increase in the application of nanotechnology in the pharmaceutical field. The application of nanotechnology involves the utilization of nanomaterials for the precise delivery of nucleic acid drugs to pancreatic tissue. Nano polymer offers unparalleled tunability in size, surface chemistry, and stimuli-responsive behavior, enabling rational design to effectively overcome lobular heterogeneity, dense desmoplastic stroma, and the stringent blood-pancreas barrier, and even navigate the heterogeneous TME[30]. Their facile adaptability for sophisticated surface engineering and stimuli-responsive design renders them particularly promising candidates to surmount the biological barriers that have historically constrained pancreatic targeting efficiency.
The nano polymer is characterized by its minute size (less than 200 nm) and physicochemical properties that render it particularly well-suited for nanomedical applications[31]. These properties have been shown to enhance membrane permeability and the retention effect. The small size and alterable surface specificity of these nanoparticles enable them to deliver therapeutic drugs to targeted cancer cells in the biological system via cellular uptake. This uptake can occur via various mechanisms, including receptor-mediated uptake and endocytosis, known as active and passive transport mechanisms[32].
The methods employed to achieve targeted delivery to pancreatic tumors can be categorized into two primary types: passive targeting and active targeting. Passive targeting enhances accumulation within pancreatic tumors by modifying nanoparticle size and composition. Active targeting achieves specificity by conjugating nanoparticles with specific ligands that bind to receptors uniquely expressed on pancreatic tumors’ cells. The subsequent table provides a selection of nanoparticle delivery systems targeted to the pancreas (Table 2).
Table 2. Polymeric nanocarriers for targeted pancreatic delivery.
| Target | Targeted delivery strategy | Polymer scaffold | Payload | Receptor | Ligand | Ref |
| Pancreatic bata cell | Specific lipid composition | DC-CHol, EPC, SM | Anti-miR-375 | - | - | [33] |
| Pancreatic cancer | Adjust the composition of nanoparticles | POEGMA, PDMAEMA | Anti-TUBB βIII-tubulin-siRNA | - | - | [34] |
| Pancreatic cancer | Convert caf into a drug depot | BPEI, DSPE-PEG-AEAA, SPC/DOPE, Cholesterol | BRD4 inhibitor, JQ1C18-ceramide, Gemcitabine, PIL-12 | - | - | [35] |
| Pancreatic cancer | Recognize and bind to the plectin-1 protein | Arg-rich motif | miR-9 | PL-1 | PL-1 | [36] |
| Pancreatic cancer | Egfr targeting peptides | Type B gelatin | Plasmid DNA of green fluorescent protein | EGFR | EGFR targeting peptide | [36] |
| Pancreatic cancer | Targeting peptide (eppt1) | Iron oxide | siPLK1 | uMUC1 | EPPT1 | [37] |
| Pancreatic cancer | Active tumor targeting of m1 cell membrane | PPA, NHS,M1 macrophage membrane | siPD-L1,type I photosensitizer (PPA-Arg9) | - | - | [38] |
| Pancreatic cancer | Pegylated g5pamam dendrimer | mPEG-COOH | miR-21i, Gemcitabine | - | - | [39] |
| Pancreatic cancer | Non-toxic delivery carrier | PEG17-PPS80-SS-DP | rrsp-mrna | - | - | [40] |
| Pancreatic islet | Epha2 targeting peptide | cystamine bisacrylamide, diamino hexane | pCMV-RAE-1γ | EphA2/A4 | CHVLWSTRC | [41] |
| Pancreatic cancer | Dna aptamer specifically | Calcium phosphosilicate | AP1153 | CCKBR | G16/AP1153 | [42] |
| Pancreatic cancer | Optimize the composition formula | mPEG-PLGA, Lipid | HIF1 siRNA Gemcitabine | - | - | [43] |
| Pancreatic cancer | Optimize the composition formula | D,L-lactide-co-glycolide | miR-150 | - | - | [44] |
| Pancreatic cancer | Human tf fab’antibody | N3-PEG-PLL | anti-PLKA sirna | TF | human TF Fab’ antibody | [45] |
| Pancreatic cancer | Epha2 and cd13 target peptide | Arginine-rich fragment | anti-STAT3 sirna | EphA2 and CD13 | EphA2 and CD13 target peptide | [46] |
| Pancreatic cancer | Bionic exosomes | PEGylated lipids, DC2.4 cell membranes | Triptolide | - | - | [47] |
| Pancreatic cancer | Alternating magnetic field | Carboxymethyl-dextran | miRNA34a, Gemcitabine | - | - | [48] |
| Pancreatic cancer | RGD peptide | PEG, P(TMC-DTC), PEI | siKRAS | NRP-1 | cRGDpeptide | [49] |
| Pancreatic cancer | Effectively treated orthotopic pancreatic cancer in mice | PEIRs | MLKL mrna | - | - | [50] |
| Pancreatic cancer | Photodynamic therapy | HA-hemin, HA-Ce6, NF | HL-sgRNA | - | - | [51] |
| Pancreatic cancer | Transthyretin (ttr14) peptide | tetrahedral framework nucleic acid | CEBPA target saRNA | hTfR | tTR14 | [52] |
| Pancreatic cancer | Glucose transporter peptide | DOTAP, DSPE-PEOz, CHEMS | NF-κB inhibitorTPCA-1 and CD71-targeted Glut1 siRNA | CD71 | GLUT1 | [53] |
| Pancreatic cancer | PTP peptide | PEG-G2 dendritic molecules | TR3 siRNA, PTX | plectin-1 | PTP peptide | [54] |
EPC: egg phosphatidylcholine; SM: sphingomyelin; BPEI: branched polyethylenimine; DSPE: 1,2-distearoyl-sn-glycero-3-phosphoethanolamine; PEG: polyethylene glycol; AEAA: aminoethyl anisamide; SPC: soy phosphatidylcholine; DOPE: dioleoylphosphatidylethanolamine; BRD4: bromodomain-containing protein 4; IL: interleukin; miR: microRNA; PL-1: plectin-1 protein; EGFR: epidermal growth factor receptor; HIF: hypoxia-inducible factor; saRNA: small activating RNA; sgRNA: single-guide RNA; mRNA: messenger RNA; TF: tissue factor; uMUC1: underglycosylasted MUC1; PPA: pyropheophorbide A; NHS: N-hydroxysuccinimide; DP: dipalmitate; TMC: trimethylene carbonate; DTC: dithiolane trimethylene carbonate; NRP: neuropilin; RGD: Arg-Gly-Asp; MLKL: mixed lineage kinase domain-like protein; HA: hyaluronic acid; PTP: protein tyrosine phosphatases; PPS: poly(propylene sulfide); PLL: poly(L-lysine); epha2: ephrin type-a receptor 2.
The disadvantages of polymeric nanocarriers are also pressing issues requiring resolution. For instance, certain polymeric components may elicit innate immune responses, accelerate clearance kinetics and introduce risks of adverse reactions. Furthermore, the long-term fate of non-biodegradable or slowly degrading nanoparticles within pancreatic tissue remains poorly characterized, with potential risks of lysosomal dysfunction or chronic inflammation[55]. To overcome these challenges, naturally biodegradable polymers should be selected, and pancreatic-targeting moieties should be conjugated to enhance specific delivery to pancreatic tissue.
3.3 Pancreatic targeted lipid nanoparticle (LNP) delivery system
LNPs are advanced delivery systems designed to encapsulate and transport nucleic acids, utilizing a self-assembly process driven by microfluidic mixing of lipid-containing organic phases and RNA-containing aqueous phases (Figure 3). Their key advantages include high encapsulation efficiency, strong protection of RNA therapeutics, with their clinical utility well exemplified by COVID-19 mRNA vaccines. LNPs also hold promise for gene therapies and cancer immunotherapies. The pathophysiological features of PDAC, including dense desmoplasia, acidic hypoxia, CAF dominance, and profound immunosuppression, present formidable yet addressable challenges for therapeutic delivery. LNPs, with their precisely tunable physicochemical properties, pH-responsive activation mechanisms, facile surface functionalization for CAF targeting, and intrinsic immunostimulatory capacity, represent a technologically mature platform, ideally suited to exploit these tumor-specific vulnerabilities.
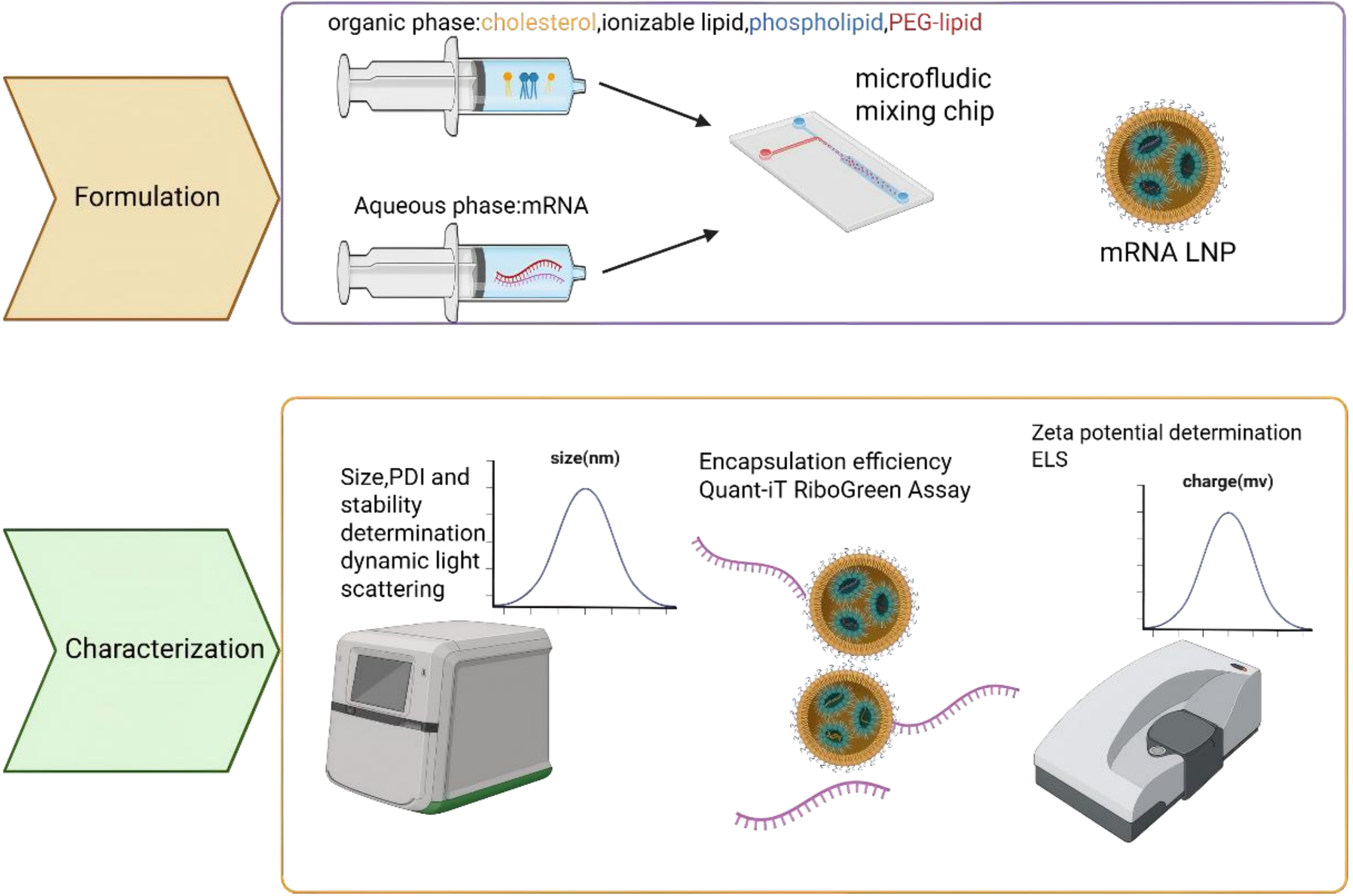{kind=link}
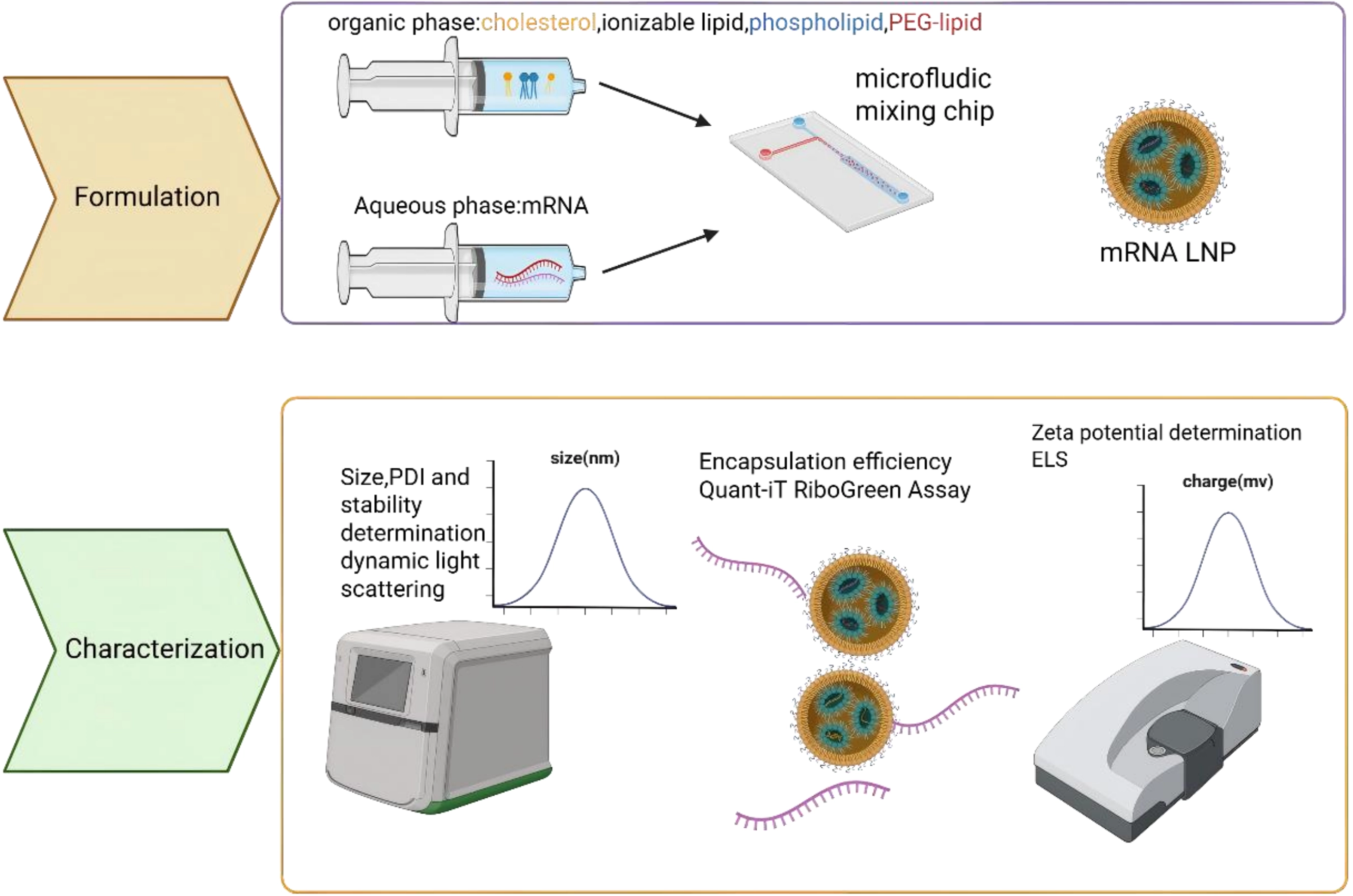
Figure 3. Flow chart of LNP encapsulated nucleic acid drugs. Formulation: After sequence design, the synthesized bare nucleotides are dissolved in the aqueous phase, while phospholipid molecules are dissolved in the organic phase. The two phases self-assemble into drug-loaded LNPs through a microfluidic device. Characterization: Examine size; PDI; stability; encapsulation efficiency; Zete potential of the formulated LNP. Created in BioRender. Chang, L. (2026) https://BioRender.com/3pu1b12. LNP: lipid nanoparticle; PDI: polydispersity index; PEG: polyethylene glycol; mRNA: messenger RNA; ELS: electrophoretic light scattering.
However, a major limitation of conventional LNPs is their strong hepatic tropism after intravenous injection, which restricts delivery to non-liver tissues such as the pancreas[56]. To improve pancreatic targeting (Table 3), strategies include optimizing lipid formulations (e.g., using novel cationic lipids like P6CIT or adjusting cholesterol content[57]), modifying surface chemistry with targeting ligands (e.g., tLyp-1 peptide for tumor homing or vitamins for enhanced pancreatic uptake[60]), and controlling particle size. For instance, smaller (~30 nm) LNPs improve general pancreatic distribution, while larger ones (~160 nm) favor islet retention[61].
Table 3. Related research on LNP delivery system.
| Component | Targeted delivery strategy | Payload | Receptor | Ligand | Ref |
| P6CIT, Phospholipid, PEG-lipid, cholesterol | Cationic lipid | IL-12 mRNA | - | - | [57] |
| 306Oi10/200Oi10/514O6,10, DOPE, cholesterol, C14-PEG2000 | Cationic lipid | Cy5-mLuc | - | - | [58] |
| DMG PEG, Cholesterol, Phospholipid, Ionizable lipid, Endogenous Vitamin | Add vitamin to LNP | Cre recombinase mRNA | VDR | Vitamin D3 | [59] |
| DSPE-PEG-Maleimide+tLyp-1, Tween-80, Cholesterol, DOPE, C12-200/cKK-E12 | LNP conjugate tlyp-1 | antiKRAS siRNA | NRP-1 | tLyp-1 | [60] |
| DOPC, Cholesterol, DSG-PEG2000 | Adjust the component ratio | - | - | - | [61] |
| DOPE, cholesterol, DMG-PEG2000, 4A2-B8-PH | New ionizable lipid:4A2-B8-PH | IL-12 mRNA | - | - | [62] |
LNP: lipid nanoparticle; mRNA: messenger RNA; IL: interleukin; DOPE: dilinoleoyl phosphatidylethanolamine; PEG: polyethylene glycol; VDR: vitamin D receptor; DSPE: 1,2-distearoyl-sn-glycero-3-phosphoethanolamine; siRNA: small interfering RNA; NRP: neuropilin; DOPC: 1,2-dioleoyl-sn-glycero-3-phosphocholine; DSG: distearoyl glycerol; DMG: dimyristoyl glycerol.
Although these methods have achieved pancreatic targeting, the targeting efficiency remains low. In particular, the delivery efficiency of intravenously administered pancreatic-targeting systems based on particle size modulation is less than 10%. How to achieve precise and efficient pancreatic targeting remains a technical barrier that needs to be overcome at this stage. Additionally, for strategies involving ligand-modified nanoparticles for pancreatic targeting, identifying suitable ligands has also become a significant challenge. The intricate multicomponent architecture of LNPs further increases the difficulty of formulation screening and optimization. Notably, several pioneering studies have demonstrated innovative strategies to achieve pancreatic targeting using LNPs. Ivan Isaac et al. reported a breakthrough approach by incorporating vitamins as a fifth component into the LNP formulation, leveraging vitamin D receptor-mediated endogenous targeting mechanisms to enable specific pancreatic delivery[59]. In parallel, Jillian R. Melamed et al. achieved pancreatic mRNA expression through intraperitoneal injection, circumventing the hepatic first-pass effect[58]. Jiaqi Lei et al. optimized pancreatic delivery by modulating LNP particle size, demonstrating that size-tuned LNPs administered via intraperitoneal route could efficiently penetrate pancreatic tissue[63]. Qiu et al. optimized an ionizable lipid (4A2-B8-PH) that achieves pancreatic mRNA expression[62]. These multifaceted approaches encompassing vitamin receptor targeting, alternative administration routes, and physicochemical optimization converge to enable clinically viable pancreatic mRNA delivery.
3.4 Viral vector delivery system
The utilization of viral vectors for nucleic acid drug delivery has several advantages. The primary advantage of viral vectors lies in their ability to target specific organs with a high degree of precision[64]. There are several methods to improve pancreatic targeting (Table 4). For instance, the serotype of a given recombinant adeno-associated virus (AAV) can be modified through the alteration of the cap gene within the packaging plasmid, thereby altering its tissue tropism or susceptible cell types[66]. As demonstrated in preceding studies, AAV6, AAV8, AAV9 and AAV pan exhibit a high degree of infectivity in the pancreas. Among these, AAV8 has the greatest prevalence, while AAV6 demonstrates optimal infectivity in the pancreatic duct[72]. Viral vectors possess the capacity for effective penetration of biological tissue, a property advantageous for their ability to traverse the BPB.
Table 4. Viral vector-based pancreatic delivery.
| Target | Virus | Payload | Targeted delivery strategy | Ref |
| Pancreas | Y447+y733f-aav8 | mCherry reporter gene | Tyrosine (Y) residues to phenylalanine (F) | [65] |
| Acinar and islet cells | Adenoviral vector | Secretable alkaline phosphatase reporter gene | Pancreatic-specific expression cassette | [66] |
| Pancreas | HSV-1 recombinant vector | Proenkephalin complementary DNA | HSV-1 recombinant vector | [67] |
| Pancreas | Double mutant coxsackievirus vector | GPL-1 | Double mutations coxsackievirus vector | [68] |
| Pancreatic islet cell | Bicistronic rAAV (serotype 2) vector | pTR-UF19 | Bicistronic raav (serotype 2) vector | [69] |
| Pancreas | Recombinant adenovirus vector | Escherichia coli LacZ gene | Recombinant adenoviral vector | [70] |
| Pancreas | A capsid-optimized AAV8 | hSPINK1 | AAV8 | [71] |
HSV: herpes simplex virus; AAV: adeno-associated virus; LacZ: beta-galactosidase.
Although viral vectors have achieved efficient pancreatic-targeted delivery, their relatively slow expression kinetics make them more suitable for long-term expression scenarios, failing to meet the requirements of mRNA for short-term, high-dose expression. Moreover, viral vectors face challenges in packaging long-chain mRNA or chemically modified mRNA molecules[73]. They also may trigger pre-existing immune responses (e.g., adenoviral vectors), leading to compromised delivery efficiency or adverse side effects.
3.5 Antibody delivery system
Antibodies have garnered significant recognition in the treatment of various diseases, particularly cancer, owing to their exceptional specificity, high affinity for target cell antigens, and prolonged circulation in the body.
With the further advancement of antibody-mediated RNA delivery technology, increasing researchers are turning their attention to the development of antibody-oligonucleotide conjugate (AOC) technology. AOC demonstrates enhanced pharmacokinetic properties and more precise biodistribution compared to traditional oligonucleotide therapy. It can be conjugated to various types of oligonucleotides, including siRNA and ASO. This flexibility allows researchers to design different AOC drugs according to different disease types[74]. Currently, clinical trials are underway for AOC drugs, showcasing their potential in therapeutic applications.
The latest research has discovered TMAB3, a tumor-targeting monoclonal antibody which exhibits robust cell penetration and the capacity to bind RNA. TMAB3 can form stable, non-covalent antibody/RNA complexes of a defined size, enabling highly specific and functional RNA delivery within tumors. Single-cell RNA sequencing and flow cytometry revealed that TMAB3/3p-hpRNA treatment evoked a robust antitumor immune response characterized by RIG-I activation and increased infiltration and activation of cytotoxic T cells. These findings establish that the TMAB3/RNA complex can deliver RNA payloads specifically to hard-to-treat tumor cells, achieving anti-tumor effects and providing an antibody-based platform to advance RNA therapy research in cancer patients[75].
4. Treatment Strategies for Pancreatic Cancer
4.1 Conventional treatment methods for pancreatic cancer
Current standard-of-care for pancreatic cancer encompasses cytotoxic chemotherapy, immunotherapy, and supportive interventions. Gemcitabine, fluorouracil, nab-paclitaxel, and paclitaxel constitute first-line antiproliferative agents. Oxaliplatin and irinotecan are reserved for advanced disease in combination protocols. Checkpoint inhibitors such as nivolumab and pembrolizumab reactivate antitumor immunity, though durable responses remain subtype specific. Pancreatic enzyme replacement therapy addresses exocrine insufficiency and improves quality of life. Oral capecitabine-based regimens offer palliative options for late-stage disease, yet variable bioavailability necessitates careful patient selection[76].
Although conventional therapies have achieved modest efficacy in pancreatic cancer treatment, chemotherapy is constrained by off-target toxicity, stromal barrier exclusion, and acquired drug resistance[76]. Immune checkpoint inhibitors demonstrate limited clinical benefit due to the immune-desert phenotype and immunosuppressive microenvironment[77]. Oral formulations suffer from variable bioavailability and heterogeneous patient responses. RNA therapeutics, as an emerging technology platform, offer substantial promise by enabling sequence-specific gene regulation, de novo protein synthesis, and rapid therapeutic adaptation, thereby presenting a transformative paradigm to overcome the limitations that have historically plagued pancreatic cancer management.
4.2 RNA-based therapeutic for the treatment of pancreatic cancer
4.2.1 mRNA therapeutics
mRNA therapy represents a therapeutic strategy wherein in vitro-transcribed mRNA is introduced into host cells to direct the transient production of therapeutic proteins[78]. Key structural components of synthetic mRNA ensure its stability and efficient translation: the 5’ cap protects against exonuclease degradation and initiates translation, while the optimized 5’ and 3’ UTRs enhance ribosome binding and prolong mRNA half-life[79]. A long poly-A tail further stabilizes the molecule and strengthens translational persistence[80].
The major advantage of mRNA drugs rests with their ability to utilize the host’s cellular machinery to produce complex or secreted proteins without risking genomic integration, enabling rapid and transient protein expression. This makes them highly versatile for treating various conditions. For example, cancer occurs when genes in normal cells mutate, leading to abnormal cell proliferation. Some unique proteins are produced on the surface of cancer cells, known as ‘tumor antigens’[81]. The design of mRNA vaccines is based on these tumor antigens. By delivering mRNA that encodes tumor antigens to human cells, the cells synthesize the antigens according to the mRNA’s instructions[82]. Once these antigens have been synthesized, immune cells recognize them and activate the immune system, triggering a series of immune reactions. T cells are activated and can recognize and attack cancer cells specifically[83], while B cells produce antibodies to further enhance the immune response. Additionally, the mRNA vaccine activates the immune system’s memory function, enabling it to attack cancer cells faster and more effectively if they are encountered again in the future, thereby preventing recurrence.
mRNA tumor vaccines fall into two categories. One category is individualized cancer vaccine, which recognizes specific antigens according to the tumor tissues of patients (Figure 4). mRNA technology is used to produce specific cancer antigens in vivo and activate the T-cell immune response to kill tumor cells. The other is the universal cancer vaccine, which uses mRNA technology to encode a broad spectrum of tumor-associated antigens and activate the immune system to kill tumor tissue[84]. The subsequent table will present recent advances in clinical (Table 5) and preclinical (Table 6) research on mRNA technologies for pancreatic cancer treatment.
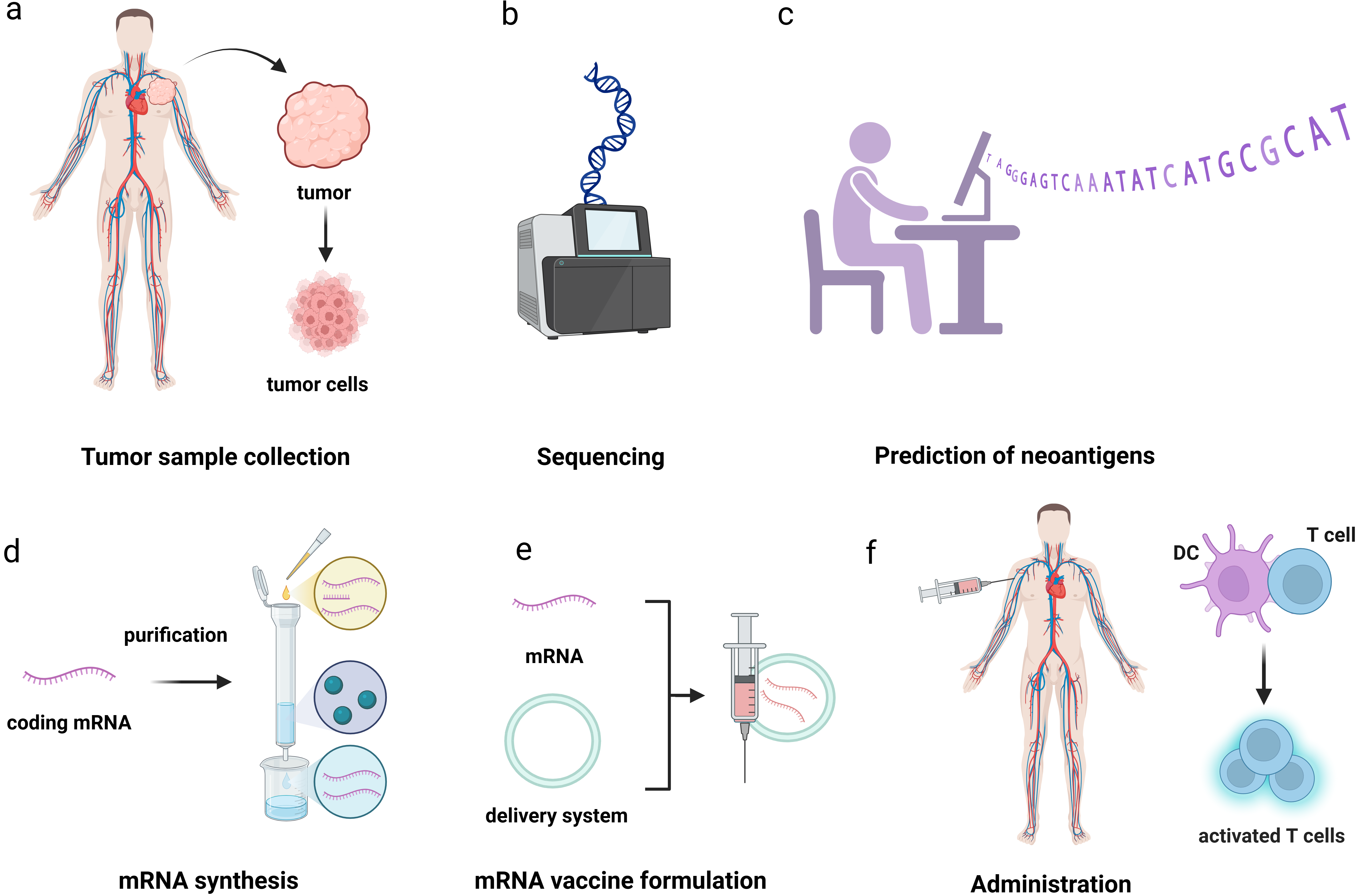{kind=link}
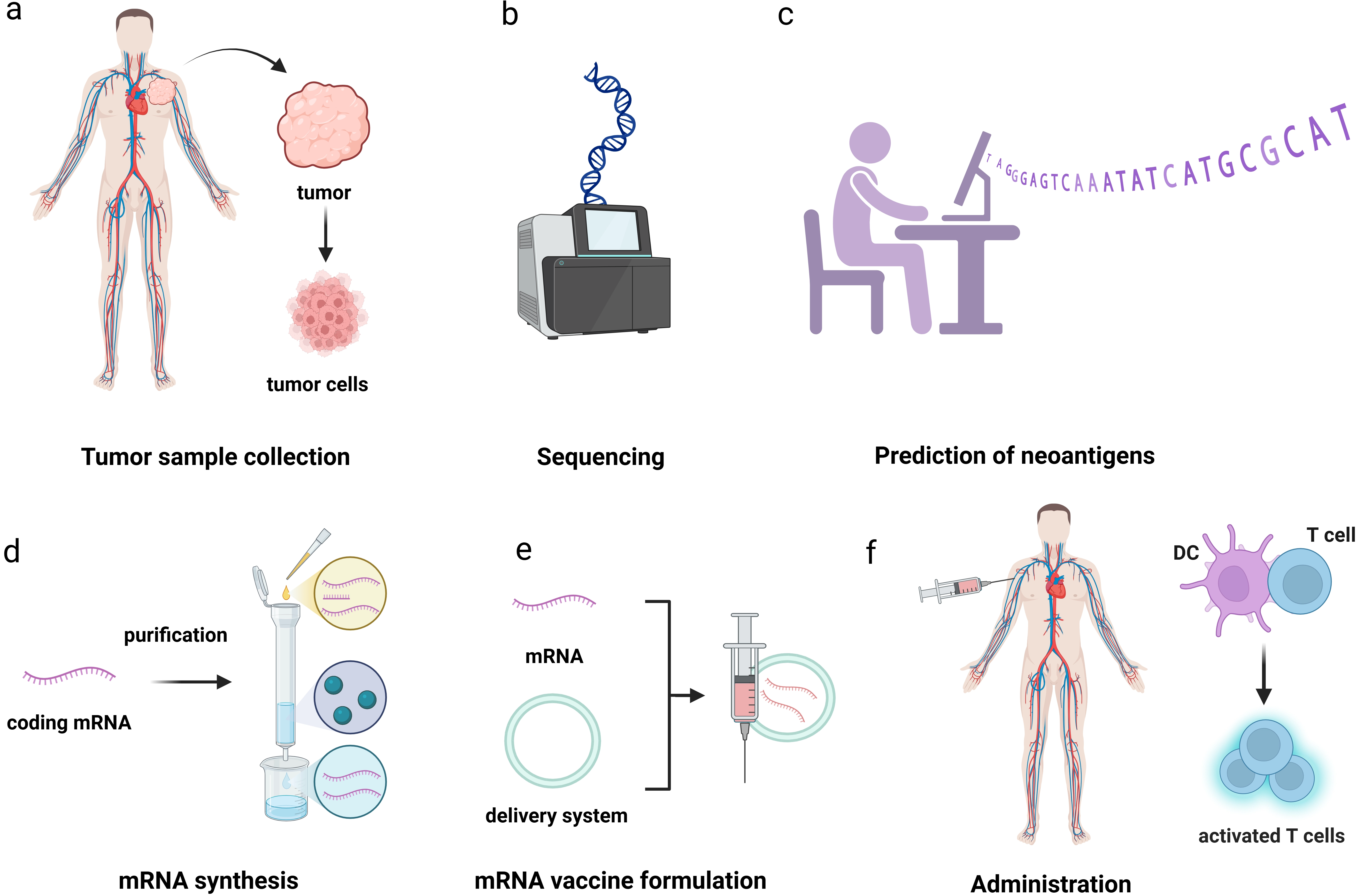
Figure 4. The workflow of PCV. (a) Collect tumor tissue samples and peripheral blood samples from patients; (b) Compare the DNA of tumors and normal cells to identify tumor specific non synonymous mutations; (c) Determine the patient’s HLA subtype through second-generation sequencing to ensure that the subsequently screened antigens can be recognized by the patient’s immune system; (d) Design mRNA sequences that can translate specific antigens through a certain codon encoding method; (e) Preparation of mRNA vaccines; (f) Activate the body’s immune response. Created in BioRender. Chang, L. (2026) https://BioRender.com/ygrqe4f. PCV: personalized cancer vaccine; mRNA: messenger RNA; HLA: human leukocyte antigen.
Table 5. Clinical trials on mRNA vaccine for cancer treatment.
| Pipeline | Catagory | Clinical | Clinical number | Enrollment | Start time | End time | Indication |
| BNT122 | PCV | II stage | NCT04486378 | 327 | 2021.03 | 2030.08 | colon cancer |
| LK101 | PCV | I stage (China/USA) | NCT06054932 | 18 | 2023.09 | 2026.03 | incurable advanced solid tumors |
| AK154 | PCV | I stage | NCT06913218 | 60 | 2025.07 | 2028.11 | PDAC |
| ABO2102 | universal cancer vaccine | I stage (China/USA) | NCT07455617 | 101 | 2026.02 | 2030.02 | KRAS Mutant solid tumor |
| EVM16 | PCV | I stage | NCT06541639 | 78 | 2025.03 | 2028.06 | solid tumor |
| EVM14 | universal cancer vaccine | I/IIa stage | NCT07095868 | 94 | 2025.11 | 2031.11 | NSCLC |
| mRNA-4157 | PCV | III stage | NCT03313778 | 161 | 2017.08 | 2027.11 | solid tumor |
mRNA: messenger RNA; PCV: personalized cancer vaccine; PDAC: pancreatic ductal adenocarcinoma; NSCLC: non-small cell lung cancer.
Table 6. Related research on mRNA therapy for pancreatic cancer.
| Antigen | Time | Therapy strategy | Ref |
| KRAS G12D | 2025 | STING agonists and KRAS tumor antigens mRNA | [85] |
| LYPFPLAL | 2025 | specific CD8+T cell epitopes (LYPFPLAL) | [86] |
| IL-12 | 2024 | IL-12 mRNA | [87] |
| CAFs | 2025 | reprogrammed M2 macrophages-FAP-CAR-M | [88] |
mRNA: messenger RNA; STING: stimulator of interferon genes; IL: interleukin; CAFs: cancer-associated fibroblasts.
Protein replacement therapy represents another widely employed strategy for pancreatic cancer treatment, among which cytokine supplementation constitutes a representative approach. Qiu et al. reported the treatment of pancreatic cancer via intraperitoneal injection of 4A2-B8-PH LNPs encapsulating IL-12 mRNA[62]. Once released into the TME, IL-12 binds to the its receptor on the surface of T cells and NK cells, activating downstream signaling pathways such as the JAK-STAT pathway. This activation promotes the proliferation, differentiation, and cytotoxicity of T cells (particularly CD8+ cytotoxic T lymphocytes) and NK cells. Anti-PD-1 antibodies are widely used clinically as immune checkpoint inhibitors. mRNA encoding PD-1 antibodies enables transient expression of PD-1 antibodies within host cells, blocking the interaction between PD-1 and PD-L1 and preventing T cell exhaustion, thereby achieving therapeutic efficacy in pancreatic cancer[89].
Furthermore, mRNA technology can be employed to weaken the fibrotic barrier on the surface of tumor tissue. Using a technique involving a mannose-modified mRNA-LNP, M2 macrophages can be reprogrammed in vivo into FAP-CAR-M. Following adoptive transfer, FAP-CAR-Ms infiltrate the desmoplastic stroma and mediate selective depletion of CAFs through antibody-dependent cellular phagocytosis, thereby alleviating physical compression of tumor vasculature and enhancing drug penetration. Concurrently, CAR-M polarization shifts toward pro-inflammatory M1 phenotypes, reversing immunosuppression and fostering T cell infiltration, thereby enhancing the efficacy of immunotherapy[88].
4.2.2 siRNA therapeutic
Structural characteristics of siRNA drugs: siRNA is composed of two complementary strands, the guide strand (antisense strand) and the passenger strand (sense strand). Its 3’ end has two hydroxylated nucleotide overhangs (such as 3’ Oh dinucleotide), enhancing stability and RNA induced silencing complex (RISC) recognition efficiency[90].
Mechanism of action of siRNA drugs: siRNA is loaded into RISC, in which argonaute2 protein unwinds the siRNA, degrades the passenger strand, and retains guide strand as targeting guide[91]. siRNA can not only guide RISC to cleave homologous single-stranded mRNA, but also serve as a primer to bind to target RNA and synthesize more dsRNA under the action of RNA-dependent RNA polymerase (RdRP)[92]. The newly synthesized dsRNA is then cleaved by Dicer, generating many secondary siRNA, thereby amplifying the effect of RNAi.
In the process of growth and metastasis of pancreatic cancer, there will be many genes with abnormal expression and upregulation. siRNA drugs can selectively silence these genes with high abnormal expression to play an anti-tumor role. The primary targets of these drugs concentrate on three types of pathways: the tumor proliferation and metastasis pathway, the tumor resistance pathway, and the immune pathway. For example, the following tables will introduce some related targets for siRNA therapeutic (Table 7) and clinical trials for pancreatic treatment (Table 8).
Table 7. Related research on siRNA target for pancreatic cancer.
| Target | Mechanism | Time | Ref |
| PKN3 | Promoting tumor cell migration and invasion. | 2008 | [93] |
| TGF-b | Promote metastasis through EMT | 2013 | [94] |
| Mcl-1 | Inhibit the Bax/Bak-mediated mitochondrial apoptosis pathway, thereby sustaining tumor cell survival | 2010 | [95] |
| Bcl-xL | Block the apoptotic signaling pathway | 2020 | [96] |
| Twist1 | Enhance tumor cell migration and invasion | 2025 | [97] |
| ABCA12 | Regulate lipid metabolism and drug efflux, influencing tumor cell survival and drug resistance | 2022 | [98] |
| PRC1 | Regulate mitotic spindle assembly and cell cycle progression, promoting tumor cell proliferation. | 2024 | [99] |
| RAP80 | Alter tumor cells’ sensitivity to chemotherapy and radiotherapy | 2012 | [100] |
| NS‑398 | Inhibit prostaglandin synthesis, blocking inflammatory signaling pathways | 2016 | [101] |
| survivin | Inhibit the activity of caspase-3 and caspase-7, apoptosis is blocked | 2009 | [102] |
| PLK1 | Promotes EMT and tumor metastasis | 2016 | [38] |
| EPAS1 | Promote angiogenesis and glycolysis, helping tumor cells adapt to hypoxic microenvironments | 2015 | [103] |
| NRF-2 | Activate antioxidant genes (such as HO-1 and NQO1) to eliminate ROS | 2024 | [104] |
| KRAS G12D | Activate the MAPK and PI3K pathways, promoting cell proliferation, migration and survival | 2023 | [105] |
| GFAT1 | Promote glycosylation modifications that activate EGFR and RAS signaling pathways, enhancing tumor cell proliferation and metastasis | 2025 | [106] |
| Mcl-1 | Inhibit the Bax/Bak-mediated mitochondrial apoptosis pathway, sustaining tumor cell survival | 2010 | [95] |
| CEACAM6 | Accelerates tumor progression by inhibiting apoptosis, promoting drug resistance and angiogenesis | 2025 | [107] |
| GP73 | Regulate cell cycle and apoptosis pathways, promoting tumor cell proliferation and metastasis | 2018 | [108] |
| GINS2 | Maintain replication fork stability, promotes tumor cell proliferation | 2020 | [109] |
| PHGDH | Promote rapid proliferation of tumor cells | 2019 | [110] |
| COX-2 | Accelerates tumor growth and metastasis by promoting inflammation and angiogenesis | 2012 | [111] |
EMT: epithelial-mesenchymal transition; ROS: reactive oxygen species; MAPK: mitogen-activated protein kinase; EGFR: epidermal growth factor receptor; RAS: rat sarcoma viral oncogene homolog family.
Table 8. Clinical trials on siRNA for cancer treatment.
| Pipeline | Clinical stage | Clinical number | Enrollment | Start time | End time | Target |
| STP707 | Phase I | NCT05037149 | 50 | 2021.11 | 2024.01 | TGF-b1+COX-2 |
| Sig12d LODER | Phase II | NCT01676259 | 80 | 2018.03 | 2023.08 | KRAS G12D |
| APN401 | Phase I | NCT03087591 | 11 | 2017.04 | 2020.12 | Cbl-b |
| CALAA-01 | Phase I | NCT00689065 | 24 | 2008.05 | 2012.09 | RRM2 |
| EphA2 | Phase I | NCT01591356 | 49 | 2015.07 | 2027.04 | EphA2 |
siRNA: siRNA; TGF: transforming growth factor.
4.2.3 ASO therapeutic
ASO therapy is a small-nucleic acid drug technology that regulates gene expression by specifically binding to target mRNA. The core mechanism of this process involves the formation of complementary base pairing between chemically modified oligonucleotide sequences and target RNA. A DNA-RNA hybrid duplex can be formed through complementary base pairing with specific sequences on the target mRNA[112]. The RNase H1 enzyme specifically cleaves the RNA part of the hybrid strand, resulting in the degradation of the target mRNA. The cleaved and degraded mRNA cannot be translated into protein, thereby downregulating the expressing of the targeted RNA[113].
ASO drugs, like siRNA drugs, can also silence the expression of targeted genes. However, the difference lies in that after siRNA degrades target mRNA through the RISC complex, RISC can repeatedly bind to new targets, extending the duration of drug efficacy[114]. In addition, the released sense strand of siRNA can serve as a primer to amplify dsRNA under the action of RdRP. After degradation by Dicer, more siRNA is generated, achieving a cascade amplification effect and maintaining the effect for 3-6 months[115]. ASO mainly degrades mRNA through RNase H1 or interferes with spatial steric hindrance, lacking a similar cyclic amplification mechanism; its effect is a single event with a half-life of only 2-4 weeks, and the target level tends to rebound after drug withdrawal[116]. ASO drugs targeting pancreatic tumors currently focus on three types of pathways: tumor growth and metastasis pathway, tumor drug resistance pathway, and immune evasion pathway. The following table would show some related research on ASO therapy (Table 9 and Table 10).
Table 9. Related research on pancreatic cancer ASO therapy.
| Target | Time | Mechanism | Ref |
| miR-21 | 2019 | Promote cancer proliferation and metastasis | [117] |
| Bcl-XL | 2006 | Maintain tumor drug resistance | [118] |
| APE/ref-1 | 2004 | Activate transcription factors to promote the survival of cancer cells | [119] |
| miR-21 | 2017 | Suppressing tumor suppressor genes | [120] |
| hTERT | 2009 | Maintain telomere length | [121] |
| STAT3 | 2021 | Regulate Bcl-XL, induce immune escape | [122] |
| K-ras/IGF-IR | 2008 | Inducing tumor drug resistance | [123] |
| gastrin mRNA | 1999 | Promote tumor cell proliferation | [124] |
| TGF-β2 | 2004 | Promote the proliferation and metastasis of tumor cells | [125] |
| K-ras | 2014 | Activate the MAPK pathway to inhibit tumor drug resistance | [126] |
| PKCα | 1998 | Promote tumor metastasis | [127] |
| amphiregulin | 1997 | Promote tumor metastasis | [128] |
| kinase 1 | 1998 | Promote tumor proliferation | [129] |
| gastrin mRNA | 2025 | Activate CCK2 receptors to promote tumor proliferation | [130] |
| hTERT | 2006 | Maintaining telomere length supports the survival of tumor cells | [131] |
ASO: antisense oligonucleotide; miRNA: microRNA; hTERT: human telomerase reverse transcriptase; TGF: transforming growth factor; PKC: protein kinase C; APE: Apurinic/apyrimidinic endonuclease; IGF: insulin-like growth factor.
Table 10. Clinical trials on ASO for cancer treatment.
| Pipeline | Clinical stage | Clinical number | Enrollment | Start time | End time |
| BP1002 | Phase I | NCT04072458 | 30 | 2020.11 | 2025.04 |
| BP1001-A | Phase I | NCT04196257 | 50 | 2022.08 | 2027.10 |
| OT-101 | Phase II/III | NCT06079346 | 455 | 2024.05 | 2027.06 |
| Danvatirsen | Phase I | NCT05986240 | 38 | 2024.05 | 2030.03 |
ASO: antisense oligonucleotide.
5. Prospects
Thanks to continuous breakthroughs in delivery technology, sequence screening and nucleic acid structure modification techniques, the field of RNA-based drugs is expanding beyond traditional viral vaccines to include areas such as cancer and metabolic diseases[132]. This article provides a comprehensive overview of the latest research on the use of nucleic acid drugs in treating pancreatic cancer. However, several issues still need to be addressed before pancreatic-targeted nucleic acid drugs are clinically translated. The production cost of exosomes is high, and their large-scale production remains difficult to achieve[133]. Nano polymers are not stable enough, and their long-term safety in the human body is unknown[134]. Although pancreatic-targeted liposomes improve drug targeting to the pancreas, production cost also increases[135]. Viral vectors can trigger human immune storms and inflammatory reactions[136]. Polymeric nanocarriers and LNPs enable precise, scalable manufacturing with well-defined quality attributes compared to exosomes and viral vectors. Polymeric systems offer exceptional chemical tunability, as monomer composition, molecular weight, and crosslink density can be precisely manipulated, whereas LNP lipid formulations are constrained by phase behavior and stability requirements. Nevertheless, LNPs demonstrate superior endosomal escape efficiency through the proton sponge mechanism and benefit from established clinical infrastructure, providing regulatory precedent and manufacturing scalability that polymer platforms currently lack.
Overcoming the BPB remains a critical challenge for effective RNA therapeutics in pancreatic diseases. The BPB comprises capillary endothelial cells, basement membranes, and pancreatic stellate cells that restrict macromolecular entry[137]. Nanoparticles can exploit small-size effects, typically below 200 nm, to penetrate intercellular tight junctions and breach this barrier. Surface functionalization with ligands targeting highly expressed pancreatic receptors (such as vitamin D), can further enhance nanoparticle tropism. In tumor-bearing contexts, pancreatic cancer cell-associated antibodies or acid-sensitive formulations can improve enrichment within the TME. Alternatively, intraperitoneal administration offers a promising strategy to bypass the BPB entirely and achieve direct pancreatic delivery[138].
Current research into nucleic acid drug therapy mostly focuses on single target. Multi-drug combination therapy strategies require further exploration. Due to the limitations of current delivery technologies, drug accumulation in other organs can easily lead to adverse reactions unrelated to therapeutic purpose. Future development should focus on enhancing drug targeting to the pancreas and exploring multi-target combination therapy.
6. Conclusions
siRNA, mRNA, and AOC have emerged as promising therapeutic tools for PDAC. However, the unique biological barriers of PDAC and inherent limitations of delivery systems have severely constrained their clinical applications. The distinctive biological barriers of PDAC present multi-layered challenges for nucleic acid delivery. The blood-pancreas barrier, characterized by tight junctions and selective permeability, restricts the delivery of nucleic acid drugs to pancreatic tumor sites. The immunosuppressive TME, featuring M2-type TAMs and MDSCs, further compromises the efficacy of immunomodulation-dependent nucleic acid therapeutics. Additionally, the dense desmoplastic stroma creates a physical barrier that impedes therapeutic agent penetration into the tumor parenchyma. Efficient delivery systems capable of overcoming PDAC barriers are essential for realizing the therapeutic potential of nucleic acid-based treatments in pancreatic cancer.
Existing nucleic acid delivery systems demonstrate varying capabilities in addressing PDAC-specific delivery challenges, each with distinct advantages and inherent limitations. Cell-derived exosomes, as natural intercellular communication vehicles, possess inherent biocompatibility and the ability to traverse biological barriers. However, the low yield of native exosomes and difficulties in large-scale production have hindered their clinical translation. Polymeric carriers and LNPs offer tunable physicochemical properties, and their tumor-targeting capability can be enhanced through the modification with ligands (such as peptides or antibodies) targeting PDAC-specific antigens. These carriers can protect nucleic acids from nuclease degradation in the circulation and TME. Yet their penetration into the dense PDAC stroma remains limited, and non-specific accumulation may lead to off-target toxicity. Viral vectors, despite their high transfection efficiency, face significant immunogenicity concerns in the immunosuppressive TME of PDAC, potentially triggering adverse immune responses and compromising long-term therapeutic efficacy.
To improve delivery efficiency, future strategies must integrate pathological insights of PDAC with targeted delivery system optimization. First, enhancing stromal penetration is critical. For instance, modifying delivery vehicles with enzymes such as hyaluronidase can degrade extracellular matrix components and reduce stromal density to facilitate carrier penetration into the tumor parenchyma. Second, precise tumor targeting can be achieved by functionalizing carriers with ligands that recognize PDAC-specific molecules. Alternatively, designing pH-sensitive or enzyme-responsive polymeric/LNPs enables specific triggering of nucleic acid release in the acidic TME or upon encountering stroma-specific enzymes, thereby improving intracellular drug bioavailability.
In conclusion, nucleic acid drug delivery for PDAC treatment faces unique challenges arising from tumor biological barriers and pathological complexity. Existing delivery systems have made progress in addressing specific barriers. However, further optimization is needed to enhance stromal penetration, tumor targeting, and intracellular bioavailability. Future research should focus on integrating pathological insights with innovative delivery technologies and personalized medicine approaches. With continuous advancements in delivery system design and deepening understanding of PDAC biology, nucleic acid therapeutics hold promise for overcoming the limitations of conventional therapies.
Authors contribution
Cong H: Writing-original draft, software, data curation.
Chang L: Writing-review & editing, software, data curation.
Li Y, Zou WB: Writing-review & editing, validation.
Xu C: Writing-review & editing, validation, funding acquisition.
Conflicts of interest
Congcong Xu is a Junior Executive Editor of BME Horizon. The other authors declare no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
This work was supported by the National Natural Science Foundation of China (Grant No. U25C2006), Jiangsu Funding Program for Excellent Postdoctoral Talent (Grant No. 2025ZB320), Academic Degree and Postgraduate Education Reform Project of Jiangsu Province (Grant No. KYCX24_3300), Science and Technology Program of Suzhou City (Grant No. SYW2025195), Jiangsu Province Youth Science and Technology Talent Support Program (Grant No. JSTJ-2025-128), Chronic Disease Management Research Project of National Health Commission Capacity Building and Continuing Education Center (Grant No. GWJJMB202510022107), GenScript Life Science Research Grant Program and Natural Science Foundation of Jiangsu Province (Grant No. SBK20250405084).
Copyright
© The Author(s) 2026.
References
-
2. Hull A, Li Y, Bartholomeusz D, Hsieh W, Allen B, Bezak E. Radioimmunotherapy of pancreatic ductal adenocarcinoma: A review of the current status of literature. Cancers. 2020;12(2):481.[DOI]
-
3. Jagrosse ML, Dean DA, Rahman A, Nilsson BL. RNAi therapeutic strategies for acute respiratory distress syndrome. Transl Res. 2019;214:30-49.[DOI]
-
6. Wang D, Wang S, Liu J, Shi X, Xiong T, Li R, et al. Nanomedicine penetrating blood-pancreas barrier for effective treatment of acute pancreatitis. Adv Sci. 2025;12(13):2413925.[DOI]
-
7. Ning Y, Lin K, Fang J, Ding Y, Zhang Z, Chen X, et al. Gastrointestinal pan-cancer landscape of tumor matrix heterogeneity identifies biologically distinct matrix stiffness subtypes predicting prognosis and chemotherapy efficacy. Comput Struct Biotechnol J. 2023;21:2744-2758.[DOI]
-
8. Li Y, Li F, Bai X, Li Y, Ni C, Zhao X, et al. ITGA3 is associated with immune cell infiltration and serves as a favorable prognostic biomarker for breast cancer. Front Oncol. 2021;11:658547.[DOI]
-
9. Gkretsi V, Stylianopoulos T. Cell adhesion and matrix stiffness: Coordinating cancer cell invasion and metastasis. Front Oncol. 2018;8:145.[DOI]
-
10. Fernando W, MacLean E, Monro S, Power Coombs MR, Marcato P, Rupasinghe HPV, et al. Phloridzin docosahexaenoate, an omega-3 fatty acid ester of a flavonoid precursor, inhibits angiogenesis by suppressing endothelial cell proliferation, migration, and differentiation. Biomolecules. 2024;14(7):769.[DOI]
-
13. Biffi G, Tuveson DA. Diversity and biology of cancer-associated fibroblasts. Physiol Rev. 2021;101(1):147-176.[DOI]
-
15. Cheng PSW, Zaccaria M, Biffi G. Functional heterogeneity of fibroblasts in primary tumors and metastases. Trends Cancer. 2025;11(2):135-153.[DOI]
-
16. Stott MC, Oldfield L, Hale J, Costello E, Halloran CM. Recent advances in understanding pancreatic cancer. Fac Rev. 2022;11:9.[DOI]
-
17. Gaggianesi M, Di Franco S, Pantina VD, Porcelli G, D’Accardo C, Verona F, et al. Messing up the cancer stem cell chemoresistance mechanisms supported by tumor microenvironment. Front Oncol. 2021;11:702642.[DOI]
-
18. Tschumperlin DJ, Lagares D. Mechano-therapeutics: Targeting mechanical signaling in fibrosis and tumor stroma. Pharmacol Ther. 2020;212:107575.[DOI]
-
19. Sneller L, Mathur K, Kottilil S, Mathur P. Hot and cold HCC: Uncoupling viral oncogenesis and therapy. Viruses. 2025;17(9):1255.[DOI]
-
20. Khalyfa A, Castro-Grattoni AL, Gozal D. Cardiovascular morbidities of obstructive sleep apnea and the role of circulating extracellular vesicles. Ther Adv Respir Dis. 2019;13:1753466619895229.[DOI]
-
22. Shi L, Zhou Y, Yin Y, Zhang J, Chen K, Liu S, et al. Advancing tissue damage repair in geriatric diseases: Prospects of combining stem cell-derived exosomes with hydrogels. Int J Nanomed. 2024;19:3773-3804.[DOI]
-
23. Zhou W, Zhou Y, Chen X, Ning T, Chen H, Guo Q, et al. Pancreatic cancer-targeting exosomes for enhancing immunotherapy and reprogramming tumor microenvironment. Biomaterials. 2021;268:120546.[DOI]
-
24. Kamerkar S, LeBleu VS, Sugimoto H, Yang S, Ruivo CF, Melo SA, et al. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature. 2017;546(7659):498-503.[DOI]
-
25. McAndrews KM, Xiao F, Chronopoulos A, LeBleu VS, Kugeratski FG, Kalluri R. Exosome-mediated delivery of CRISPR/Cas9 for targeting of oncogenic KrasG12D in pancreatic cancer. Life Sci Alliance. 2021;4(9):e202000875.[DOI]
-
26. Mendt M, Kamerkar S, Sugimoto H, McAndrews KM, Wu CC, Gagea M, et al. Generation and testing of clinical-grade exosomes for pancreatic cancer. JCI Insight. 2018;3(8):e99263.[DOI]
-
28. Creeden JF, Sevier J, Zhang JT, Lapitsky Y, Brunicardi FC, Jin G, et al. Smart exosomes enhance PDAC targeted therapy. J Control Release. 2024;368:413-429.[DOI]
-
29. Hsu SK, Jadhao M, Liao WT, Chang WT, Lin IL, Chiu CC. The role of exosomes in pancreatic ductal adenocarcinoma progression and their potential as biomarkers. Cancers. 2023;15(6):1776.[DOI]
-
31. Rosalba TPF, Matos GDR, Salvador CEM, Andrade CKZ. Rational design and multicomponent synthesis of lipid–peptoid nanocomposites towards a customized drug delivery system assembly. Molecules. 2023;28(15):5725.[DOI]
-
32. Zhang P, Guo H, Liu C. Fabrication of carboxylmethyl chitosan nanocarrier via self-assembly for efficient delivery of phenylethyl resorcinol in B16 cells. Polymers. 2020;12(2):408.[DOI]
-
40. Escher TE, Yuk SA, Qian Y, Qiang W, Almunif S, Sharma S, et al. Expression of a RAS degrader via synthetic nanocarrier-mediated mRNA delivery reduces pancreatic tumors. ACS Appl Bio Mater. 2025;8(6):4805-4814.[DOI]
-
41. Blevins KS, Jeong JH, Ou M, Brumbach JH, Kim SW. EphA2 targeting peptide tethered bioreducible poly(cystamine bisacrylamide–diamino hexane) for the delivery of therapeutic pCMV-RAE-1γ to pancreatic islets. J Control Release. 2012;158(1):115-122.[DOI]
-
43. Zhao X, Li F, Li Y, Wang H, Ren H, Chen J, et al. Co-delivery of HIF1α siRNA and gemcitabine via biocompatible lipid-polymer hybrid nanoparticles for effective treatment of pancreatic cancer. Biomaterials. 2015;46:13-25.[DOI]
-
44. Singh A, Arora S, Swaminathan SK, Kirtane A, Srivastava S, Bhardwaj A, et al. Synthesis, characterization, and evaluation of poly (D,L-lactide-co-glycolide)-based nanoformulation of miRNA-150: Potential implications for pancreatic cancer therapy. Int J Nanomed. 2014;2933.[DOI]
-
48. Lafuente-Gómez N, Martínez-Mingo M, Díaz-Riascos ZV, García-Prats B, de la Iglesia I, Dhanjani M, et al. Gemcitabine and miRNA34a mimic codelivery with magnetic nanoparticles enhanced anti-tumor effect against pancreatic cancer. J Control Release. 2025;383:113791.[DOI]
-
49. Huang R, Du H, Cheng L, Zhang P, Meng F, Zhong Z. Targeted nanodelivery of siRNA against KRAS G12D inhibits pancreatic cancer. Acta Biomater. 2023;168:529-539.[DOI]
-
52. Wang L, Yao Q, Guo X, Wang B, Si J, Wang X, et al. Targeted delivery of CEBPA-saRNA for the treatment of pancreatic ductal adenocarcinoma by transferrin receptor aptamer decorated tetrahedral framework nucleic acid. J Nanobiotechnol. 2024;22(1):392.[DOI]
-
54. Li Y, Wang H, Wang K, Hu Q, Yao Q, Shen Y, et al. Targeted co-delivery of PTX and TR3 siRNA by PTP peptide modified dendrimer for the treatment of pancreatic cancer. Small. 2017;13(2):1602697.[DOI]
-
55. Zhu J, Xia F, Wang S, Guan Y, Hu F, Yu F. Recent advances in nanomaterials and their mechanisms for infected wounds management. Mater Today Bio. 2025;31:101553.[DOI]
-
57. Shen Q, Liu J, Zeng L, Ren Y, Liao J, Chen S, et al. Pancreas-targeted lipid nanoparticles for relatively non-invasive interleukin-12 mRNA therapy in orthotopic pancreatic ductal adenocarcinoma. J Control Release. 2025;381:113588.[DOI]
-
60. Anthiya S, Öztürk SC, Yanik H, Tavukcuoglu E, Şahin A, Datta D, et al. Targeted siRNA lipid nanoparticles for the treatment of KRAS-mutant tumors. J Control Release. 2023;357:67-83.[DOI]
-
61. Oguma T, Kanazawa T, Kaneko YK, Sato R, Serizawa M, Ooka A, et al. Effects of phospholipid type and particle size on lipid nanoparticle distribution in vivo and in pancreatic islets. J Control Release. 2024;373:917-928.[DOI]
-
63. Lei J, Yang K, Cao W, Qi S, Du X, Li H, et al. Pancreatic-targeted lipid nanoparticles based on organ capsule filtration. Nature. 2026;652(8108):220-229.[DOI]
-
72. Quirin KA, Kwon JJ, Alioufi A, Factora T, Temm CJ, Jacobsen M, et al. Safety and efficacy of AAV retrograde pancreatic ductal gene delivery in normal and pancreatic cancer mice. Mol Ther Methods Clin Dev. 2018;8:8-20.[DOI]
-
76. Rebelo R, Polónia B, Santos LL, Vasconcelos MH, Xavier CPR. Drug repurposing opportunities in pancreatic ductal adenocarcinoma. Pharmaceuticals. 2021;14(3):280.[DOI]
-
77. Centonze G, Maisonneuve P, Mathian É, Grillo F, Sabella G, Lagano V, et al. Digital immunophenotyping of lung atypical carcinoids and large cell neuroendocrine carcinomas identifies three subtypes with specific tumor-immune microenvironment features. Endocr Pathol. 2025;36(1):39.
-
78. Pan X, Zhang YWQ, Dai C, Zhang J, Zhang M, Chen X. Applications of mRNA delivery in cancer immunotherapy. Int J Nanomed. 2025;20:3339-3361.[DOI]
-
79. Chaudhary N, Weissman D, Whitehead KA. mRNA vaccines for infectious diseases: Principles, delivery and clinical translation. Nat Rev Drug Discov. 2021;20(11):817-838.[DOI]
-
81. Haque S, Yellu M, Randhawa J, Hashemi-Sadraei N. Profile of pembrolizumab in the treatment of head and neck squamous cell carcinoma: Design development and place in therapy. Drug Des Devel Ther. 2017;11:2537-2549.[DOI]
-
82. Vohra J, Barbosa GR, Reis LO. mRNA and DNA-based vaccines in genitourinary cancers: A new frontier in personalized immunotherapy. Vaccines. 2025;13(9):899.[DOI]
-
83. Calabrò L, Bronte G, Grosso F, Cerbone L, Delmonte A, Nicolini F, et al. Immunotherapy of mesothelioma: The evolving change of a long-standing therapeutic dream. Front Immunol. 2024;14:1333661.[DOI]
-
84. Deng Z, Tian Y, Song J, An G, Yang P. mRNA vaccines: The dawn of a new era of cancer immunotherapy. Front Immunol. 2022;13:887125.[DOI]
-
86. Chang J, Zheng K, Li Y, Shan W, Zheng L, Mao HQ, et al. Preclinical development of the lipid nanoparticle-based mRNA vaccine with multiple natural T cell epitopes for pancreatic cancer. Cancer Res. 2025;85:848.[DOI]
-
88. Wang W, Hu K, Xue J, Chen J, Du X, Zhao T, et al. In vivo FAP-CAR macrophages enhance chemotherapy and immunotherapy against pancreatic cancer by removing the fibrosis barrier. J Control Release. 2025;384:113888.[DOI]
-
90. Montgomery RL, Yu G, Latimer PA, Stack C, Robinson K, Dalby CM, et al. microRNA mimicry blocks pulmonary fibrosis. EMBO Mol Med. 2014;6(10):1347-1356.[DOI]
-
91. Czarnecki O, Bryan AC, Jawdy SS, Yang X, Cheng ZM, Chen JG, et al. Simultaneous knockdown of six non-family genes using a single synthetic RNAi fragment in Arabidopsis thaliana. Plant Methods. 2016;12:16.[DOI]
-
92. Au JLS, Yeung BZ, Wientjes MG, Lu Z, Wientjes MG. Delivery of cancer therapeutics to extracellular and intracellular targets: Determinants, barriers, challenges and opportunities. Adv Drug Deliv Rev. 2016;97:280-301.[DOI]
-
95. Xiang G, Wang H, Chen K, Liu H. Adenovirus-mediated siRNA targeting Mcl-1 gene increases radiosensitivity of pancreatic carcinoma cells in vitro and in vivo. Surgery. 2010;147(4):553-561.[DOI]
-
96. Morimoto Y, Takada K, Takeuchi O, Watanabe K, Hirohara M, Hamamoto T, et al. Bcl-2/Bcl-xL inhibitor navitoclax increases the antitumor effect of Chk1 inhibitor prexasertib by inducing apoptosis in pancreatic cancer cells via inhibition of Bcl-xL but not Bcl-2. Mol Cell Biochem. 2020;472:187-198.
-
98. Zheng S, Liu D, Wang F, Jin Y, Zhao S, Sun S, et al. ABCA12 promotes proliferation and migration and inhibits apoptosis of pancreatic cancer cells through the AKT signaling pathway. Front Genet. 2022;13:906326.[DOI]
-
99. Lee H, Bae AN, Yang H, Lee JH, Park JH. Modulation of PRC1 promotes anticancer effects in pancreatic cancer. Cancers. 2024;16(19):3310.[DOI]
-
103. Pan X, Zhu Q, Sun Y, Li L, Zhu Y, Zhao Z, et al. PLGA/poloxamer nanoparticles loaded with EPAS1 siRNA for the treatment of pancreatic cancer in vitro and in vivo. Int J Mol Med. 2015;35(4):995-1002.[DOI]
-
105. Almeida C, Teixeira AL, Dias F, Morais M, Medeiros R. Extracellular vesicles as potential therapeutic messengers in cancer management. Biology. 2023;12(5):665.[DOI]
-
107. Kim H, Woo CG, Son SM, Lee YP, Kim HK, Yang Y, et al. Targeted suppression of CEACAM6 via pHLIP-delivered RNAs in pancreatic ductal adenocarcinoma. Medicina. 2025;61(4):598.[DOI]
-
110. Zheng M, Guo J, Xu J, Yang K, Tang R, Gu X, et al. Ixocarpalactone A from dietary tomatillo inhibits pancreatic cancer growth by targeting PHGDH. Food Funct. 2019;10(6):3386-3395.[DOI]
-
112. Le Gall L, Sidlauskaite E, Mariot V, Dumonceaux J. Therapeutic strategies targeting DUX4 in FSHD. J Clin Med. 2020;9(9):2886.[DOI]
-
113. Murphy AJ, Wilton SD, Aung-Htut MT, McIntosh CS. Down syndrome and DYRK1A overexpression: Relationships and future therapeutic directions. Front Mol Neurosci. 2024;17:1391564.[DOI]
-
114. Shivatare SS, Shivatare VS, Wong CH. Glycoconjugates: Synthesis, functional studies, and therapeutic developments. Chem Rev. 2022;122(20):15603-15671.[DOI]
-
115. Grassi G, Del Pinto R, Agabiti Rosei C, Agnoletti D, Borghi C, Cicero AFG, et al. Reduction of high cholesterol levels by a preferably fixed-combination strategy as the first step in the treatment of hypertensive patients with hypercholesterolemia and high/very high cardiovascular risk: A consensus document by the Italian society of hypertension. High Blood Press Cardiovasc Prev. 2022;29(2):105-113.
-
116. Caddeo A, Romeo S. Precision medicine and nucleotide-based therapeutics to treat steatotic liver disease. Clin Mol Hepatol. 2025;31:S76-S93.[DOI]
-
117. Ge JH, Zhu JW, Fu HY, Shi WB, Zhang CL. An antisense oligonucleotide drug targeting miR-21 induces H1650 apoptosis and caspase activation. Technol Cancer Res Treat. 2019;18:1533033819892263.[DOI]
-
122. Oweida AJ, Mueller AC, Piper M, Milner D, Van Court B, Bhatia S, et al. Response to radiotherapy in pancreatic ductal adenocarcinoma is enhanced by inhibition of myeloid-derived suppressor cells using STAT3 anti-sense oligonucleotide. Cancer Immunol Immunother. 2021;70(4):989-1000.[DOI]
-
124. Smith JP, Verderame MF, Zagon IS. Antisense oligonucleotides to gastrin inhibit growth of human pancreatic cancer. Cancer Lett. 1998;135(1):107-112.[DOI]
-
125. Stauder G, Bischof A, Egger T, Hafner M, Herrmuth H, Jachimczak P, et al. TGF-β2 suppression by the antisense oligonucleotide AP 12009 as treatment for pancreatic cancer: Preclinical efficacy data. J Clin Oncol. 2004;22:4106.[DOI]
-
126. Wang Y, Gao L, Shao Q. Apoptosis of human pancreatic carcinoma PC-2 cells by an antisense oligonucleotide specific to point mutated K-ras. Pathol Oncol Res. 2014;20(1):81-85.[DOI]
-
127. Denham DW, Franz MG, Denham W, Zervos EE, Gower WR, Rosemurgy AS, et al. Directed antisense therapy confirms the role of protein kinase C–α in the tumorigenicity of pancreatic cancer. Surgery. 1998;124(2):218-224.[DOI]
-
129. Gray PJ, Bearss DJ, Han H, Nagle R, Tsao MS, Dean N, et al. Identification of human polo-like kinase 1 as a potential therapeutic target in pancreatic cancer. Mol Cancer Ther. 2004;3(5):641-646.[DOI]
-
132. Han Q, Fu H, Chu X, Wen R, Zhang M, You T, et al. Research advances in treatment methods and drug development for rare diseases. Front Pharmacol. 2022;13:971541.[DOI]
-
133. Chen Y, Liu H, He Y, Yang B, Lu W, Dai Z. Roles for exosomes in the pathogenesis, drug delivery and therapy of psoriasis. Pharmaceutics. 2025;17(1):51.[DOI]
-
134. Shrestha B, Wang L, Brey EM, Uribe GR, Tang L. Smart nanoparticles for chemo-based combinational therapy. Pharmaceutics. 2021;13(6):853.[DOI]
-
135. Kinoshita R, Ishima Y, Chuang VTG, Watanabe H, Shimizu T, Ando H, et al. The therapeutic effect of human serum albumin dimer-doxorubicin complex against human pancreatic tumors. Pharmaceutics. 2021;13(8):1209.[DOI]
-
137. Yao Y, Ji P, Chen H, Ge J, Xu Y, Wang P, et al. Ferroptosis-based drug delivery system as a new therapeutic opportunity for brain tumors. Front Oncol. 2023;13:1084289.[DOI]
-
138. Chen Z, Deng X, Lu Y, Lu L, Qin Y, Qu H, et al. Role of miRNA-214-3p in cancer (Review). Oncol Rep. 2025;54(4):1-15.[DOI]
Copyright
© The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Publisher’s Note
Science Exploration remains a neutral stance on jurisdictional claims in published
maps
and institutional affiliations. The views expressed in this article are solely those
of
the author(s) and do not reflect the opinions of the Editors or the publisher.
Share And Cite



Science Exploration Style
Cong H, Chang L, Li Y, Zou WB, Xu C. Delivering RNA-based therapeutics for pancreatic cancer treatment: Current progress and challenges. BME Horiz. 2026;4:202609. https://doi.org/10.70401/bmeh.2026.0028
Tips
Copy completed.
Submit a Manuscript
Author Instructions
Cite this Article
Article Metrics
0
View
0
Download
Cited
Article Updates

- Abstract
- Keywords
- 1. Introduction
- 2. Challenges of Pancreatic Cancer Treatment
- 3. Pancreatic Targeted Delivery Strategy
- 4. Treatment Strategies for Pancreatic Cancer
- 5. Prospects
- 6. Conclusions
- Authors contribution
- Conflicts of interest
- Ethical approval
- Consent to participate
- Consent for publication
- Availability of data and materials
- Funding
- References
- Copyright
Science Exploration Style
Cong H, Chang L, Li Y, Zou WB, Xu C. Delivering RNA-based therapeutics for pancreatic cancer treatment: Current progress and challenges. BME Horiz. 2026;4:202609. https://doi.org/10.70401/bmeh.2026.0028
copy
Share Link
copy